分析个体两类状态的免疫差异、辅助确定个体状态的方法与流程
本发明属于生物检测领域,具体的,本发明涉及一种分析个体两类状态的免疫差异的方法、一种分析个体两类状态的免疫差异的装置、一种辅助确定个体状态的方法和一种辅助确定个体状态的装置。
背景技术:
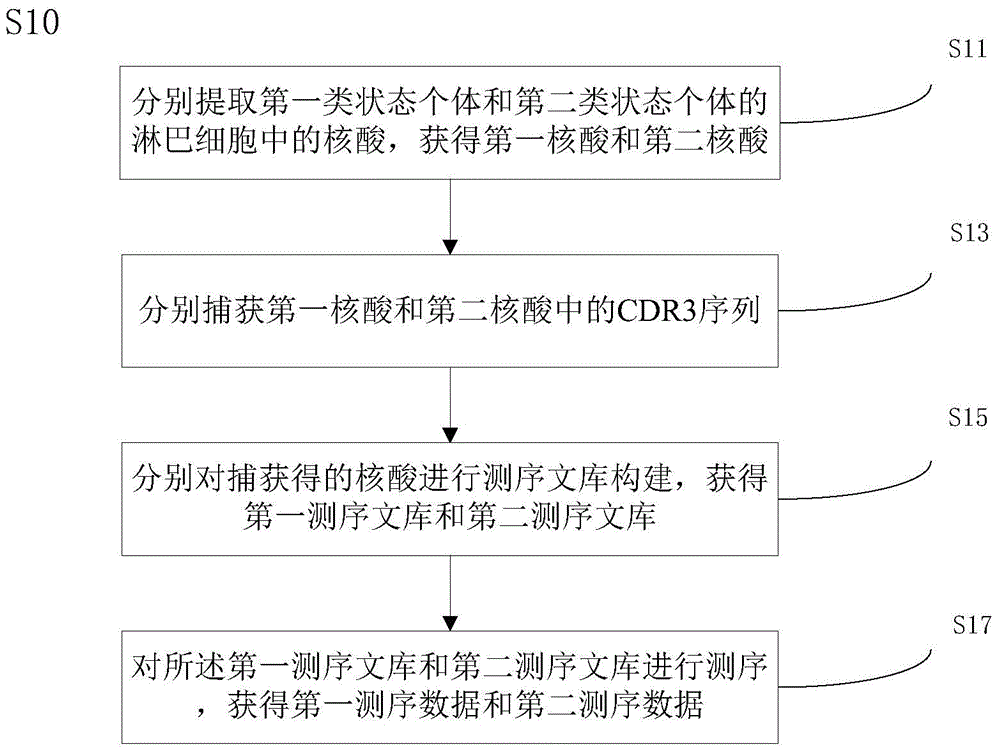
:结直肠癌(ColorectalCancer,CRC)属于消化道恶性肿瘤,是人类最常见的恶性肿瘤之一,近年来发病率呈明显上升趋势,其发病率和死亡率分别占我国恶性肿瘤的第三位和第四位。2008年我国新发结直肠癌病例22万例,死亡10.9万例。WHO最新数据显示,我国结直肠癌的发病率为14.2/10万人,死亡率6.9/10万人[McMillanDC,McArdleCS.Epidemiologyofcolorectallivermetastases.Surgoncol2007;l6(1):3-5]。结直肠癌远处转移是导致死亡的主要原因,而肝脏是结直肠癌最常见的,也常是唯一的转移部位。结直肠癌肝转移(Colorectallivermetastasis,CLM)的发生率可高达50%,即大约50%的结直肠癌患者在疾病过程中发生肝脏转移,其中15%~25%在结直肠癌确立诊断时已存在肝转移[WeberSM,JarnaginWR,DeMatteoRP,BlumgartLH,FongY.Survivalafterresectionofmultiplehepaticcolorectalmetastases.AnnSurgOncol2000;7(9):643-650]。目前我国筛查结直肠癌常见的初筛方案是进行大便隐血试验(fcecaloccultbloodtesting,FOBT)[LIEBERMANDA.Screening,surveillanceandpreventionofcolorectalcancer.GastrointestEndoscClinNAm2008;18:595-605],以肿瘤的伴随症状-出血为检测对象,但FOBT断的灵敏度和特异性差异较大,影响因素复杂。FOBT阳性待检者则进行结肠镜检查。结肠镜检查也是目前CRC最基本的诊断手段。通过放大结肠镜能够在活体情况下看到亚细胞结构,结合三维重建技术,可以判定病变的性质,其与活检病理诊断的符合率高达95%,但该检测手段持续时间较长,容易造成患者不适。尽管测序技术已经在肿瘤检测中得到了有效应用,但也主要基于一定数量的基因标志物进行检测,还没有研究是通过评估免疫组库受体库表达谱来对结直肠癌进行或者辅助进行诊断检测。免疫组库是指在任何指定时间,某个个体的循环系统中所有功能多样性B细胞和T细胞的总和。在机体的多种疾病进程中,都有免疫过程参与,而这些疾病特异性的免疫反应, 能被机体及时记录下来。通过检测这些表达的B细胞或T细胞受体基因,就能准确的将其反映出来,用来评估或者辅助评估个体的免疫状态,疾病的发生,发展和预后,甚至指导治疗。T细胞受体(Tcellreceptor,TCR)是T细胞表面特异性识别抗原和介导免疫应答的分子,是人类基因组中多态性最高的区域之一,决定着人的免疫系统如何适应环境的变化。T细胞受体库的多样性直接反映了机体免疫应答的状态。TCR可分为TCRα/β和TCRγ/δ两种类型,外周血T细胞主要为TCRα/β的T细胞,是介导机体特异性细胞免疫反应的主要细胞[DavisMM,BjorkmanPJ.T-cellantigenreceptorgenesandT-cellrecognition.Nature1988;334:395-402.;WangC,SandersCM,YangQ,eta1.HighthroughputsequencingrevealscomplexpatternofdynamicinterrelationshipsamonghumanTcellsubsets.ProcNatlAcadSciUSA,2010;107(4):1518-23.]。在T细胞发育过程中CDR3区由V、D和J进行重排而形成具有功能的TCR编码基因(T细胞克隆)。正常个体在无抗原刺激时,TCR基因重排是随机的,因此正常人外周T细胞呈多家族、多克隆性特点。不同抗原(如肿瘤)刺激后,TCRV区基因可对该抗原产生特异性识别,并使带有这类基因的T细胞得到优势扩增,可用于分析不同TCRV亚家族T细胞的表达和利用[WoodsworthDJ,CastellarinM,HoltRA.SequenceanalysisofT-cellrepertoiresinhealthanddisease.GenomeMed.2013;5(10):98.;KrangelMS.GenesegmentselectioninV(D)Jrecombination:Accessibilityandbeyond.NatImmunol2003;4:624–630.]。技术实现要素:本发明旨在至少解决上述问题之一或者提出一种商业选择手段。依据本发明的一方面,本发明提供一种分析个体两类状态的免疫差异的方法,包括:获取第一测序数据和第二测序数据,所述第一测序数据为第一类状态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第一读段,所述第二测序数据为第二类状态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第二读段,所述淋巴细胞基因组的至少一部分包括CDR3序列的至少一部分;分别对第一测序数据中的第一读段和第二测序数据中的第二读段进行拼接,获得第一拼接序列和第二拼接序列;将第一拼接序列和第二拼接序列分别与多种CDR3参考序列比对,获得第一CDR3序列和第二CDR3序列,所述多种CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;比较第一CDR3序列和第二CDR3序列中的各种VJ组合亚型的使用频率的差异,确定差异具有统计意义的VJ组合亚型对第一类状态和第二类状态的区分效果,第一CDR3 序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第一CDR3序列的种类数目与支持所有VJ组合亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第二CDR3序列的种类数目与支持所有VJ组合亚型的第二CDR3序列的种类总数的比值。所称的个体的两类状态可以是一个或者一群生物个体的不同时间点和/或不同空间位置的两类状态,也可以是不同个体或者不同群体在某个时间点和/或空间的各自的状态,这里的状态指免疫状态,包括核酸和/或氨基酸水平上反映出的生物体免疫状态。根据本发明的一个实施例,该方法中的第一测序数据和第二测序数据获取,包括:分别提取第一类状态个体和第二类状态个体的淋巴细胞中的核酸,获得第一核酸和第二核酸;分别捕获第一核酸和第二核酸中的CDR3序列;分别对捕获得的核酸进行测序文库构建,获得第一测序文库和第二测序文库;对所述第一测序文库和第二测序文库进行测序,获得第一测序数据和第二测序数据。在本发明的一个实施例中,所述捕获利用多重PCR实现。减少非目的区域例如非免疫相关区域数据的带入,利于提高目标区域分析效率。根据本发明的一个实施例,利用双末端测序获得成对读段,该方法中的第一测序数据包括多对第一读段对,每对第一读段对由两个第一读段组成,该方法中的第二测序数据包括多对第二读段对,每对第二读段对由两个第二读段组成。在该实施例中,所述拼接是依据有重叠的第一读段或第二读段,以及第一读段对或者第二读段对中一对读段对的两个读段之间的距离来进行的。拼接也称为组装,所得的拼接序列也称为重叠群(contigs)。根据本发明的一个实施例,所述多种CDR3参考序列包括V基因参考序列和J基因参考序列。所述将第一拼接序列和第二拼接序列分别与多种CDR3参考序列比对,包括:将所述第一拼接序列和第二拼接序列分别与所述多种CDR3参考序列进行比对,获得第一比对结果和第二比对结果,其中,所述第一比对结果包括能够与至少一种V基因参考序列和至少一种J基因参考序列都比对上的第一拼接序列,所述第二比对结果包括能够与至少一种V基因参考序列和至少一种J基因参考序列都比对上的第二拼接序列;基于所述第一比对结果,确定其中的第一拼接序列上的CDR3序列的起始位置,基于所述第二比对结果,确定其中的第二拼接序列上的CDR3序列的起始位置;分别将第一比对结果中的第一拼接序列上的CDR3序列起始位置之后的部分和第二比对结果中的第二拼接序列上的CDR3序列起始位置之后的部分与所述多种CDR3参考序列进行重新比对,获得第一重新比对结果和第二重新比对结果。在本发明的一个实施例中,上述重新比对的比对条件设置为:与所述V基因参考序列的TRB基因参考序列区进行所述重新比对所允许的错配碱基数为0,与所述V基因参考序列的IGH基因参考序列区进行所述重新比对所允许的错配碱基数为2, 和/或与所述J基因参考序列的TRB基因参考序列区进行所述重新比对所允许的错配碱基数为0,与所述J基因参考序列的IGH基因参考序列区进行所述重新比对所允许的错配碱基数为2。将拼接序列上的CDR3序列起始位置确定出,且以不同的比对条件例如相对更严格的比对条件将CDR3序列起始位置之后的部分进行重新比对,利于获得这些拼接序列的准确信息,利于提高后续基于这些contigs的免疫差异分析的准确性。根据本发明的一个实施例,在获得第一重新比对结果和第二重新比对结果后,还包括:分别对所述第一重新比对结果和所述第二重新比对结果进行过滤,以获得所述第一CDR3序列和所述第二CDR3序列,其中包括,分别去除第一重新比对结果和第二重新比对结果中的符合以下任一描述的拼接序列:其所属的CDR3序列种类的拼接序列支持数为1,即该种CDR3序列只包含这一条拼接序列,未能比对上V基因参考序列或者J基因参考序列,比对上所述CDR3参考序列的假基因参考序列区,比对上V基因参考序列和J基因参考序列、且比对上二者的方向相反,无法确定其上的CDR3的起始位置,含终止密码子或者不含开放阅读框。去除符合以上任意之一的contigs,去除这些contigs信息不明确、难以明确、无义、错误或者低可靠性的contigs的干扰,利于提高后续免疫差异分析的准确性和效率。根据本发明的一个实施例,利用ROC分析评估是否所述区分效果。ROC分析指ROC曲线(receiveroperatingcharacteristiccurve,接收者操作特征曲线),是一种二元分类模型,即输出结果只有两种类别的模型。考虑一个二分问题,即将实例分成正类(positive)或负类(negative),对一个二分问题来说,会出现四种情况:如果一个实例是正类并且也被预测成正类,即为真正类(Truepositive,TP),如果实例是负类被预测成正类,称之为假正类(Falsepositive,FP),相应地,如果实例是负类被预测成负类,称之为真负类(Truenegative,TN),正类被预测成负类则为假负类(falsenegative,FN)。TP:正确肯定的数目;FN:漏报,没有正确找到的匹配的数目;FP:误报,给出的匹配是不正确的;TN:正确拒绝的非匹配对数。在一个二分类模型中,对于所得到的连续结果,这边的连续结果指高频CDR3序列比例对多个第一类状态和第二类状态个体的分类结果,假设已确定差异具有统计意义的高频CDR3序列比例的阈值,比如说0.3,大于这个值的个体划归为第一类状态(正类),小于这个值则划到第二类状态(负类)。如果减小阈值,减到0.2,固然能识别出更多的第一类状态个体,也就是提高了识别出的正类占所有正类的比例,即TPR(truepositiverate,真正类率),但同时也将更多的负类当作了正类,即提高了FPR(falsepositiverate,负正类率)。为了形象化这一变化,引入ROC,ROC曲线可以用于评价一个分类器,即评价这一差异具有统计意义的高频CDR3序列比例的阈值。AUC(AreaUnderrocCurve)为ROC曲线下方的面积,AUC值介于0.5到1.0之间,AUC越大,分类器分类效果越好。根据本发明的一些实施例,该分析个体两类状态的免疫差异的方法还包括:比较第一CDR3序列和第二CDR3序列中的各种V亚型的使用频率的差异,确定差异具有统计意义的V亚型对第一类状态和第二类状态的区分效果,第一CDR3序列的V亚型的使用频率为支持该V亚型的第一CDR3序列的种类数目与支持所有V亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的V亚型的使用频率为支持该V亚型的第二CDR3序列的种类数目与支持所有V亚型的第二CDR3序列的种类总数的比值;和/或,比较第一CDR3序列和第二CDR3序列中的各种V合并亚型的使用频率的差异,确定差异具有统计意义的V合并亚型对第一类状态和第二类状态的区分效果,第一CDR3序列中的V合并亚型的使用频率为支持该V合并亚型的第一CDR3序列的种类数目与支持所有V合并亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的V合并亚型的使用频率为支持该V合并亚型的第二CDR3序列的种类数目与支持所有V合并亚型的第二CDR3序列的种类总数的比值;和/或,比较第一高频CDR3序列比例和第二高频CDR3序列比例的差异,确定差异具有统计意义且能够区分所述第一类状态和所述第二类状态的高频CDR3序列比例的数值范围,所述第一高频CDR3序列比例为所述第一CDR3序列的种类数中高频CDR3序列的种类数所占的比例,所述第二高频CDR3序列比例为所述第二CDR3序列的种类数中高频CDR3序列的种类数所占的比例,所述第一高频CDR3序列为在所述第一CDR3序列中频率不小于0.05%的CDR3序列,所述第二高频CDR3序列为在所述第二CDR3序列中频率不小于0.05%的CDR3序列。在该实施例中,通过扩增T细胞受体β链CDR3并进行高通量测序,对直肠癌患者及正常人组织和血液中的TCRβ链CDR3的多样性及特异性进行比较分析,发现使用血液样品就能对正常人和结直肠癌患者进行有效的区分。因此,检测待测者外周血TCRβ链CDR3的表达特征,可辅助结合临床用于直肠癌的无创早期诊断检测。需要说明的是,确定出的能够区分开第一类状态和第二类状态的V亚型、V合并亚型、VJ组合亚型和/或高频CDR3序列比例的范围能够作为区分开结直肠癌和健康人群的一个结直肠癌的免疫差异因素或者用于辅助判断个体属于哪一类状态,但仅依此还未能用于诊断确定个体是否为结直肠癌患者。根据本发明的一个实施例,所述第一高频CDR3序列为在所述第一CDR3序列中频率不大于0.5%的CDR3序列,第二高频CDR3序列为在所述第二CDR3序列中频率不大于0.5%的CDR3序列。增加对高频CDR3序列的频率的上限的限定,去除离群的高频CDR序列,使统计分析结果更具有意义。根据本发明的一个实施例,当第一类状态和第二类状态个体分别为结直肠癌和健康人群,确定差异具有统计意义的VJ组合亚型包括以下至少之一、至少之二、至少之三、至少 之四、至少之五、至少之六、至少之七、至少之八、至少之九或者全部十种:TRBV19&TRBJ1-1、TRBV25-1&TRBJ1-1、TRBV25-1&TRBJ1-4、TRBV25-1&TRBJ2-1、TRBV27&TRBJ1-4、TRBV6-2&TRBJ1-1、TRBV6-3&TRBJ1-1、TRBV6-6&TRBJ1-1、TRBV6-9&TRBJ1-1和TRBV7-9&TRBJ2-3。通过扩增T细胞受体β链CDR3并进行高通量测序,对结直肠癌患者及正常人血液中的TCRβ链CDR3的多样性及特异性进行研究,发现通过血液样品找到了一些健康人和结直肠癌患者存在明显的VJ使用差异。检测血液中TCRβ链CDR3多样性及特异性具有无创、可随时监控的特点,因此通过TCR-seq检测外周血TCRβ链CDR3的表达特征,可辅助临床检测,以辅助实现结直肠癌的早期诊疗。根据本发明的一个实施例,所述确定差异具有统计意义的V亚型对第一类状态和第二类状态的区分效果,包括:利用主成分分析方法(PrincipalComponentAnalysis,PCA)确定能够区分开第一状态和第二状态的V亚型,以及,利用ROC分析确定所述能够区分开第一状态和第二状态的V亚型对第一状态和第二状态的区分效果。PCA把原先的n个特征用数目更少的m个特征取代,新特征是旧特征的线性组合。CDR3V基因有几十个,将各个V基因称为V亚型或者V区基因,一般会得到的具有统计意义的多个V亚型,PCA能对高维数据进行降维,即得出权重较大的V亚型,权重较大的V亚型对分类起了主要作用,经过降维同时也除去了噪声。根据本发明的一个实施例,所述确定差异具有统计意义的V合并亚型对第一类状态和第二类状态的区分效果,包括:利用主成分分析方法确定能够区分开第一状态和第二状态的V合并亚型,以及,利用ROC分析确定所述能够区分开第一状态和第二状态的V合并亚型对第一状态和第二状态的区分效果。V合并亚型指合并的V区基因,例如,根据IMGT数据库(http://www.imgt.org/),48个V区基因片段可合并成23个进行分析,当获得的差异具有统计意义的V合并亚型有多个,利用PCA能够进行降维,确定主成分,即对分类起主要作用的V合并亚型。进行ROC分析,依据ROC曲线及其AUC值,能够评估分类器即主成分的分类效果。根据本发明的一个实施例,所述确定差异具有统计意义的VJ组合亚型对第一类状态和第二类状态的区分效果,包括:利用主成分分析方法确定能够区分开第一状态和第二状态的VJ组合亚型,以及,利用ROC分析确定所述能够区分开第一状态和第二状态的VJ组合亚型对第一状态和第二状态的区分效果。VJ组合亚型指V区基因和/或V合并亚型与J区基因的组合,当获得的差异具有统计意义的VJ组合亚型有多个,利用PCA能够进行降维,确定主成分,即确定对分类起主要作用的VJ组合亚型。而进行ROC分析,依据ROC曲线及其AUC值,能够评估分类器即主成分的分类效果。依据本发明的另一方面,本发明提供一种分析个体两类状态的免疫差异的装置,该装置可以用以实施上述本发明任一实施方式的分析个体两类状态的免疫差异的方法,装置包括:测序数据获取单元,用于获取第一测序数据和第二测序数据,所述第一测序数据为第一类状态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第一读段,所述第二测序数据为第二类状态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第二读段,所述淋巴细胞基因组的至少一部分包括CDR3序列的至少一部分;拼接单元,与所述测序数据获取单元连接,用于分别对第一测序数据中的第一读段和第二测序数据中的第二读段进行拼接,获得第一拼接序列和第二拼接序列;比对单元,与所述拼接单元相连,用于将第一拼接序列和第二拼接序列分别与多种CDR3参考序列比对,获得第一CDR3序列和第二CDR3序列,所述多种CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;免疫差异分析单元,与所述比对单元相连,用于比较第一CDR3序列和第二CDR3序列中的各种VJ组合亚型的使用频率的差异,确定差异具有统计意义的VJ组合亚型对第一类状态和第二类状态的区分效果,第一CDR3序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第一CDR3序列的种类数目与支持所有VJ组合亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第二CDR3序列的种类数目与支持所有VJ组合亚型的第二CDR3序列的种类总数的比值。本领域普通技术人员可以理解,通过对该装置增加相应功能单元或者子单元能够实现上述本发明任一具体实施方式的方法。前述对本发明任一具体实施方式中的分析个体两类状态的免疫差异的方法的技术特征和效果的描述,同样适用本发明的这一方面的装置,在此不再赘述。依据本发明的再一方面,本发明提供一种辅助确定个体状态的方法,该方法包括:提取待测个体的淋巴细胞中的核酸;对所述核酸中的CDR3序列进行捕获;对捕获得的核酸进行序列测定,获得测序结果,所述测序结果包括多个读段;对所述测序结果中的读段进行拼接,获得拼接片段;将所述拼接片段分别与多种CDR3基因参考序列进行比对,获得CDR3序列,所述CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;基于获得的CDR3序列,确定待测个体的各种VJ组合亚型的使用频率,所述VJ组合亚型的使用频率为支持该VJ组合亚型的CDR3序列的种类数目与支持所有VJ组合亚型的CDR3序列的种类总数的比值;比较所述VJ组合亚型的使用频率与其对应阈值的差异,以辅助确定个体状态,所述阈值的确定包括利用上述本发明任一具体实施方式中的分析个体两类状态的免疫差异的方法。所述与VJ组合亚型的使用频率对应的阈值即为上述的差异具有统计意义的VJ组合亚型及其各自的使用频率,这些组合亚型及各自的使 用频率数值范围能够用以区分开所述第一类状态和/或所述第二类状态。根据本发明的一些实施例,辅助确定个体状态的方法还包括:确定以下(a)-(c)至少之一:(a)CDR3序列中的各种V亚型的使用频率,所述V亚型的使用频率为支持该V亚型的CDR3序列的种类数目与支持所有V亚型的CDR3序列的种类总数的比值,(b)CDR3序列中的各种V合并亚型的使用频率,所述V合并亚型的使用频率为支持该V合并亚型的CDR3序列的种类数目与支持所有V合并亚型的CDR3序列的种类总数的比值,(c)高频CDR3序列比例,所述高频CDR3序列比例为所述CDR3序列种类总数中高频CDR3序列种类数所占的比例,所述高频CDR3序列为在所述CDR3序列中频率不小于0.05%的CDR3序列;比较所述确定的(a)-(c)至少之一与其对应阈值的差异,以辅助确定个体状态。前述对本发明一方面的分析个体两类状态的免疫差异的方法的技术特征和优点的描述,同样适用本发明这一方面的辅助确定个体状态的方法,在此不再赘述。依据本发明的又一方面,本发明提供一种辅助确定个体状态的装置,该装置可以实施上述本发明一方面的辅助确定个体状态的方法。该装置包括:核酸提取部,用于提取待测个体的淋巴细胞中的核酸;捕获部,与核酸提取部相连,用于对所述核酸中的CDR3序列进行捕获;测序部,与捕获部相连,用于对捕获得的核酸进行序列测定,获得测序结果,所述测序结果包括多个读段;拼接部,与测序部相连,用于对所述测序结果中的读段进行拼接,获得拼接片段;比对部,与拼接部相连,用于将所述拼接片段分别与多种CDR3基因参考序列进行比对,获得CDR3序列,所述CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;免疫因素确定部,与比对部相连,用于基于获得的CDR3序列,确定待测个体的各种VJ组合亚型的使用频率,所述VJ组合亚型的使用频率为支持该VJ组合亚型的CDR3序列的种类数目与支持所有VJ组合亚型的CDR3序列的种类总数的比值;差异比较部,与免疫因素确定部相连,用于比较所述VJ组合亚型的使用频率与其对应阈值的差异,以辅助确定个体状态,所述阈值的确定包括利用上述本发明任一具体实施方式中的分析个体两类状态的免疫差异的方法。本领域普通技术人员可以理解,通过对该装置增加相应功能单元或者子单元能够实现上述本发明任一具体实施方式的方法。前述对本发明一方面的辅助确定个体状态的方法的技术特征和优点的描述,同样适用本发明这一方面的装置,在此不再赘述。本发明提供基于T细胞受体和/或B细胞受体的高变区域CDR3测序数据,进行免疫相关分析、辅助确定个体状态的方法和/或装置,有效解决目前对免疫高通量数据分析及对鉴定出的CDR3区域进行后续分析的局限和匮乏。本发明提供了基于鉴定出的CDR序列的分析方案及分析手段,能够便于挖掘潜在可利用的生物信息,为免疫组库的临床应用与科学 研究提供助力。附图说明本发明的上述和/或附加的方面和优点从结合下面附图对实施方式的描述中将变得明显和容易理解,其中:图1是本发明一个实施例中的分析个体两类状态的免疫差异的方法的步骤示意图。图2是本发明一个实施例中的分析个体两类状态的免疫差异的方法的步骤示意图。图3是本发明一个实施例中的分析个体两类状态的免疫差异的装置示意图。图4是本发明一个实施例中的辅助确定个体免疫状态的方法的步骤示意图。图5是本发明一个实施例中的辅助确定个体免疫状态的装置示意图。图6是本发明一个实施例中的结直肠癌人群和健康人群的TRBV19&TRBJ1-1的使用频率差异示意图,其中,**表示p<0.01。图7是本发明一个实施例中的结直肠癌人群和健康人群的TRBV25-1&TRBJ1-1的使用频率差异示意图,其中,***表示p<0.001。图8是本发明一个实施例中的结直肠癌人群和健康人群的TRBV25-1&TRBJ1-4使用频率差异示意图,其中,***表示p<0.001。图9是本发明一个实施例中的结直肠癌组和健康组的差异显著的VJ组合亚型的使用频率的主成分分析结果示意图。具体实施方式下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中,自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。需要说明的,本文中所使用的术语“第一”、“第二”、“第一类”、“第二类”或者“第一部分”等仅为方便描述,不能理解为指示或暗示相对重要性,也不能理解为之间有先后顺序关系。在本发明的描述中,除非另有说明,“多个”的含义是两个或两个以上。在本文中,除非另有明确的规定和限定,术语“相连”、“连接”等术语应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通。如图1所示,依据本发明的一个实施例,提供一种分析个体两类状态的免疫差异的方法,该方法包括:S10获取第一测序数据和第二测序数据,所述第一测序数据为第一类状 态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第一读段,所述第二测序数据为第二类状态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第二读段,所述淋巴细胞基因组的至少一部分包括CDR3序列的至少一部分;S20分别对第一测序数据中的第一读段和第二测序数据中的第二读段进行拼接,获得第一拼接序列和第二拼接序列;S30将第一拼接序列和第二拼接序列分别与多种CDR3参考序列比对,获得第一CDR3序列和第二CDR3序列,所述多种CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;S40比较第一CDR3序列和第二CDR3序列中的各种VJ组合亚型的使用频率的差异,确定差异具有统计意义的VJ组合亚型对第一类状态和第二类状态的区分效果,第一CDR3序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第一CDR3序列的种类数目与支持所有VJ组合亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第二CDR3序列的种类数目与支持所有VJ组合亚型的第二CDR3序列的种类总数的比值。所称的个体的两类状态可以是一个或者一群个体的不同时间点和/或不同空间位置的两类状态,也可以是不同个体或者不同群体在某个时间点和/或空间的各自的状态,这里的状态指免疫状态,包括核酸和/或氨基酸水平上反映出的生物体免疫状态。免疫差异指核酸和/或氨基酸水平上反映出的免疫状态差异。频率指出现的次数的比例,不同种类的CDR3序列不同,一种CDR3序列至少包含一条拼接序列,即一种CDR3序列至少有一条拼接序列的支持,亦即至少有一条拼接序列比对上该种CDR3序列的参考序列,例如,有三种CDR3序列分别表示为A序列、B序列和C序列,如果A序列的拼接序列支持数有70条,B序列的拼接序列支持数有20条,C序列的拼接序列支持数有10条,则其中A序列的频率是70/(70+20+10),若定义超过50%的为高频CDR3序列,则高频CDR3序列的比例为1/3。所称的区分效果,包括区分开两类状态的准确率、精确度、特异性以及任意其它的可用以评估分类器分类效果的方法中的相关值。所称第一和第二测序数据是通过测序获得的,根据本发明的一个实施例,如图2所示,该方法中的S10第一测序数据和第二测序数据获取,包括:S11分别提取第一类状态个体和第二类状态个体的淋巴细胞中的核酸,获得第一核酸和第二核酸;S13分别捕获第一核酸和第二核酸中的CDR3序列;S15分别对捕获得的核酸进行测序文库构建,获得第一测序文库和第二测序文库;S17对所述第一测序文库和第二测序文库进行测序,获得第一测序数据和第二测序数据。文库的构建方法根据所选择的测序方法的要求进行,测序方法依据测序平台的不同可选择但不限于Illumina公司的Hisq2000/2500测序平台、LifeTechnologies公司的IonTorrent平台和单分子测序平台,测序方式可以选择单端测序,也可 以选择双末端测序,获得的下机数据是测读出来的片段,称为读段(reads)。在本发明的一个实施例中,所述捕获利用多重PCR实现,例如利用IMGT数据库中的已知CDR3序列自己设计或者委托设计合成多重引物或者使用市售试剂盒,利用这些引物使核酸中的CDR3序列富集,减少非目的区域例如非免疫相关区域数据的带入或比例,利于提高目标区域分析效率。根据本发明的一个实施例,利用双末端测序获得成对读段,该方法中的第一测序数据包括多对第一读段对,每对第一读段对由两个第一读段组成,该方法中的第二测序数据包括多对第二读段对,每对第二读段对由两个第二读段组成。在该实施例中,所述拼接是依据有重叠的第一读段或第二读段,以及第一读段对或者第二读段对中两个读段之间的距离来进行的。拼接也称为组装,组装可使用soapdenovo等软件进行,所得的拼接序列也称为重叠群(contigs)。所称比对可以利用已知比对软件,例如利用SOAP、BWA和TeraMap等使用或调整其默认参数进行。根据本发明的一个实施例,所述多种CDR3参考序列包括V基因参考序列和J基因参考序列,优选的,V基因参考序列包括全部各个V区基因参考序列,J基因参考序列包括全部各个J区基因参考序列。所称的参考序列指预先确定的序列,可以是预先获得的待测样本所属或者所包含的生物类别的任意参考模板,例如,若待测样本来源的个体为人类,参考序列可选择NCBI数据库提供的HG19,进一步地,也可以预先配置包含更多参考序列的资源库,例如依据待测样本来源个体的状态、地域等因素选择或是测定组装出更接近的序列作为参考序列。在本发明的一个实施例中,所述将第一拼接序列和第二拼接序列分别与多种CDR3参考序列比对,包括:将所述第一拼接序列和第二拼接序列分别与所述多种CDR3参考序列进行比对,获得第一比对结果和第二比对结果,其中,所述第一比对结果包括能够与至少一种V基因参考序列和至少一种J基因参考序列都比对上的第一拼接序列,所述第二比对结果包括能够与至少一种V基因参考序列和至少一种J基因参考序列都比对上的第二拼接序列;基于所述第一比对结果,确定其中的第一拼接序列上的CDR3序列的起始位置,基于所述第二比对结果,确定其中的第二拼接序列上的CDR3序列的起始位置;分别将第一比对结果中的第一拼接序列上的CDR3序列起始位置之后的部分和第二比对结果中的第二拼接序列上的CDR3序列起始位置之后的部分与所述多种CDR3参考序列进行重新比对,获得第一重新比对结果和第二重新比对结果。在本发明的一个实施例中,上述重新比对的比对条件设置为:与所述V基因参考序列的TRB基因参考序列区进行所述重新比对所允许的错配碱基数为0,与所述V基因参考序列的IGH基因参考序列区进行所述重新比对所允许的错配碱基数为2,和/或与所述J基因参考序列的TRB 基因参考序列区进行所述重新比对所允许的错配碱基数为0,与所述J基因参考序列的IGH基因参考序列区进行所述重新比对所允许的错配碱基数为2。依据拼接序列比对上参考序列的位置以及CDR3序列的特点,将拼接序列上的CDR3序列起始位置确定出,且以不同的比对条件例如相对更严格的比对条件将CDR3序列起始位置之后的部分进行重新比对,利于获得这些拼接序列的准确信息,利于提高后续基于这些contigs的免疫差异分析的准确性。根据本发明的一个实施例,在获得第一重新比对结果和第二重新比对结果后,还包括:分别对所述第一重新比对结果和所述第二重新比对结果进行过滤,以获得所述第一CDR3序列和所述第二CDR3序列,其中包括,分别去除第一重新比对结果和第二重新比对结果中的符合以下描述至少之一的拼接序列:其所属的CDR3序列种类的拼接序列支持数为1,即这种CDR3序列中只包含这一条拼接序列,这种CDR3序列可靠性低,未能比对上V基因参考序列或者J基因参考序列,比对上所述CDR3参考序列的假基因参考序列区,比对上一个V基因参考序列和一个J基因参考序列、且比对上二者的方向相反,无法确定其上的CDR3的起始位置,含终止密码子或者不含开放阅读框。所称的比对上,指在比对过程中一般对比对参数进行设置,例如设置一条拼接序列最多允许有s个碱基错配(mismatch),如设置为s≤3,若该拼接序列中有超过s个碱基发生错配,则视该序列无法比对到(比对上)参考序列。比对上假基因区的拼接序列对后续分析意义不大。比对上V基因参考序列和J基因参考序列、但比对上二者的方向相反的拼接序列多数是由于组装错误去除的,所说的方向可以以参考序列的方向为参照。去除以上这些contigs信息不明确、难以明确、无义、错误或者低可靠性的contigs的干扰,利于提高后续免疫差异分析的准确性和效率。根据本发明的一个实施例,利用ROC分析确定所说的区分效果。ROC分析指ROC曲线(receiveroperatingcharacteristiccurve,接收者操作特征曲线),是一种二元分类模型,即输出结果只有两种类别的模型。考虑一个二分问题,即将实例分成正类(positive)或负类(negative),对一个二分问题来说,会出现四种情况:如果一个实例是正类并且也被预测成正类,即为真正类(Truepositive,TP),如果实例是负类被预测成正类,称之为假正类(Falsepositive,FP),相应地,如果实例是负类被预测成负类,称之为真负类(Truenegative,TN),正类被预测成负类则为假负类(falsenegative,FN)。TP:正确肯定的数目;FN:漏报,没有正确找到的匹配的数目;FP:误报,给出的匹配是不正确的;TN:正确拒绝的非匹配对数。在一个二分类模型中,对于所得到的连续结果,这边的连续结果指VJ组合亚型的使用频率对多个第一类状态和第二类状态个体的分类结果,假设已确定差异具有统计意义的VJ组合亚型的使用频率的阈值,比如说0.3,大于这个值的个体划归为第一类状态(正 类),小于这个值则划到第二类状态(负类)。如果减小阈值,减到0.2,固然能识别出更多的第一类状态个体,也就是提高了识别出的正类占所有正类的比例,即TPR(truepositiverate,真正类率),但同时也将更多的负类当作了正类,即提高了FPR(falsepositiverate,假正类率)。为了形象化这一变化,引入ROC,ROC曲线可以用于评价一个分类器,即评价这一差异具有统计意义的VJ组合亚型的使用频率的阈值。AUC(AreaUnderrocCurve)为ROC曲线下方的面积,AUC值介于0.5到1.0之间,AUC越大,分类器分类效果越好。根据本发明的一个实施例,当第一类状态和第二类状态个体分别为结直肠癌和健康人群,确定差异具有统计意义的VJ组合亚型包括以下至少之一、至少之二、至少之三、至少之四、至少之五、至少之六、至少之七、至少之八、至少之九或者全部十种:TRBV19&TRBJ1-1(等同于TRBV19TRBJ1-1)、TRBV25-1&TRBJ1-1、TRBV25-1&TRBJ1-4、TRBV25-1&TRBJ2-1、TRBV27&TRBJ1-4、TRBV6-2&TRBJ1-1、TRBV6-3&TRBJ1-1、TRBV6-6&TRBJ1-1、TRBV6-9&TRBJ1-1和TRBV7-9&TRBJ2-3。通过扩增T细胞受体β链CDR3并进行高通量测序,对结直肠癌患者及正常人血液中的TCRβ链CDR3的多样性及特异性进行研究,发现通过血液样品找到了一些健康人和结直肠癌患者存在明显的VJ使用差异。检测血液中TCRβ链CDR3多样性及特异性具有无创、可随时监控的特点,因此通过TCR-seq检测外周血TCRβ链CDR3的表达特征,可辅助临床检测,以辅助实现结直肠癌的早期诊疗。在本发明的一个实施例中,确定出的能够区分出结直肠癌的差异VJ组合亚型的使用频率(%)为以下至少之一或者全部或者部分的组合,TRBV19&TRBJ1-1:0.01-0.20、TRBV25-1&TRBJ1-1:0.100.60、TRBV25-1&TRBJ1-4:0.01-0.20、TRBV25-1&TRBJ2-1:0.10-0.40、TRBV27&TRBJ1-4:0.10-0.50、TRBV6-2&TRBJ1-1:0.50-2.00、TRBV6-3&TRBJ1-1:0.50-2.00、TRBV6-6&TRBJ1-1:0.08-0.50、TRBV6-9&TRBJ1-1:0.06-0.30、TRBV7-9&TRBJ2-3:0.10-0.50。需要说明的是,这个(些)确定出的VJ组合亚型及其各自的使用频率的数值范围能够作为区分开结直肠癌和健康人群的一个免疫差异因素或者辅助判断个体属于哪一类状态,但难以直接作为区分结直肠癌与另一类非直肠癌患者的因素或范围,即仅依此还未能用于诊断判断个体是否为结直肠癌患者。根据本发明的一些实施例,该分析个体两类状态的免疫差异的方法还包括:比较第一CDR3序列和第二CDR3序列中的各种V亚型的使用频率的差异,确定差异具有统计意义的V亚型对第一类状态和第二类状态的区分效果,第一CDR3序列的V亚型的使用频率为支持该V亚型的第一CDR3序列的种类数目与支持所有V亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的V亚型的使用频率为支持该V亚型的第二CDR3序列的种 类数目与支持所有V亚型的第二CDR3序列的种类总数的比值;和/或,比较第一CDR3序列和第二CDR3序列中的各种V合并亚型的使用频率的差异,确定差异具有统计意义的V合并亚型对第一类状态和第二类状态的区分效果,第一CDR3序列中的V合并亚型的使用频率为支持该V合并亚型的第一CDR3序列的种类数目与支持所有V合并亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的V合并亚型的使用频率为支持该V合并亚型的第二CDR3序列的种类数目与支持所有V合并亚型的第二CDR3序列的种类总数的比值;和/或,比较第一高频CDR3序列比例和第二高频CDR3序列比例的差异,确定差异具有统计意义并且能够区分所述第一类状态和所述第二类状态的高频CDR3序列比例的数值范围,所述第一高频CDR3序列比例为所述第一CDR3序列的种类总数中高频CDR3序列种类数所占的比例,所述第二高频CDR3序列比例为所述第二CDR3序列的种类总数中高频CDR3序列种类数所占的比例,所述第一高频CDR3序列为在所述第一CDR3序列中频率不小于0.05%的CDR3序列,所述第二高频CDR3序列为在所述第二CDR3序列中频率不小于0.05%的CDR3序列。进一步比较两类状态个体的V亚型、V合并亚型的使用频率和/或高频CDR3序列的比例的差异,以进一步分析两类状态的免疫差异。在本发明的一个实施例中,第一高频CDR3序列为在所述第一CDR3序列中频率不大于0.5%的CDR3序列,第二高频CDR3序列为在所述第二CDR3序列中频率不大于0.5%的CDR3序列。增加对高频CDR3序列的频率的上限的限定,去除离群的高频CDR序列,使统计分析结果更具有意义。对应的,根据本发明的一些实施例,所述确定差异具有统计意义的V亚型对第一类状态和第二类状态的区分效果,包括:利用主成分分析方法(PrincipalComponentAnalysis,PCA)确定能够区分开第一状态和第二状态的V亚型,以及,利用ROC分析确定所述能够区分开第一状态和第二状态的V亚型对第一状态和第二状态的区分效果;当第一状态和第二状态分别为肝癌人群和正常人群时,利用PCA确定所述能够区分开第一状态和第二状态的主成分1包括的V亚型为TRBV18、TRBV4-1、TRBV4-2和TRBV6-9,这四个V亚型对这两状态的区分能力能够代表反映所有的差异具有显著性的V亚型对这两状态的区分能力的95%,或者利用PCA,确定所述能够区分开第一状态和第二状态的主成分1包括的V亚型为TRBV4-1、TRBV18和TRBV6-9,这三个V亚型能够代表反映所有的差异具有显著性的V亚型对这两状态的区分能力的90%;主成分分析(PCA)是多元统计分析中用来分析数据的一种方法,它是用一种较少数量的特征对样本进行描述以达到降低特征空间维数的方法,它的本质实际上是K-L变换。PCA把原先的n个特征用数目更少的m个特征取代,新特征是旧特征的线性组合。CDR3V基因有几十个,各个V基因也称为V亚型或者 V区基因,一般会得到的具有统计意义的多个V亚型,PCA能对高维数据进行降维,即得出权重较大(特征值)的V亚型,权重较大的V亚型对分类起了主要作用,经过降维同时也除去了噪声。在本发明的一个实施例中,TRBV18、TRBV4-1、TRBV4-2和TRBV6-9这四个V亚型的特征值占到所有确定出的V亚型的特征值之和的95%,可将这四个V亚型作为主成分,这里的特征值为PCA中的概念,若AX=λX,则称λ是矩阵A的特征值,X是对应的特征向量,可以这样理解:矩阵A作用在它的特征向量X上,仅仅使得X的长度发生了变化,缩放比例就是相应的特征值λ。根据本发明的一个实施例,所述确定差异具有统计意义的V合并亚型对第一类状态和第二类状态的区分效果,包括:利用主成分分析方法确定能够区分开第一状态和第二状态的V合并亚型,以及,利用ROC分析确定所述能够区分开第一状态和第二状态的V合并亚型对第一状态和第二状态的区分效果。V合并亚型指合并的V区基因,例如,根据IMGT数据库(http://www.imgt.org/),48个V区基因片段可合并成23个进行分析,当获得的差异具有统计意义的V合并亚型有多个,利用PCA能够进行降维,确定主成分,即对分类起主要作用的V合并亚型。进行ROC分析,依据ROC曲线及其AUC值,能够评估分类器即主成分的分类效果。根据本发明的一个实施例,所述确定差异具有统计意义的VJ组合亚型对第一类状态和第二类状态的区分效果,包括:利用主成分分析方法确定能够区分开第一状态和第二状态的VJ组合亚型,以及,利用ROC分析确定所述能够区分开第一状态和第二状态的VJ组合亚型对第一状态和第二状态的区分效果;当第一状态和第二状态分别为肝癌组织和肝癌旁组织,利用PCA降维确定出所述能够区分开第一状态和第二状态的主成分包括的VJ组合亚型为TRBV6-4&TRBJ1-1和TRBV6-4&TRBJ2-2,这两个VJ组合亚型能够反映代表所有的差异具有显著性的VJ组合亚型对这两状态的区分能力的95%。VJ组合亚型指V区基因和/或V合并亚型与J区基因的组合,当获得的差异具有统计意义的VJ组合亚型有多个,利用PCA能够进行降维,确定主成分,即确定对分类起主要作用的VJ组合亚型。而进行ROC分析,依据ROC曲线及其AUC值,能够评估分类器即主成分的分类效果。如图3所示,依据本发明的另一方面,本发明提供一种分析个体两类状态的免疫差异的装置100,该装置100可以用以实施上述本发明任一实施方式的分析个体两类状态的免疫差异的方法,装置100包括:测序数据获取单元10,用于获取第一测序数据和第二测序数据,所述第一测序数据为第一类状态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第一读段,所述第二测序数据为第二类状态个体的淋巴细胞基因组的至少一部分的序列测定数据,包括多个第二读段,所述淋巴细胞基因组的至少一部分包括CDR3 序列的至少一部分;拼接单元20,与所述测序数据获取单元10连接,用于分别对第一测序数据中的第一读段和第二测序数据中的第二读段进行拼接,获得第一拼接序列和第二拼接序列;比对单元30,与所述拼接单元20相连,用于将第一拼接序列和第二拼接序列分别与多种CDR3参考序列比对,获得第一CDR3序列和第二CDR3序列,所述多种CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;免疫差异分析单元40,与所述比对单元30相连,用于比较第一CDR3序列和第二CDR3序列中的各种VJ组合亚型的使用频率的差异,确定差异具有统计意义的VJ组合亚型对第一类状态和第二类状态的区分效果,第一CDR3序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第一CDR3序列的种类数目与支持所有VJ组合亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的VJ组合亚型的使用频率为支持该VJ组合亚型的第二CDR3序列的种类数目与支持所有VJ组合亚型的第二CDR3序列的种类总数的比值。在本发明的一些实施例中,免疫差异分析单元40还用于进行以下(a)-(c)至少之一:(a)比较第一CDR3序列和第二CDR3序列中的各种V亚型的使用频率的差异,确定差异具有统计意义的V亚型对第一类状态和第二类状态的区分效果,第一CDR3序列的V亚型的使用频率为支持该V亚型的第一CDR3序列的种类数目与支持所有V亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的V亚型的使用频率为支持该V亚型的第二CDR3序列的种类数目与支持所有V亚型的第二CDR3序列的种类总数的比值,(b)比较第一CDR3序列和第二CDR3序列中的各种V合并亚型的使用频率的差异,确定差异具有统计意义的V合并亚型对第一类状态和第二类状态的区分效果,第一CDR3序列中的V合并亚型的使用频率为支持该V合并亚型的第一CDR3序列的种类数目与支持所有V合并亚型的第一CDR3序列的种类总数的比值,第二CDR3序列中的V合并亚型的使用频率为支持该V合并亚型的第二CDR3序列的种类数目与支持所有V合并亚型的第二CDR3序列的种类总数的比值,(c)比较第一高频CDR3序列比例和第二高频CDR3序列比例的差异,确定差异具有统计意义的高频CDR3序列比例对第一类状态和第二类状态的区分效果,所述第一高频CDR3序列比例为所述第一CDR3序列的种类总数中高频CDR3序列种类数所占的比例,所述第二高频CDR3序列比例为所述第二CDR3序列的种类总数中高频CDR3序列种类数所占的比例,所述第一高频CDR3序列为在所述第一CDR3序列中频率不小于0.05%的CDR3序列,所述第二高频CDR3序列为在所述第二CDR3序列中频率不小于0.05%的CDR3序列。本领域普通技术人员可以理解,通过对该装置增加相应功能单元或者子单元能够实现上述本发明任一具体实施方式的方法。前述对本发明任一具体实施方式中的分析个体两类状态的免疫差异的方法的技术特征和效果的描述,同样适用本发明的这一方面的装置,在此不 再赘述。如图4所示,依据本发明的再一方面,提供一种辅助确定个体状态的方法,该方法包括步骤:S100提取待测个体的淋巴细胞中的核酸;S200对所述核酸中的CDR3序列进行捕获;S300对捕获得的核酸进行序列测定,获得测序结果,所述测序结果包括多个读段;S400对所述测序结果中的读段进行拼接,获得拼接片段;S500将所述拼接片段分别与多种CDR3基因参考序列进行比对,获得CDR3序列,所述CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;S600基于获得的CDR3序列,确定待测个体的各种VJ组合亚型的使用频率,所述VJ组合亚型的使用频率为支持该VJ组合亚型的CDR3序列的种类数目与支持所有VJ组合亚型的CDR3序列的种类总数的比值;S700比较所述VJ组合亚型的使用频率与其对应阈值的差异,以辅助确定个体状态,所述阈值的确定包括利用上述本发明任一具体实施方式中的分析个体两类状态的免疫差异的方法,阈值即为上述确定出的数值范围或者为数值范围的上下限。在本发明的一些实施例中,该方法的S600还包括进行以下(1)-(3)至少之一:(1)CDR3序列中的各种V亚型的使用频率,所述V亚型的使用频率为支持该V亚型的CDR3序列的种类数目与支持所有V亚型的CDR3序列的种类总数的比值,(2)CDR3序列中的各种V合并亚型的使用频率,所述V合并亚型的使用频率为支持该V合并亚型的CDR3序列的种类数目与支持所有V合并亚型的CDR3序列的种类总数的比值,(3)高频CDR3序列的比例,所述高频CDR3序列的比例为高频CDR3序列种类数目在所述CDR3序列种类总数中所占的比例,所述高频CDR3序列为在所述CDR3序列中频率不小于0.05%的CDR3序列;相应的,S700还包括比较S600中确定的(1)-(3)的至少之一与其相应阈值的差异,以辅助确定个体状态。前述对本发明一方面的分析个体两类状态的免疫差异的方法的技术特征和优点的描述,同样适用本发明这一方面的辅助确定个体状态的方法,在此不再赘述。如图5所示,依据本发明的又一方面,提供一种辅助确定个体状态的装置1000,该装置1000可以实施上述本发明一方面的辅助确定个体状态的方法。该装置1000包括:核酸提取部100,用于提取待测个体的淋巴细胞中的核酸;捕获部200,与核酸提取部100相连,用于对所述核酸中的CDR3序列进行捕获;测序部300,与捕获部200相连,用于对捕获得的核酸进行序列测定,获得测序结果,所述测序结果包括多个读段;拼接部400,与测序部300相连,用于对所述测序结果中的读段进行拼接,获得拼接片段;比对部500,与拼接部400相连,用于将所述拼接片段分别与多种CDR3基因参考序列进行比对,获得CDR3序列,所述CDR3参考序列包括V基因参考序列、D基因参考序列和J基因参考序列中的至少两种;免疫因素确定部600,与比对部500相连,用于基于获得的CDR3序列, 确定待测个体的各种VJ组合亚型的使用频率,所述VJ组合亚型的使用频率为支持该VJ组合亚型的CDR3序列的种类数目与支持所有VJ组合亚型的CDR3序列的种类总数的比值;差异比较部700,与免疫因素确定部600相连,用于比较所述VJ组合亚型的使用频率与其相应阈值的差异,以辅助确定个体状态,所述阈值的确定包括利用上述本发明任一具体实施方式中的分析个体两类状态的免疫差异的方法。在本发明的一些实施例中,免疫因素确定部600还用于进行以下(i)-(iii)至少之一:(i)CDR3序列中的各种V亚型的使用频率,所述V亚型的使用频率为支持该V亚型的CDR3序列的种类数目与支持所有V亚型的CDR3序列的种类总数的比值,(ii)CDR3序列中的各种V合并亚型的使用频率,所述V合并亚型的使用频率为支持该V合并亚型的CDR3序列的种类数目与支持所有V合并亚型的CDR3序列的种类总数的比值,(iii)高频CDR3序列比例,所述高频CDR3序列比例为所述CDR3序列种类总数中高频CDR3序列种类数所占的比例,所述高频CDR3序列为在所述CDR3序列中频率不小于0.05%的CDR3序列;相应的,差异比较部700还用于比较所述(i)-(iii)至少之一与其对应阈值的差异,以辅助确定个体状态。前述对本发明一方面的辅助确定个体状态的方法的技术特征和优点的描述,同样适用本发明这一方面的装置,在此不再赘述。为了使本发明技术方案及优点更加清楚明白,以下结合实施例对本发明的分析个体两类状态的免疫差异的方法和/或装置、辅助确定个体免疫状态的方法和/或装置进行详细的描述。应当理解,下面示例用于解释本发明,不是对本发明的限制。需要说明的是在本文中所使用的术语“第一”、“第二”等仅为方便描述,不能理解为指示或暗示相对重要性,也不能理解为之间有先后顺序关系。在本发明的描述中,除非另有说明,“多个”的含义是两个或两个以上。除另有交待,以下实施例中涉及的未特别交待的试剂、序列(接头、标签和引物)、软件及仪器,都是常规市售产品或者开源的,例如购买Illumina的测序文库构建试剂盒。实施例一一般方法,包括:首先,对CDR3进行测序与鉴定:用淋巴细胞分离液分离外周血T/B淋巴细胞,提取DNA(或RNA),采用多重PCR或者5'RACE对CDR3进行捕获,通过Hiseq2000或Hiseq2500或Miseq平台进行高通量测序。对所测数据进行质控后比对到IMGT数据库(http://www.imgt.org/),确定其CDR3序列。其次,对免疫结果的分析:高频CDR3序列即为高增殖克隆(highlyexpandedclone),定义HEC比率——高增殖克隆比率(highlyexpandedclone-rate,HECrate)为频率超过0.05%,较佳的,频率不超过0.5%的CDR3的种类数目占CDR3种类总数的比例。对差异使用的V亚型、V合并亚型(Vmerge)和/或VJ组合亚型进行PCA分析。涉及的细节与步骤如下:常规统计量部分说明:1、CDR3丰度,通过测序出的免疫数据,质控纠错后通过比对软件与IMGT网站免疫参考序列进行比对,确定CDR3支持的reads数(支持CDR3的reads即为比对上该CDR3的reads),并计算出每种CDR3克隆所占比率。2、CDR3长度,即对鉴定出的CDR3序列长度进行统计。3、VJ使用(VJ组合亚型使用频率),即通过对确定的CDR3序列所比对上的VJ情况进行VJ连用的所占比率。单独统计V亚型或者J亚型使用频率。4、HECrate,统计分析高频CDR3序列的丰度(如0.1%~0.5%)占总体序列的种类数的比率达到某个阈值或落入某个范围。具体分析内容说明:1.HECrate比较统计频率超过0.1%(或者0.1%~0.5%)的CDR3占UniqueCDR3(CDR3种类)总数的比例。用T检验等检验两组个体之间是否存在差异,例如检验某疾病组与正常组之间是否存在差异。2.V、J亚型分析2.1V亚型以及VJ组合亚型关联分析统计不同V亚型下样本的相对丰度,并对疾病组和对照组样本进行T检验、Wilcox检验等,来找到P值<0.01的V亚型。或者依据不同V亚型区分疾病组和对照组的最小错误率,找出最小错误率最低的V亚型,这些V亚型即有可能与研究目的相关。或者对训练集挑选出的相关亚型在测试集中进行ROC分析并计算AUC值,对于区分效果明显者亦可使用全部亚型进行区分,不进行P值挑选。VJ使用或V合并亚型分析类似。2.2对V亚型或VJ亚型进行PCA分析统计不同V亚型下样本的相对丰度,然后用PCA(主成分分析)的方法算出各个样本的第一主成分和第二主成分的值作图,看是否有疾病组和对照组的分开聚集现象,如是否使两类状态达到线性可分。如果某个主成分可以很好的区分疾病组和对照组,对训练集找出有差异的V亚型,在测试集中进行验证,并对测试集进行ROC分析并计算AUC值。多次随 机抽取训练集与测试集,求出AUC均值,以判断挑选出的亚型在疾病差别中是否稳定。VJ组合亚型,合并V型同理分析。通过此方法,可寻找不同指标来对人群进行区分,进而可找出或者辅助找出某此疾病潜在的Bio-mark,利于达到无创检测目的,亦有利于辅助对疾病的治疗进行预后的监控。由于免疫反应的特性,免疫的研究对早期检测可能优于现有技术水平,对免疫数据的积累,后期可能达到一次测序,检查多项疾病的目的,能极大的提高人民健康水平。实施例二以T淋巴细胞为研究目标,采用优化的多重PCR的技术对T细胞受体β链最具多样性的互补决定区CDR3区进行扩增,扩增引物、扩增方法、文库构建测序等可按照CN103205420A中描述的进行,获得下机数据,全面分析TCR组成,评估免疫系统的多样性,挖掘免疫组库与肝癌、肝炎、直肠癌的发生和发展的关系信息。该方法包括如下步骤:(一)根据T细胞受体CDR3序列,设计Vsegment和Jsegment引物如CN103205420A,以及参考序列构建,包括从数据库中获得已知CDR3序列集合。(二)样本制备1.抽取待检者外周血5mL,存于EDTA抗凝管中,使用Ficoll淋巴细胞分离液在3h内进行外周血PBMC分离;2.trizol法提取总RNA;3.RNA定量检测;(三)文库制备及测序1.RNA逆转录为cDNA;2.多重PCR扩增T细胞受体β链CDR3序列,切胶回收目的片段;3.对T细胞受体β链CDR3片段进行末端修复;4.对T细胞受体β链CDR3片段末端加A;5.连接接头(Adapter);6.连接产物PCR扩增;7.连接产物磁珠纯化;8.文库定量及质控;9.IlluminaHiSeq2500/2000上机测序;(四)下机数据进行生物信息分析1.SOAPnuke过滤:去除低质量reads;2.利用拼接程序,将PEreads进行拼接合并;3.拼接好的数据与参考序列比对;4.重新比对;5.重比对结果过滤;6.相关统计及作图分析。个体在无抗原刺激时,TCR基因重排是随机的,因此正常人外周T细胞呈多家族、多克隆性特点。当抗原刺激后,TCRV区基因可对该抗原产生特异性识别,并使带有这类基因的T细胞得到优势扩增,通过对待检者外周血PBMC中的T细胞受体β链CDR3进行扩增及高通量测序,对TCRV区基因多样性分布及变化进行分析,进而分析不同TCRV亚家族T细胞的表达和利用,从而可以发现差异,这些差异可能能够应用或者辅助应用于另一种状态,另一种正常或异常状态,如肝癌、肝炎、直肠癌等的早期无创诊断检测、发病进展监测、指导肿瘤术后效果检测评估等。例如,通过对待检者的细胞免疫水平进行综合评价,进行肿瘤的早期无创诊断;进一步通过比较患者手术/用药前后的免疫组库变化来监测疾病发展,评估预后效果,指导选择合适的治疗方案,预防肿瘤复发。若用于辅助临床检测,具有如下优势:1)微创性:受检者只需要提供5-10mL外周血样本;2)实时性:可对受检者进行多次实时采血,辅助早期筛查时的定期检测,监控肿瘤发病风险,肿瘤患者可在手术后、化疗后随时检测,以分析手术预后情况及化疗效果;3)高通量:基于新一代测序技术的免疫组库测序,能够在很短的时间内同时进行多例样本检测。一次测序得到百万级别条数的序列信息。实施例三对来自医院的10例结直肠癌外周血样本及20例健康人外周血样本进行了TCRβ链CDR3的测序检测。免疫组库测序检测以外周血中分离的PBMC作为研究对象,内容如下:1.外周血取样1)取患者外周血样本5ml于EDTA抗凝管中。上下轻轻颠倒4-6次充分混匀后,室温放置,并在2小时以内完成PBMC分离工作;2)加入3倍体积的无菌生理盐水,上下颠倒混匀;3)取3ml细胞分层液于15ml离心管中,并小心的吸取2)步稀释的全血细胞4ml沿管壁叠加于分层液面上,体积大于4ml的分多管进行。水平离心,400g,室温条件下离心30分钟;4)小心吸取淋巴细胞层,置于另一离心管中,加入5倍以上体积的无菌生理盐水,400g室温条件下离心10分钟;5)倒掉上清液,加入1mlTRIzol。用吸头反复吹打细胞直至看不见成团的细胞块,整个溶液呈清亮而不粘稠的状态;转移至2ml离心管。6)液氮速冻后-80°保存,干冰盒运输,避免反复冻融。2.RNA的提取1)每管PBMC(组织样本经液氮研磨后)加入1mlTrizol,混均,冰上放置5min。2)加入氯仿0.2ml/管,振摇15s。15-30℃孵育2-3min,4℃,12000g,离心15min。3)吸取上层无色液体转移至新的EP管中。4)加入等体积异丙醇,混匀,15-30℃孵育10-30min,4℃,12000g,离心10min。5)去上清,加入75%乙醇1ml,涡旋振荡30s,4℃,7500g,离心5min。6)吸净上清,管内沉淀在超净台中鼓风静置3-5min。7)加入20ulDEPC水溶解,-80℃冰箱保存。3.RNA反转录(RNAreversetranscripsion)RNA(补DEPCH2O)10ul(RNA总量200ng)ReversePrimer1ul65℃变性5min后立即置于冰上,依次加入以下体系:4.文库构建4.1多重PCR(multiplexpolymerchainreaction)扩增T细胞受体CDR3区4.1.1使用QIAGEN公司的MultiplexPCR试剂盒,配置PCR的反应体系,进行PCR。PCR反应条件:4.1.2多重PCR产物,QIAquickGelPurificationKit纯化胶回收产物1)配置2%的回收胶。2)将多重PCR产物进行电泳,400mA,100V,电泳2h。3)EB染胶。4)片段选择:100-200bp。5)使用30ul超纯水进行回溶。4.2末端修复1)在1.5ml的离心管中配制末端修复反应体系:2)上述100μL反应混合物轻微振荡混合均匀,瞬时离心,在Thermomixer中20℃温浴30min。3)用QIAquickPCRPurificationKit纯化产物,34μL回溶。4.3末端加“A”(A-Tailing)1)在1.5ml的离心管中配制末端加“A”反应体系:DNA32μL10xbluebuffer5μLdATP(1mM)10μLKlenow(3’-5’exo-)3μL2)上述50μL反应混合物轻微振荡混合均匀,瞬时离心后置于Thermomixer中37℃温浴30min。3)用QIAquickMinElutePCRPurificationKit纯化产物,17μL回溶。4.4Adapter的连接(AdapterLigation)1)在1.5ml的离心管中配制Adapter连接反应体系:DNA15μL2xRapidligationbuffer25μLPEAdapteroligomix(1μM)5μLT4DNALigase(Rapid)5μL2)上述50μL反应混合物轻微振荡混匀,瞬时离心后置于Thermomixer中20℃温浴15min。3)QIAquickMinElutePCRPurificationKit纯化产物,25μL回溶。4.5连接产物PCRDNA23μLPrimer1公用(10μm)1μLPrimerindexX(10μm)1μL2×phusionmastermix25μL总体积50μLPCR反应条件:4.6连接产物的纯化(AGENCOURTAMPureXPbeads)在50μL连接产物中,加入1.2倍体积的磁珠(60μL),进行磁珠纯化,加入20μLUltraPureWater,进行回溶。5.文库检测使用Agilent2100Bioanalyzer检测文库产量;使用qPCR定量检测文库产量。6.上机测序TCR-seq采用IlluminaHiSeq2500PE101+8+101(双末端测序,读段长度101bp)程序进行上机测序,测序实验操作按照制造商提供的操作说明书进行上机测序操作。7.下机数据生物信息分析及免疫组库测序结果分析7.1生物信息分析1)测序数据的预处理:去除Nrate(N比例)大于或等于5%的reads;去除含有adapter污染的reads;去除平均质量值低于15的reads;一对读段对reads1和reads2,reads1与reads2尾部质量值小于10的碱基逐个进行切除,切除后reads1长度需满足60bp以上,reads2长度需满足50bp以上。2)PairedReads合并:利用COPE和FqMerger(华大基因,BGI),将PEreads进行拼接合并为contigs。3)contigs数据与参考序列进行比对:拼接好的序列(contigs)与构建好的CDR3V/D/J参考序列(CDR3V/D/J参考序列来源于http://www.imgt.org/download/GENE-DB/)分别进 行BLAST比对。4)重新比对:根据以上合并的blast比对结果,将CDR3起始位置后的序列依照CDR3区域比对标准进行重新比对:对blast比对部分的V,D,J两端进行延伸比对至contig两端为止,并对CDR3区域进行mismatch设置,例如采用的设置标准为:V区允许的mismatch数TRB的为0、IGH的为2,J区允许的mismatch数TRB的为0、IGH的为2,D区允许的mismatch数目TRB的为0、IGH为4,过滤参数可依据mismatch数参考IMGT工具进行设置。重新计算identity(比对率),比对率的计算方式为比对上的碱基数除以该contig的比对至CDR3参考序列达到所允许的mismatch数的位置的碱基数目,对计算出的identity进行过滤:V区比对率大于或等于80%,J区大于或等于80%的最终比对结果分别作为V,D,J的型别。5)比对结果过滤:去除Contigs重复为1的比对结果,去除未比上V基因或者J基因的Contigs,去除比对V,J基因相反方向的Contigs,去除比上假基因的Contigs。根据参考序列CDR3起始位置,确定Contig的CDR3位置,去除无法确定CDR3位置的Contigs,去除含终止密码子或者无ORF的Contigs。6)相关统计与作图:将20例健康人及10例结直肠癌患者外周血样本数据进行以上初步分析后,对每个样本TCRβ链上48个V区基因片段和13个J区基因片段进行V-J使用频率统计,其中为了便于统计,48个V区基因片段合并成23个进行分析。我们利用高增殖克隆的比率(highlyexpandedclone-rate,HEC-rate)分析及V区使用的主成分分析(V-usage-PrincipalComponentAnalysis,V-usagePCA)等方法对健康人及肝癌患者进行分类分析。1)统计频率超过0.1%的高频CDR3(HEC)占UniqueCDR3总数的比例。用T检验等检验患者与健康人数据之间是否存在差异。T检验,亦称studentt检验,是用t分布理论来推论差异发生的概率,从而比较两个平均数的差异是否显著。2)统计样本VJ亚型使用频率:通过统计各样本不同VJ亚型使用的相对丰度,并对10例结直肠癌患者外周血样本和20例健康人外周血样本的VJ组合亚型使用频率进行T检验或Wilcox检验等,找到P值<0.01的VJ组合亚型,或者基于不同VJ亚型区分两组样本的最小错误率,找出最小错误率最低的VJ组合亚型。3)对VJ组合亚型进行PCA分析根据CDR3-VJ使用频率采用主成分分析(PrincipalComponentAnalysis,PCA)的方法对两组进行分类分析:用主成分分析的方法算出各个样本的第一主成分和第二主成分的值 作图,看是否有疾病组和对照组的分开聚集现象。如果某些主成分(VJ组合亚型)可以很好的区分患者和健康人,对该主成分进行受试者工作特征曲线分析(receiveroperatingcharacteristiccurve,ROC)并统计ROC曲线下的面积即AUC值。ROC曲线能很容易地查出任意界限值时的对疾病的识别能力。通过计算ROC曲线下的面积(AUC)判别识别效果,AUC越大(接近1),则分类器的识别诊断价值越佳。结合以上分析方法,从20例健康人与10例结直肠癌人群中找到了9个或者说找到了10个差异显著的VJ配对亚型(VJ组合亚型),如表1所示。表1对从20例健康人与10例结直肠癌人群中找到的9个差异VJ亚型的频率进行了箱线图分析,图6、图7和图8分别显示以TRBV19TRBJ1-1,TRBV25-1TRBJ1-1和TRBV25-1TRBJ1-4为例的箱线图分析。TRBV19TRBJ1-1的使用频率在两组样本中存在明显差异(p<0.01);RBV25-1TRBJ1-1和TRBV25-1TRBJ1-4的使用频率在两组样本中存在极显著差异(p<0.001)。利用找到的这些差异VJ组合亚型进行PCA分析,结果显示两组样本有分开聚集趋势,如图9所示。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1