基于双端读数insertsize分布的contig错误连接区域识别方法与流程
本发明属于生物信息学领域,涉及基于双端读数insertsize分布的contig错误连接区域识别方法。
背景技术:
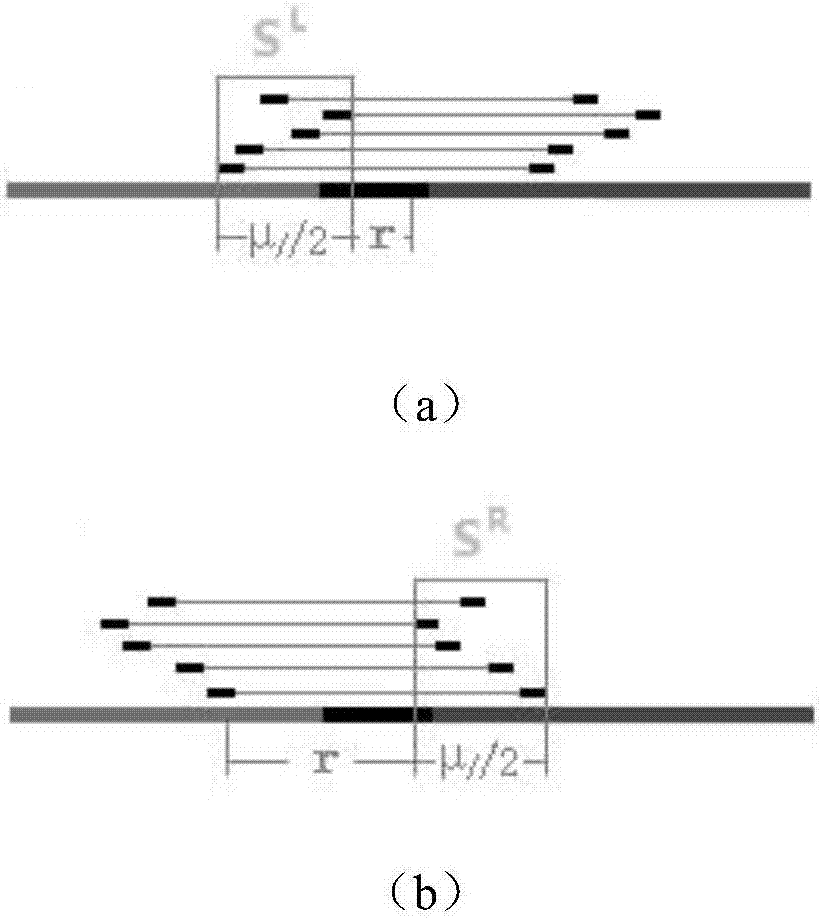
:获得一个物种的基因组是许多生命科学领域研究的基本前提,由于受物种基因组的复杂性以及测序技术未解决的测序长度制约,当前,对测序出的短序列进行组装是必不可少的任务。第三代测序技术由于测序成本高且错误率高,目前主流的测序还是采用以illumina基因测序仪为代表的第二代技术,或是二三代混合测序再拼接。第二代测序技术的特点是通量大,读长短,测序错误率低。illumina基因测序仪可以产生pairedend或matepair双端测序读数,解决一部分由读长过短带来的问题。Pairedend读数和matepair读数的每一对由两条读数组成,左读数的左端点到右读数的右端点的距离称为insertsize(插入片段长度)。已有研究表明,一个文库的insertsize服从正态分布。采用第二代读数的组装算法可以分为两类:基于overlap–layout–consensus(OLC,重叠-排列-生成一致序列)的方法和基于deBruijn图的方法。OLC方法第一步先计算读数与读数之间的所有交叠,接着根据交叠关系构造一个图,最后从图中获得长序列。deBruijn图采用一种违反直觉的方法,首先将读数切成更短的k-mer,再利用k-mer的交叠构造deBruijn图,接着从deBruijn图中得到拼接序列。OLC方法在测序倍数低、读长长的小型基因组上能很好地工作,但随着倍数和基因组规模增加,OLC方法需要的计算时间和内存太大,很难在普通机器上运转。而deBruijn图方法则可以在测序倍数高、读长(readlength)短的大型基因组上很好地工作,而随着测序成本的降低,数据的通量一直在提高,并且随着生物实验的需要,越来越复杂的物种也需要进行基因组测序。因此,在当前环境下,基于deBruijn图的方法被更为广泛地采用。基于deBruijn图方法,DanielR.Zerbino等人于2008发布了组装工具Velvet,通过双端读数,解决了一些长度小于insertsize(双端读数中每对读数之间的间距)的repeat(重复)问题,提高了拼接质量。而后,JaredT.Simpson等人发布了ABySS,提出了一种并行化方法,使复杂物种的大规模测序数据的组装成为可能。SanteGnerre等人于2011在ALLPATHS,ALLPATHS2的基础上发布了ALLPATHS-LG,针对哺乳动物基因组规模大,重复区复杂的特点,结合overlapping文库、jumpping文库以及PacBio数据,在高倍的哺乳动物数据集上取得了很好的结果。2011年,PramilaNuwanthaAriyaratne等人发布了PE-Assembler,提出了一种使用双端读数进行扩展的方法,取代传统的图遍历方法,在控制内存耗用的同时,提高了准确性,并使并行化变得简单。2014年,JunweiLuo等人发布了组装算法EPGA,通过双端读数的分布,提高了序列扩展时的准确性,提高了拼接质量。尽管可用的拼接工具越来越多,序列组装依然存在几个挑战:一个是物种自身基因组的复杂性,包括两个因素:重复区和多倍体的杂合度。对于重复区问题,一旦重复区长度大于双端数据的insertsize,则无法可靠地对重复区附近的序列进行链接,对于杂合度问题,杂合度对基因组组装的影响主要体现在不能合并同源染色体,杂合度高的区域,会把多条同源染色体都组装出来,同时也增加了deBruijn图的复杂性,使得获取高质量contig更加困难。另一个是测序不均问题,这是第二代测序技术的测序过程中PCR阶段引入的问题。这种不均会导致一些区域的覆盖非常低,以至于这些少量读数很可能被当成错误丢弃;而在另一些区域非常高,导致拼接过程中很可能产生误判。由于这些问题的存在,目前许多复杂物种的序列组装结果存在拼接碎片化,长度过短,拼接中带有错误等问题。此外,由于生物本身的复杂性和测序数据的不同规模,一个拼接工具往往在某些物种、某些数据上拼接结果很好,在另一些物种的数据上拼接结果就很差。为此,一些致力于解决这个问题的contig集成工具也陆续发布出来。包括GAM,MAIA,minimus2,CISA等,这些contig整合工具的初衷是集各个拼接工具输出的contig之所长,得到更好的contig。理想情况下,通过集成确实可以有效提高contig的质量,而现实中,由于拼接工具不能保证拼出的结果完全正确无误,一旦错误累积,将导致contig质量严重下降,因此,如何避免contig中的错误积累到整合结果中是不得不考虑的问题。技术实现要素:本发明提出了一种新的基于双端读数insertsize分布的contig异常区域识别方法,通过将双端读数比对到contigs【基因组连续片段】上,找到双端支持稀疏的区域作为种子区域(候选错误区域),并通过双端读数insertsize的分布检验对种子区域进行延伸,最终通过区域长度确定异常位点。本发明对contig中的错误识别具有较高的准确度,通过错误位点切割能够明显减少contig中的拼接错误,有效地提高了contig的质量。本发明的技术方案为:一种基于双端读数insertsize分布的contig错误连接区域识别方法,包括以下步骤:步骤一:输入contigs集合和双端读数文库,使用序列比对工具将双端读数文库的双端读数比对到contigs集合上,得到比对结果;对每一对双端读数(RL,RR),记它们比对到contig上的位置为(PL,PR),若PL或PR未比对到任何位置,记位置为NA;根据比对结果,在每条contig的每个位点p维持3个集合,分别记为SMp、SLp和SRp:SMp={PR-PL|RL,RRmappedaccordantly,and(PR+PL)/2=p},表示以p为中点,一致地比对到该contig上的所有双端支持中,每对双端读数之间的距离,即insertsize;SLp={PR-PL|RL,RRmappedaccordantly,andPL=p},表示以p为左顶点,一致地比对到该contig上的所有双端支持中,每对双端读数之间的距离,即insertsize;SRp={PR-PL|RL,RRmappedaccordantly,andPR=p},表示以p为右顶点,一致地比对到该contig上的所有双端支持中,每对双端读数之间的距离,即insertsize;并记DLp=|{(PL,PR)|PL=pand(PR=NAorRRmappedinothercontig)}|,表示左读数比对到p位置,右读数没有比对到这条contig的读数数量;DRp=|{(PL,PR)|PR=pand(PL=NAorRLmappedinothercontig)}|,表示右读数比对到p位置,左读数没有比对到这条contig的读数数量;步骤二:根据比对结果,得到双端支持稀疏的区域;将这些区域作为错误连接的候选区域;如果一对双端读数跨过某个contig区域,则称该对读数是对这个区域的一个支持;定义集合检查集合Z中的两两区间,若两个区间的距离小于较小区间的1/5,则将两个区间所包含的范围进行合并,得到更新后的集合Z’;取集合Z’中区间长度大于指定长度的元素,组成候选区域的集合C;C中的每个区间都是双端支持相对其他位置薄弱的区域,因此有更大的可能性是错误连接的位置。步骤三:根据比对结果,计算每一个候选区域比对的不一致率,并使用假设检验计算候选区域附近的双端读数insertsize服从已知正态分布的概率,根据计算出的不一致率和假设检验结果决定是否对各候选区域r向左或向右延伸μ/2;根据各候选区间的最终长度判定候选区域是否是错误连接位置,如果不是,则将其从候选区域集合C中去除;步骤四、根据比对结果确定错误连接区域的边界,边界确定后,即确定了所有错误连接区域;将所有错误连接区域剪除。所述步骤三包括以下步骤:3.1)如图2(a),对C中的每个区间r=[a,b],在区间左边,定义集合:SL={x|x∈SLa-μ/2∪SLa-μ/2+1∪..∪SLa-2∪SLa-1},表示所有左端落在候选区间外面并且离候选区间距离小于μ/2的双端读数中,每对双端读数之间的距离;计算不一致比对率其中,|SL|表示所有左端落在候选区间外面并且离候选区间距离小于μ/2的双端读数数量;若u小于指定阈值,则放弃延伸左边;否则,使用Kolmogorov-Smirnov检验计算SL服从N(μ,σ2)的概率(p-value);若概率小于指定概率阈值,则认为SL不服从N(μ,σ2),支持了这个区域是错误连接位置,将区间r向左延伸μ/2,μ为已知的正态分布均值;3.2)同样,如图2(b),对C中的每个区间r=[a,b],在区间右边,定义集合:SR={x|x∈SRb+1∪SRb+2∪..∪SRb+μ/2-1∪SRb+μ/2},表示所有右端落在候选区间外面并且离候选区间距离小于μ/2的双端读数中,每对双端读数之间的距离;计算不一致比对率其中,|SR|表示所有右端落在候选区间外面并且离候选区间距离小于μ/2的双端读数数量;若u小于指定阈值,则放弃延伸右边;否则,使用Kolmogorov-Smirnov检验计算SR服从N(μ,σ2)的概率(p-value);若概率小于指定概率阈值,则认为SR不服从N(μ,σ2),支持了这个区域是错误连接位置,将区间r向右延伸μ/2,μ为已知的正态分布均值;根据各候选区间的最终长度判定候选区域是否是错误连接位置,如果候选区间的最终长度小于μ,说明该候选区域不是错误连接位置,则将其从候选区域集合C中去除;基于假设,一个错误连接至少将导致长度μ的范围双端分布出现问题。所述步骤四包括以下步骤:为了确定错误连接区域的边界,依然使用双端读数分布;4.1)如图3所示,在区间r=[a,b]的左端,取μ/2的长度,定义集合BL={x|x∈SRa∪SRa+1∪..∪SRa+μ/2-1∪SRa+μ/2},执行下列步骤:i.检查BL是否服从N(μ,σ2),若服从,则跳过步骤ii,进入步骤iii;否则,初始化平移步长为μ/4,将BL向左移动一步,同时将步长减半,进入步骤ii;ii.检查BL是否服从N(μ,σ2),若服从,则将BL向右平移一步,并将步长减半;否则,将BL向左平移一步,并将步长减半;重复ii直到步长减小到步长指定值,进入步骤iii;iii.在r中移除被BL包含的部分;4.2)对于区间r的右边,也使用同样的方法确定边界。在区间r=[a,b]的右端,取μ/2的长度,定义集合BR={x|x∈SLb∪SLb-1∪..∪SLb-μ/2+1∪SLb-μ/2},执行下列步骤:i.检查BR是否服从N(μ,σ2),若服从,则跳过步骤ii,进入步骤iii;否则,初始化平移步长为μ/4,将BR向右移动一步,同时将步长减半,进入步骤ii;ii.检查BR是否服从N(μ,σ2),若服从,则将BR向左平移一步,并将步长减半;否则,将BR向右平移一步,并将步长减半;重复ii直到步长减小到步长指定值,进入步骤iii;iii.在r中移除被BR包含的部分。区间r中剩余的部分即为确定的错误连接区域;所述步骤一中的序列比对工具采用现有序列比对工具bowtie2。所述步骤一中的双端读数文库为jumpinglibrary,即跳查文库,双端距离较大的文库。所述步骤二中,取集合Z’中区间长度大于μ/10的元素,组成候选区域的集合C,其中μ为已知的正态分布均值。所述步骤3.1)和3.2)中,指定阈值取全局不一致比对率ug的4倍;全局不一致比对率ug的计算方法为:ug=1-Na/N其中,N表示全部能比对到contigs上的读数数量(单端能比对到contigs上的双端读数数量);Na表示能一致比对到contigs上的读数数量(双端能比对到contigs上的双端读数数量),即不仅读数本身比对到contig上,读数的对端读数(mate)也比对到同一条contig上,并且比对的方向和距离符合双端读数的方向和insertsize范围。所述步骤3.1)和3.2)中,指定概率阈值设为0.01。所述步骤四中,步长指定值取1。本发明:a)提出了一种新的基于合并双端支持稀疏区间的候选区域筛选方法,如果一对双端读数跨过某个contig区域,则称该对读数是对这个区域的一个支持,在该方法中以双端读数的中点为代表保存这个支持。根据比对结果,在每个contig位置上保存以该位置为中点的双端支持数量,将连续支持数为零的位点合并成区间,再根据区间与区间的距离大小将邻近的区间进行合并,得到候选区域。该方法有效提高了错误识别的效率,并提高了识别准确度。b)提出了一种新的基于双端支持分布的错误位置判定方法,对每一个候选区间,在候选区间的左边,收集所有左端落在候选区间外面并且离区间距离小于μ/2的双端读数,这些双端读数有单端比对上的,也有双端比对上的,计算二者的比值为不一致比对率;再利用Kolmogorov-Smirnov检验计算双端比对上的读数服从已知分布的p-value(概率)。若不一致比对率大于阈值且p-value(概率)大于阈值,则将候选区间往左延伸μ/2。在候选区域的右边做同样的计算。根据候选区间的最终长度对其进行取舍,得到待切除区间。提出了一种新的基于双端支持分布的边界确定方法,对每一个待切除区间,在区间的左边选取μ/2长度的子区间,收集所有右端落在该子区间的双端支持,折半地左右平移该区间,直到区间内的双端支持服从已知分布,将这部分序列在切除区间去除,在待切除区间的右边做同样的计算,最终确定剪切边界。有益效果:本发明的方法不仅通过错误位点切割有效地减少contig中的拼接错误,对基于contig集成的序列拼接方法,本发明也能有效地避免这些错误积累到集成过程中,在高倍数据集上有效减少了contig连接错误的数量,为下游处理提供了更纯净的contig,有助于正确的contig进行合并,通过实验验证,有效提高了最终拼接的长度(N50)并减少了错误,提高序列拼接的质量和准确率。附图说明图1:Contig连接错误实例;图2:双端读数分布检验,图2(a)为区间左边检验,图2(b)为区间左边检验;图3:错误剪切边界确定;具体实施方式一、Contig错误分析无论是基于deBruijn图的de-novo拼接方法,还是基于OLC的拼接方法,从deBruijn图和overlap图中获得的长序列(contig)通常很难保证完全正确,尤其是在基因组复杂、repeat(重复)富集的物种中,错误更是难以避免。这些错误以contig的错误连接为主,即两段来自基因组不同位置的序列,被错误地拼在了一起,这种错误通常会在后续处理过程中保留下来,更严重的是,由于后续步骤对这种错误敏感,因此,contig中的错误很可能被放大。例如图1所示,使用某一拼接软件得到a,<b1,b2>,c,d,e5条contigs,这些contigs在基因组上的顺序为a,b1,c,d,b2,e,即,<b1,b2>是错误连接的contig。这将导致几个问题:1)contig中的错误被继承到最终拼接的结果中,形成更长的带错误的序列,降低了拼接结果的可靠性;2)错误的连接阻断了正确的连接,如contigb1和c本应是紧邻的,由于b1错误地和b2接在了一起,使得b1无法连接到c,阻碍了更长序列的形成,降低了拼接结果的质量;3)Contig中的错误还可能在scaffolding(超长序列片段组装)阶段导致二次错误,例如,contigd的后续contig应为b2,但b2已经被contigb1粘住,这可能导致contigd选择另一个有少量双端支持的错误的后续contig,扩大了序列拼接错误。因此,减少contig中的错误连接具有重要意义。本文提出了一种基于双端读数分布的contig错误连接识别和纠正方法,有效地减少contig中的错误连接数,提高了contig的质量。经contig集成工具和scaffolding工具验证,有效提高了最终拼接出的序列长度并较少了最终结果的错误数。二、双端读数mapping使用现有序列比对工具bowtie2将jumpinglibrary(跳查文库,即双端距离较大的文库)的双端读数比对到contig集合上,对每一对读数(RL,RR),记它们比对到contig上的位置为(PL,PR),若PL或PR未比对到任何位置,记位置为NA。根据比对结果,在每条contig的每个位点p维持3个集合:SMp={PR-PL|RL,RRmappedaccordantly,and(PR+PL)/2=p},表示以p为中点,一致地比对到该contig上的所有双端支持,这里只需要记录双端的距离,下同;SLp={PR-PL|RL,RRmappedaccordantly,andPL=p},表示以p为左顶点,一致地比对到该contig上的所有双端支持;SRp={PR-PL|RL,RRmappedaccordantly,andPR=p},表示以p为右顶点,一致地比对到该contig上的所有双端支持;并记DLp=|{(PL,PR)|PL=pand(PR=NAorRRmappedinothercontig)}|,表示左读数比对到p位置,右读数没有比对到这条contig的双端读数数量;DRp=|{(PL,PR)|PR=pand(PL=NAorRLmappedinothercontig)}|,表示右读数比对到p位置,左读数没有比对到这条contig的双端读数数量。三、确定候选区域定义集合检查集合中的两两区间,若两个区间的距离小于较小区间的1/5,则将两个区间所包含的范围进行合并。取区间长度大于指定长度(预设值μ/10,其中μ为已知的正态分布均值)的元素,组成候选区域的集合C。C中的每个区间都是双端支持相对其他位置薄弱的区域,因此有更大的可能性是错误连接的位置。如图2(a),对C中的每个区间r=[a,b],在区间左边,定义集合:SL={x|x∈SLa-μ/2∪SLa-μ/2+1∪..∪SLa-2∪SLa-1}计算不一致比对率若u小于指定阈值,则放弃延伸左边,其中阈值按比对结果的全局不一致率计算,预设值取全局不一致率的4倍。否则,使用Kolmogorov-Smirnov检验计算SL服从N(μ,σ2)的p-value。若p-value小于指定阈值(取0.01),则认为SL不服从N(μ,σ2),支持了这个区域是错误连接位置,将区间r向左延伸μ/2,μ为已知的正态分布均值。同样,如图2(b),在区间右边,定义集合:SR={x|x∈SRb+1∪SRb+2∪..∪SRb+μ/2-1∪SRb+μ/2}计算不一致比对率u。并同样根据u和假设检验结果决定是否对区间r向右延伸μ/2。最后去除区间长度小于μ的区间,基于假设,一个错误连接至少将导致长度μ的范围双端分布出现问题。四、确定剪切边界为了确定剪切的边界,依然使用双端读数分布。如图3所示,在区间r=[a,b]的左端,取μ/2的长度,定义集合BL={x|x∈SRa∪SRa+1∪..∪SRa+μ/2-1∪SRa+μ/2},执行下列步骤:i.检查BL是否服从N(μ,σ2),若服从,则跳过步骤ii,进入步骤iii,否则,初始化平移步长为μ/4,将BL向左移动一步,同时将步长减半。ii.检查BL是否服从N(μ,σ2),若服从,则将BL向右平移一步,并将步长减半;否则,将BL向左平移一步,并将步长减半。重复ii直到步长减小到指定值【指定值取1】。iii.在r中移除被BL包含的部分。对于区间r的右边,也使用同样的方法确定边界。边界确定后,即确定了所有异常区域。移除所有异常区域,将错误连接的contigs剪开,获得最终结果。五、实验验证为了验证本方法的有效性,我们使用PEAssembller、Velvet、SOAPdenovo和Abyss四个组装工具对三个物种的illumina测序数据进行组装产生contigs。然后用jumppinglibrary数据对产生的contigs分别进行改错,staph,ecoli,spome的jumppinglibrary倍数分别为70倍,61倍,179倍。然后,使用本发明的改错方法对其进行改错。为了检验改错的准确性,我们将改错过程中剪切下来的区域收集起来,并使用权威的序列拼接评价工具quast检查被剪除的区域是否是错误序列,并做了统计。对于每一组contigs,我们使用改错工具进行改错,统计改错的次数,同时,每次改错后我们都使用quast检查错误是否减少,即改错是否正确,结果如表1所示。表1.错误识别准确率表中每一项表示(errorconfirmedbyquast/errorcalled)staphecolispomeAbyss0/00/00/0Velvet3/32/276/76PEAssembller0/00/02/2SOAPdenovo0/00/00/0从表1中可以看出,在3个数据集的12组测试实验中,有4组改正了contig的错误,改错准确率均是100%。我们还可以看到,Spome数据集由于基因组复杂,更容易产生组装错误,并且由于jumppinglibrary倍数高,改错效果最好。为了进一步验证contigs改错的现实意义,我们将上述改错前后的contig按每个物种进行集成。集成工具使用了CISA,使用quast评估整合后的contig。表2.改错前后的contig整合结果,使用整合工具CISA从表2中可以看到,staph数据集和spome数据集上,改错之后在保持拼接长度的前提下,减少了最终集成结果的拼接错误。在基因组最为复杂的spome物种上,不仅拼接错误减少了58,拼接长度和覆盖度也有了提高。从这组实验结果可以看出,本文提出的方法对拼接容易出错的复杂物种具有较好的适应性,能有效改善contig整合结果。Contigs改错不仅对contig集成具有重要意义,对scaffolding也会产生影响。我们选择上述实验中改错工具产生作用的4组contig用scaffold工具SSPACE进行scaffolding测试,使用quast统计了改错前后的scaffolding结果,如表3所示。表3.改错前后的scaffolding结果,使用scaffold工具SSPACE从表3中可以看出,在4组实验中,最终组装结果错误数都得到降低。不仅如此,第3组和第4组实验中,由于错误的剔除,组装过程中干扰因素减少,导致拼接长度也得到提高。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1