基于模块化因子图的骨髓瘤信号通路机制确认方法与流程
本发明属于系统生物学技术领域,主要涉及生物信息学和生物数据挖掘,具体涉及一种基于模块化因子图的信号通路机制确认方法。
背景技术:
多发性骨髓瘤(multiple myeloma,MM)是一种以恶性浆细胞克隆性增殖为特点的恶性肿瘤,是目前血液系统第二大恶性肿瘤。目前MM的治疗主要包括传统化疗、新药靶向治疗及免疫治疗等。虽然新的靶向治疗明显提高了MM的疗效,但患者中位生存时间仍在3~5年,其发病机制尚不明确。因此,进一步研究影响MM细胞生长的相关机制,寻建立模型研究方法,是亟需解决的问题。
无论从生物学还是从临床表现看,MM细胞的特性不仅仅决定于其遗传学特性(如染色体重排,缺失,扩增或某些特定基因的突变)。相反,该疾病的病理生理学表现明显受MM细胞与其所处的骨髓微环境间双向相互作用的影响。Virginia Hughes指出,骨髓微环境对MM细胞的存活、生长以及耐药等重要环节有着息息相关的作用。骨髓基质细胞(bone marrow stromal cells,BMSCs)作为骨髓微环境的主要成员,与MM的发生、发展有着密切的关联。
随着对MM生物学研究的不断深入,人们发现在肿瘤发生发展过程中,信号通路控制着众多至关重要的细胞生物学过程。目前,对MM信号通路靶点的研究十分广泛,主要靶向的通路包括这些信号通路包括PI3K/Akt/mTOR/P70S6K信号通路,IKK-αF/NF-κB信号通路,Ras/Raf/MAPK信号通路和JAK/STAT3通路,它们都可以通过以下途径被激活:上游的细胞因子与相应受体的结合,或通过粘附启动的激酶途径直接由细胞粘附诱导增殖、抗凋亡信号通路的激活。
众所周知,传统的生物实验非常昂贵并且要花费大量的时间,所以近年来越来越多的人在用生物模型去模拟生物生长状况,从模拟的层面上去分析药物影响或者提取关键蛋白质。Huiming Peng等人用系统生物学的方法研究p38MAPK异型的抗药性,确定生物模型之后利用设置参数值的方式分别去探索p38的五种异型的抗药性;Xiaoqiang Sun等人基于细胞内的信号通路利用微分方程建模的方法研究在组织骨再生的过程中细胞因子的组合预测,对人体组织骨再生成时不同细胞因子对成骨细胞和破骨细胞的刺激作用进行了探索,并且筛选出较好的细胞因子组合。但是,上面的研究方法存在一定的局限性,并没有指出如何预测未知的致病因子的影响。
遗传学改变和。生化条件引起的通路激活经常发生在肿瘤恶变早期和进展期,同时它们也是患者预后的重要指标。系统生物学方法期望通过建立细胞信号转导过程的模型,找到参与此过程的各种分子之间相互作用的网络,阐明其在基因调控、疾病发生中的作用。近几年,对信号转导网络的定量分析逐日升温,通常采用一系列的方程模型描述信号转导通路的内部变化过程。
现有技术中,采用的主要方案包括以下几种:
(1)临床实验。在对于多发性骨髓瘤的研究中,目前绝大多数还是处于利用实验方法去观察肿瘤细胞的生长发育,尤其治疗期间,基本上是靠医生的经验去判断。实验成本比较昂贵。
(2)常微分方程(ordinary differential equations,ODE)。这是是描述动力学系统的常用方法,应用微分方程组可以构建一个复杂的数学模型,用以代表一系列生化反应的相互作用模式,并且模拟生物系统中各组分的时序性动态变化。常微分方程(ODE)是质量反应动力学过程的数学代表,可以用来描述连续时间范围内生物系统各组分的动态变化。对于那些不考虑空间大小,并且反应速度和反应底物的浓度成一定的比例关系的生化反应系统比较适用。Chen利用ODEs来描述ErbB信号通路的输入输出对细胞分化和增值的影响,文中用299个ODE方程表示828个级联反映,共有229个参数,计算规模很大。
(3)Petri Net的不确定性、并行性、异步性,以及对分布式系统的描述和分析能力使其在描述生物系统特性时有很大的优势。Chen和等用Petri nets建立了鸟氨酸循环的代谢模型。
上述方案主要存在的缺陷有以下几点:
(1)人为经验判断,准确率不高。
(2)ODE等数学方法仅仅是描述生物量的变化,并不能直接以图形的方式展现生物系统的结构特性。若可实现生物定量数据与图形的结合与自动转化,将能更好地刻画生物系统结构与动态性质之间的关系。
(3)标准的Petri Net常用来定性分析生物网络的结构性质,不能用于生物计算。
以下对本发明所涉及到的技术词汇/技术术语注释如下:
1、多发性骨髓瘤(multiple myeloma,MM)
2、骨髓基质细胞(bone marrow stromal cells,BMSCs)
3、常微分方程(ordinary differential equations,ODE)
4、反向蛋白质阵列(reverse phase protein arrays,RPPA)
技术实现要素:
有鉴于此,本发明在总结前人的研究基础上,提出建立一个多层次的计算系统生物学模型来研究多发性骨髓瘤细胞的生长机制,利用现有RPPA数据,结合常微分方程组和Petri网来描述信号通路,并且对骨髓基质细胞和肿瘤细胞通路之间相互作用的组合影响进行量化的探索,并且使用模块化的计算模型,降低计算成本。
具体而言,本发明所提出的技术方案如下:
本发明提供了一种基于模块化因子图的骨髓瘤信号通路机制确认方法,该方法包括:
步骤1、获取RPPA数据;
步骤2、对所述RPPA数据进行预处理,粗粒度筛选关键蛋白质;
步骤3、基于所述粗粒度筛选的关键蛋白质,构建在细胞刚性环境下,细胞内蛋白质的相互作用通路,形成信号通路;
步骤4、采用常微分方程描述所述信号通路,并将所述信号通路分解成多个小模块,针对每个所述小模块,进行参数优化,建立系统生物学模型;
步骤5、对所述系统生物学模型进行参数分析,所述参数分析包括稳定性分析和敏感性分析。
优选地,所述步骤1中,RPPA数据的获取,通过以下方式:
步骤1.1、用压强为100pa和400pa的细胞胶体模拟正常细胞和肿瘤细胞,记录不同时间点细胞内蛋白质的浓度;
步骤1.2、利用蛋白质芯片,获得正常细胞和肿瘤细胞的两组RPPA数据。
优选地,所述步骤2具体包括:
步骤2.1、对粗粒度筛选出的全部关键蛋白质数据,以t=0min为标准,进行规范化,所述规范化方法为:
其中t0表示t=0min,表示第i个蛋白质在tj时刻的浓度,表示第i个蛋白质在t0时刻的浓度,为规范化后的蛋白质浓度;
步骤2.2、计算正常细胞、肿瘤细胞内蛋白质浓度变化率,具体方式为:
将其中浓度变化大于50%的蛋白质作为有意义的表达的蛋白质,作为粗粒度筛选出的关键蛋白质。
优选地,所述步骤3具体包括:
步骤3.1、基于粗粒度筛选出的关键蛋白质,通过IPA数据库,搜索相互作用的通路;
步骤3.2、在所述步骤3.1中搜索出的通路中,选择p≤0.05的通路,作为信号通路,其中p表示某蛋白质在该通路中出现的误差率。
优选地,所述步骤4具体包括:
步骤4.1、使用常微分方程组描述信号通路,并使用RPPA数据中,高水平表达的蛋白质在不同时间点的采样数据,确定信号通路中的关键参数;使用Petri网描述整个信号通路;
步骤4.2、基于所述Petri网描述的整个信号通路,将整个信号通路分解成多个小模块;使用粒子群优化方法优化各个所述小模块参数,获得相对较小的参数范围,所述参数优化的目标函数是:
其中,表示蛋白质浓度时间序列数据,表示通过常微分方程获得的模拟的蛋白质浓度时间序列,i表示蛋白质索引,tj表示时间点,Θ表示常微分方程中的参数,M表示蛋白质数量,N表示时间点数量;
步骤4.3、把整个信号通路分解成两个子网通路,并使用因子图表示每个子网通路,为因子图中的每个因子节点构造适应函数,所述适应函数为:
步骤4.4、使用置信度传播方法,调和所述两个子网通路中共享的蛋白质参数,并以所述步骤4.2中获得的相对较小的参数范围中的参数,作为置信度传播方法的输入参数,得到一个更小的参数范围;
步骤4.5、以所述更小的参数范围中的参数,作为粒子群优化方法的输入,对所述系统生物学模型进行参数优化,获得最终的系统生物学模型。
优选地,所述步骤4.2中,整个信号通路分解成多个小模块,可依据如下分解原则:a.每个子模块中蛋白质数据尽可能少;b.每个子模块中至少有一个蛋白质浓度是有实际数据依据。
优选地,所述步骤4.3中,分解成子网通路可依据如下规则:a.从细胞表型出发,依次找出促进细胞增长或细胞死亡的蛋白质;b.如果按两条表型找出的两类蛋白质中共享蛋白质数量超过90%,则把两条子网合并为一个大网;c.如果其中一个大的子网的蛋白质数量是另外一个子网蛋白质数量的2倍或以上,则重新分解这个大的子网。
优选地,所述粒子群优化方法的具体方式如下:
vi(t+1)=wvi(t)+c1·rand()·(pi(t)-xi(t))+c2·Rand()·(pg(t)-xi(t))
xi(t+1)=xi+vi(t+1)。
优选地,所述稳定性分析通过计算变异系数对参数进行分析,所述变异系数的计算方法如下:
C·V=(标准偏差SD/平均值Mean)×100%。
优选地,所述敏感性分析中,参数的敏感性计算方法如下:
其中,ΔPi是第i个参数值的变化量,表示一个很小的变化,例如可以是增加或减少1%,[ProteinName]表示系统输出蛋白质,例如Casp3、p90RSK、CyclinD1、p21、p7056k,Pi表示优化的参数,si表示敏感性参数。
与现有技术相比,本发明技术方案具有以下的有益效果:
(1)基于肿瘤细胞信号通路的互相作用,用系统生物学的方法建立了计算模型模拟肿瘤细胞的增值和凋亡。
(2)用常微分方程去描述信号通路中的反应,用petri网描述生物信号通路网络结构,实现生物定量数据与图形的结合与自动转化,更好地刻画生物系统结构与动态性质之间的关系。然后将整个信号通路基于规则进行模块性的划分,用寻优算法获取最优值,降低了计算成本,提高了计算效率。
(3)通过模拟手段提高对肿瘤的生长过程中的分子机制的认识,实现对转移精确地进行预测。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
图1为本发明实施例的方法流程图;
图2为本发明实施例的RPPA粗粒度筛选蛋白质计算结果示例图;
图3为本发明实施例的构建与细胞刚性相关的信号通路示例图;
图4为本发明实施例的最大限度简化通路示例图;
图5为本发明实施例的用混合Petri网去描述信号通路示例图;
图6为本发明实施例的将信号通路分解成n个小模块示例图;
图7为本发明实施例的信号通路分解成的子网通路一示例图;
图8为本发明实施例的信号通路分解成的子网通路二示例图;
图9为本发明实施例的参数稳定性分析结果示例图;
图10为本发明实施例的参数敏感性分析结果示例图;
图11为本发明实施例的不同条件下蛋白质对应的参数变化示例图。
具体实施方式
下面结合附图对本发明实施例进行详细描述。应当明确,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
本领域技术人员应当知晓,下述具体实施例或具体实施方式,是本发明为进一步解释具体的发明内容而列举的一系列优化的设置方式,而该些设置方式之间均是可以相互结合或者相互关联使用的,除非在本发明明确提出了其中某些或某一具体实施例或实施方式无法与其他的实施例或实施方式进行关联设置或共同使用。同时,下述的具体实施例或实施方式仅作为最优化的设置方式,而不作为限定本发明的保护范围的理解。
本发明是通过找出影响骨髓瘤细胞生存的信号通路中的关键蛋白质,辅助对于肿瘤细胞增生的模型建立方法研究。根据骨髓瘤细胞的刚性环境(骨髓瘤细胞压强高)有助于肿瘤细胞的增生的结论,用压强为100pa和400pa的细胞分别建立正常和肿瘤细胞的模型,测得得到两组反相蛋白质阵列数据,通过实验数据(100pa VS 400pa)粗粒度筛选出可能影响肿瘤细胞增长的蛋白质,并使用常微分方程组和Petri网描述信号相关的细胞信号通路,利用模块化因子图算法优化模型中的关键参数,在最后通过合理和准确性验证,最终通过该模型得出敏感度较高的蛋白质FAK、NFκB、mTOR1。图1是本发明的总体流程图,以下结合图1对本发明的模型建立和计算方法进行详细阐述。
(1)获取RPPA(Reverse Phase Protein Arrays)数据,建立模型基础数据库
获取RPPA数据的方式可以是多样的,例如,可以采用现有的公用数据库或资料库中提供的已有的骨髓瘤细胞比对RPPA数据,即反向蛋白质阵列数据,其中对比数据模拟生长压强需设置为100pa和400pa,以此作为模型建立的对比数据,从而建立数据库。现有技术中有多种途径可以获得上述RPPA公开数据,这里不再赘述。
也可以通过做模拟对比实验,建立细胞生长的模型,设置例如压强为100pa和400pa的细胞胶体模型模拟正常和肿瘤细胞,记录在不同时间点细胞内蛋白质的浓度,在一个具体的实施方式中,该不同的时间点例如可以设置为0min,30min,60min,overnight,共4个时间点,当然,也可以根据具体的算法需要,设定不同数量的采样时间点。利用蛋白质芯片得到两组RPPA数据。该数据中包括在不同压强下173种蛋白质在不同时间点下的浓度。
当然,此处也可以采用其他的已有技术或途径,获取可以作为后续模型算法所需要的骨髓瘤细胞的RPPA数据集。
(2)数据预处理,粗粒度筛选关键蛋白质
对所有蛋白质数据以t=0min为标准对其规范化,其计算公式为:
其中t0表示t=0min,表示第i个蛋白质在tj时刻的浓度,表示第i个蛋白质在t0时刻的浓度,为规范化后的蛋白质浓度。
计算两种细胞内蛋白质浓度变化率,计算公式为:
根据此公式找出其中浓度变化大于50%的蛋白质作为有意义的表达的蛋白质。图2是根据RPPA粗粒度筛选蛋白质计算结果示例图。
(3)构建多发性骨髓瘤信号通路
信号通路是指能将细胞外的分子信号经细胞膜传入细胞内发挥效应的一系列酶促反应通路。这些细胞外的分子信号(称为配体,ligand)包括激素、生长因子、细胞因子、神经递质以及其它小分子化合物等。细胞内各种不同的生化反应途径都是由一系列不同的蛋白组成的,执行着不同的生理生化功能。各个信号通路中上游蛋白对下游蛋白活性的调节(包括激活或抑制作用)主要是通过添加或去除磷酸基团,从而改变下游蛋白的立体构象完成的。所以,构成信号通路的主要成员是蛋白激酶和磷酸酶,它们能够快速改变和恢复下游蛋白的构象。
在本发明中,根据步骤(2)粗粒度筛选的蛋白质来构造在细胞刚性环境下MIC和MSC细胞内蛋白质的相互作用通路。首先将筛选出的蛋白质通过IPA(Ingenuity Pathway Analysis)数据库寻找相互作用的重要的通路,IPA是基于云计算的一体化应用软件,它可以分析来源于基因组、microRNA、SNP、芯片、代谢组、蛋白组、RNA-Seq实验以及各类小规模实验数据。利用IPA,可以搜索基因、蛋白、化学分子、药物的各类信息,并且帮助构建相互作用模型。在高度结构化、集成丰富详实生物化学知识的Ingenuity Knowledge Base支持下,IPA的分析和搜索可以帮助所获得数据在生物体系中重要性。然后在搜索出的通路中选择p≤0.05的通路,其中p值表示某蛋白质在该pathway(即通路)中出现的误差率。由于在找出的通路中有一些其他的蛋白质,通过共享蛋白质来整合所有细胞通路并参考MM细胞的相关文献或现有的公开数据,来构建与细胞刚性相关的信号通路,如图3所示。由于估计由整个信号通路建立的常微分方程组中的所有参数是不可能的,因此我们需要重新定义信号通路,在保证通路结构完整的情况下最大限度简化通路,如图4所示。
(4)建立系统生物学模型
系统生物学模型所包含生化反应的动力学参数能够明显影响数学模拟结果,可以通过现有技术或相关文献获得,也可以通过体外的生理和生化实验测定。但是,某些生化反应的动力学参数无法直接获得,需要通过实验数据分析加以估计几乎所有描述真实生物系统的模型都过于巨大,其内部的动态变化过程无法被实验数据全部描述。因此,系统生物学模型在不断涌现的多实验数据的整合分析中能够起到关键的作用。
(4.1)在本发明中,我们使用常微分方程组描述信号通路,并采用RPPA试验数据中高水平表达的蛋白质在各个时间点的采样数据来确定信号通路中的关键参数,接上例,设定该采样时间点位4个。在一个具体的实施方式中,结合图5至图8,常微分方程可以通过以下方式进行构建:以pCylinD为例,结合图5至图8中可见,GSK3β促进CylinD的磷酸化,而pp21抑制CylinD的磷酸化,根据这些相互作用可以得到:
其中[CyclinD]、[pp21]、[pGSK3β]分别表示蛋白质CyclinD、pp21、pGSK3β的浓度;kCyclinD_pp21表示CylinD被pp21抑制的磷酸率、kCyclinD_pGSK3β表示CylinD被pGSK3β激活的磷酸率。
遵循这样的规律,根据图6,我们得到用来描述其它蛋白质的40个常微分方程,该其它蛋白质即如附图6信号通路中涉及到的蛋白质,由于其微分方程的表达式相同,此处不再一一赘述。由于这些常微分方程包含45未知参数并且蛋白质之间有相互作用,使用一般的智能算法来确定参数值是相当复杂的。本发明使用调整后的马尔科夫链模块分解方法把整个通路分解成多个小模块,然后用智能算法来优化每个小模块的参数。该方法不仅可以减少计算机的压力,也可以快速获得更优结果。
(4.2)采用信号通路的微分方程模型可以表征细胞在不同时间内分子浓度的变化,观察在不同外界条件下细胞的动态响应过程,深入的揭示信号转导的作用机制。同时,用混合Petri网去描述信号通路中的每一个生化反应及级联反应,如图5所示,把整个信号传导通路基于Petri网和分解规则分解成n个小模块,如图6所示,应用PSO算法(即粒子群优化算法)对每个小模块进行估参(即参数估计),从而在初始范围搜索空间中获得一个相对较小的参数范围。
在一个具体的实施方式中,具体的分解规则可以设置为:a.每个子模块中蛋白质数据尽可能少;b.每个子模块中至少有一个蛋白质浓度是有实验数据的,或者是有确定的数据支撑的,这样才能保证计算的高效性。并使用粒子群算法来优化各个小模块的参数。
得到的蛋白质浓度时间序列数据用表示,通过ODE方程(即常微分方程)获得的模拟的蛋白质浓度时间序列其中,i表示蛋白质索引,tj表示时间点,Θ表示ODE方程中的参数,参数优化的目标函数是:
其中,M表示蛋白质数量,N表示时间点数量,最终得到一个参数初始范围。
(4.3)然后把整个信号通路分解成两个子网通路,如图7、图8所示,为每一个子通路构造因子图,并为每一个因子节点构造适应函数,即公式(1)。
在一个具体的实施方式中,可以为分解成子网通路设定规则:a.从细胞表型出发(细胞增长、细胞死亡),依次找出促进细胞增长或细胞死亡的蛋白质;b.如果按两条表型找出的两类蛋白质中共享蛋白质数量超过90%,则把两条子网合并为一个大网;c.如果其中一个大的子网的蛋白质数量是另外一个子网蛋白质数量的2倍或以上,则重新分解这个大的子网。另外,因子图是指将一个具有多变量的全局函数因子分解,得到几个局部函数的乘积,以此为基础得到的一个双向图,叫做因子图,因子图是由变量节点和因子节点,以及连接两个节点的边构成。
(4.4)应用置信度传播(Belief Propagation,BP)方法调和两个子通路中共享的蛋白质参数,解决两个子网通路相同蛋白质对应的冲突的参数,从而得到一更优的参数范围,以(4.2)步中输出的相对较小的参数作为BP算法的输入参数,然后得到一个更小的参数范围,最后把这个范围作为最后应用PSO算法进行系统估参的输入。从而实现整个模型形成一个不断缩小搜索范围的过程。
其中表示适应度函数,表示模拟结果与实验结果的误差,分别表示参数集合和与因子节点对应的分子浓度水平的集合。表示通过包括参数集Θ的ODEs方程计算得到的蛋白质m在时间点tj模拟浓度。表示蛋白质m在时间点tj的实验水平的浓度。
(4.5)估参方法中,本发明采用粒子群优化算法PSO,以步骤(4.4)中得到的参数范围作为初始搜索空间继续对参数进行优化,从而得到最优值。
在一具体的实施方式中,可在该步中,重复运行例如5遍或更多遍,保证算法的稳定性。
PSO中,每个优化问题的解都是搜索空间中的一个粒子。所有的粒子都有一个由被优化的函数决定的适应值(fitness value),每个粒子还有一个速度决定他们飞翔的方向和距离。在本发明中,PSO初始化为一群随机粒子(随机解)。然后通过迭代找到最优解。在每一次迭代中,粒子通过跟踪两个"极值"来更新自己。第一个就是粒子本身所找到的最优解,这个解叫做个体极值。另一个极值是整个种群目前找到的最优解,这个极值是全局极值。这个过程一直迭代,直到找到比较满意的解为止。此解作为整个模型估参的最优解。
(5)参数分析
a.稳定性分析
某些生物系统(如代谢网络和信号通路等)具有特殊的潜能,能够聚合于特定的平衡态(或称为稳定状态)。当生物系统达到稳定状态时,系统内所有组分浓度改变速率为零。稳态分析可以鉴定并合理描述这些稳定状态,这有助于阐释生物系统内部的稳态维持机制,以及细胞凋亡响应等生物学过程所涉及的状态快速转换机制。在本发明中,稳定性分析指分析参数是否稳定,对于某个参数,如果其变异系数大于1,则认为不稳定,否则就是稳定的。变异系数的计算公式为:C·V=(标准偏差SD/平均值Mean)×100%
根据此公式,对本文中的所有参数进行稳定性分析。如图9所示。其中有10个参数的变异系数大于1,即所占比例为22%,意味着在这个模型里将近80%的参数是稳定的。
b.敏感性分析
在系统生物学模型构建完成之后,简单的结构化和参数化并不能很好地解释生物系统的内在调控机制,因此对模型特性的整体分析是非常重要的。敏感性分析(Sensitivity Analysis)是指从定量分析的角度研究在一定范围内扰动系统组分的初始浓度或反应动力学参数,对模型输出结果影响的大小(特定组分的浓度变化或模型整体状态的改变)。生物学实验上可以理解为敲除或过表达特定基因产物,之后测定生物体或细胞特定表型的变化。敏感性分析能够阐释系统输出对特定参数值的依赖性,这通常是研究者最为感兴趣的。值得注意的是,从理论分析的角度进行敏感性分析时,初始浓度或反应参数的扰动范围可以随意选取。但是从实践的视角出发,如果期望获得的敏感性分析结果是可靠的,那么扰动的选取范围必须符合真实的生理生化状态。此外,敏感性分析方法对于寻找生物网络的关键调控位点具有重要的指导意义。
在本发明中,敏感性分析是一种衡量某一特定参数对输出的影响,分析参数的敏感性,即参数变化是否会引起整个系统输出值的变化,简单来说,给每一个参数增加或减少1%,观察对系统输出的影响,从而找出敏感的蛋白质。根据信号通路可知输出为蛋白质Casp3、p90RSK、CyclinD1、p21、p7056k浓度。参数的敏感性计算公式:
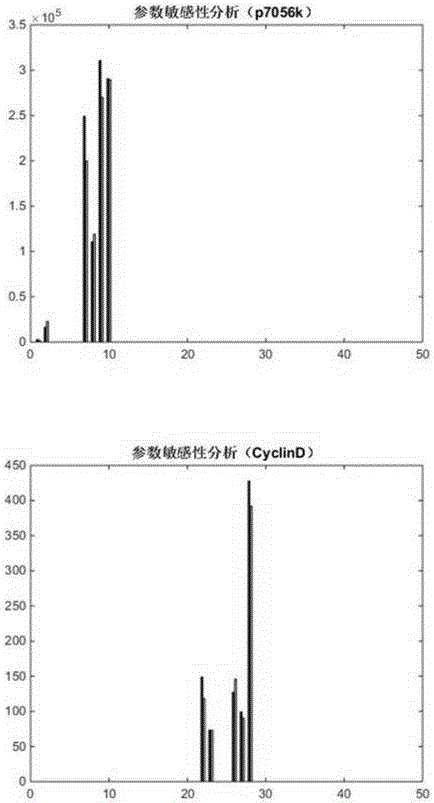
其中,ΔPi是第i个参数值的变化连,表示一个很小的变化(增加或减少1%),[ProteinName]表示系统输出蛋白质Casp3、p90RSK、CyclinD1、p21、p7056k,Pi表示优化的参数,si表示敏感性参数。其中,每个参数通过增加或减少1%,观察Casp3、p90RSK、CyclinD1、p21、p7056k变化百分比,示例如图10,通过分析可得系统输出变化较大的参数占所有参数的比例不超过2%,该结果表示所有参数中只有5-7个参数比较敏感,这说明系统输出蛋白质的浓度变化受这几个比较敏感的参数影响。
综合上述结果,对比正常细胞和肿瘤细胞对应的参数变化值,如图11所示,由上图可以看出几种蛋白质浓度变化范围比较大,则认为FAK、NFκB、mTOR1这几种蛋白质可能是通路中对肿瘤细胞生长或增生影响比较大的,从而实现对蛋白质的筛选。
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以权利要求的保护范围为准。
- 还没有人留言评论。精彩留言会获得点赞!