能够在多个平台上区分胎儿性别和胎儿性染色体异常的新方法与流程
本发明涉及一种使用通过进行下一代测序(ngs)从多个样品获得的数据的胎儿性别和胎儿性染色体(例如,删除和复制胎儿基因的性染色体x和y区域中的一些区域)的非整倍性(aneuploidy)分析的方法,及其发现和验证,其中即使输入来自各种平台的异类数据(heterogeneousdata)也可能获得相同的结果。
背景技术:
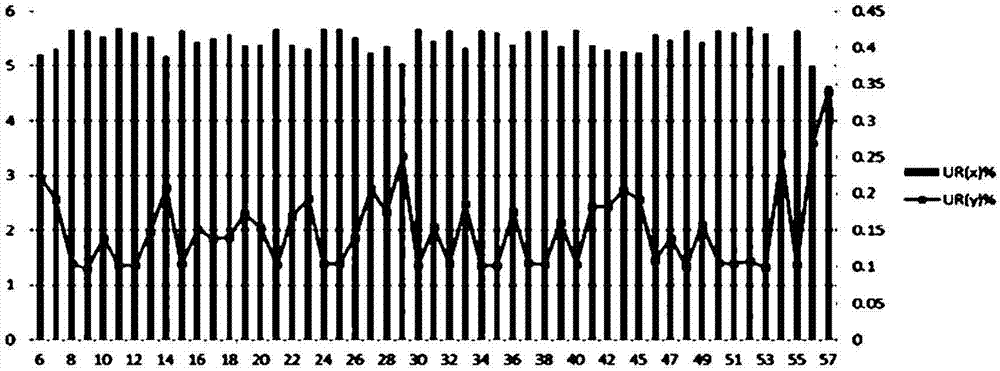
:从1970年代到最近,与染色体数值异常有关的疾病的诊断方法已经不断发展。过去,当母亲是35岁或更大年纪时,推荐进行羊膜检查或绒膜绒毛检查。已知这些技术是侵入性方法,其中胎儿丢失率为1/100至1/300,准确性不高,并且假阳性率和假阴性率高。被分类为高危人群的95%或更多的孕妇需要更准确和安全的诊断测试方法,以生出健康的婴儿。为了诊断染色体异常和性染色体异常,使用下一代测序分析技术的具有安全性和高准确性的技术从2000年代后期引入(韩国专利申请第10-2010-7003969号,第10-2012-7034453号),其中通过测序无细胞dna(cfdna)诊断非整倍性与传统分析方法相比具有高准确性和低假阴性率的优点。最近,正在进行基于下一代序列分析(ngs)技术的非侵入性产前测试(nipt)技术,即一种最新的技术(bianchidwetal.,(2014)dnasequencingversusstandardprenatalaneuploidyscreening.n.engl.j.ofmed.370:799–808.,sparksabetal.,(2012)noninvasiveprenataldetectionandselectiveanalysisofcell-freednaobtainedfrommaternalblood:evaluationfortrisomy21andtrisomy18.am.j.obstet.gynecol.206:319.e1–9.,ehrich,m.etal.,(2011)noninvasivedetectionoffetaltrisomy21bysequencingofdnainmaternalblood:astudyinaclinicalsetting.am.j.obstet.gynecol.204:205.e1–11.),并通过使用illumina和iontorrent公司的两个平台进行快速和有效的临床分析测试。开始时使用illumina平台的非侵入性产前测试(nipt)分析的灵敏度和特异性约为98%(lau,t.k.etal.,(2014)non-invasiveprenataltestingforfetalchromosomalabnormalitiesbylow-coveragewholegenomesequencingofmaternalplasmadna:reviewof1982consecutivecasesinasinglecenter.ultrasoundobstet.gynecol.43:254–264.,ashoorgetal.,(2012)chromosome-selectivesequencingofmaternalplasmacell-freednaforfirsttrimesterdetectionoftrisomy21andtrisomy18.am.j.obstet.gynecol.206:322.e1–5.)。然而,最近的研究显示了使用hiseq(illumina)和ionproton(iontorrent)的胎儿染色体13、18和21的非整倍性诊断灵敏度高于99.5%(liao,c.,etal.,(2014)noninvasiveprenataldiagnosisofcommonaneuploidiesbysemiconductorsequencing.proc.natl.acad.sci.usa.111:7415–7420.)。许多染色体相关的临床疾病与性染色体的拷贝数以及常染色体改变相关。最常见的性染色体非整倍性(sca)是特纳综合征(turnersyndrome)[45,x]、xxx综合征(三x[47,xxx])、克兰费尔特综合征(klinefeltersyndrome)[47,xxy]和超雄综合征(supermalesyndrome)([47,xyy]综合征)。已知这些疾病具有相对罕见的发病率,但报道了这些疾病引起约0.3%的累积性染色体非整倍性(morris,j.k.,etal.,istheprevalenceofklinefeltersyndromeincreasing;eur.j.hum.genet.2007;16:163-70.)。在庆北国立大学附属医院(kyungpooknationaluniversityhospital)儿科的115例患者的20年研究中,克兰费尔特综合征的性染色体异常的频率为52%,特纳综合征为42%,xxx综合征为3%,并且混合型性腺发育不良为3%。克兰费尔特综合征具有97%的典型xxy型和3%的镶嵌性(mosaicism),特纳综合征具有67%的典型x型,23%的镶嵌性,和10%的x染色体的结构异常。xxx综合征具有67%的xxx型和33%的镶嵌性,并且混合型性腺发育不良全部是45,x/46,xy(seo,hyunjietal.,acytogeneticstudyinpatientswithsexchromosomeabnomalities,koreajournalofpediatrics,vol48,no.12,2005)。最近,通过使用胎儿细胞dna(无细胞胎儿dna,cffdna)和ngs可检测到性染色体非整倍性,但通过定量比常染色体非整倍性的准确性更低的染色体数目来检测。此外,存在染色体x和y之间的序列相似性以及与gc组成相关的测序偏差,并且染色体y和其他染色体之间的相似性在信噪比处降低,并且具有小尺寸的染色体y具有许多变异。大多数病例不容易被检测到,并且在性染色体非整倍性的分类中总是有问题。不同于13、18和21染色体上的非整倍性,全球趋势是xo、xxx和xxy评判的准确性不高。事实上,在verifi(illumina)和nifty(bgi)的情况下,对13、18和21染色体的灵敏度为98.15%或更高。但是,verifi对性染色体的单体x(monosomyx)具有95%的灵敏度,并且对于xxx/xxy/xyy,数据的量小,并且因此不表示准确性。在nifty的情况下,基于修正的内部染色体z-分数而不是基于现有的z-分数,将单体x的灵敏度表示为75%,并且其余由于少量数据而未被表示。即使数据的量小,目前的算法也不具有高准确性,并且因此有必要开发一种优化的新算法。另外,当前的方法已经在每个特定平台的基础上开发,并且因此不可能应用其他平台的数据并且使用两个或更多个数据执行标准化。另外,已知能够准确检测到高达4%的胎儿dna,其与胎儿常染色体非整倍性相似(chiurwetal.,maternalplasmadnaanalysiswithmassivelyparallelsequencingbyligationfornoninvasiveprenataldiagnosisoftrisomy21.clin.chem.2009;56:459–63.,palomaki,g.e.,etal.,dnasequencingofmaternalplasmatodetectdownsyndrome:aninternationalclinicalvalidationstudy.genetmed.2011;13:913–20.),并且因此需要更好的方法和数据集的分类。因此,为了解决性别分类和性染色体非整倍性的问题,本发明的发明人发现当进行gc比例和映射率(mappability)的loess标准化时,使用chrx和chry的百分比确定chrx和chry值,无论平台的种类和数据如何,通过使用内部染色体参考调节参考线以确定最终z分数,可能以高准确性和灵敏度区分性别和性染色体异常,并且完成本发明。技术实现要素:本发明的目的是提供一种无论样品和平台的类型如何通过一次同时标准化和分析所有输入的数据来区分胎儿性别和胎儿性染色体的方法。为了实现前述目的,本发明提供了一种区分胎儿性别和胎儿性染色体异常的方法,其包括:(a)通过从母血中提取dna而在两个或更多个平台上进行下一代测序(ngs);(b)将对每个平台测序的序列(读长(reads))与参考基因组数据库进行比对;(c)提取每个平台在染色体上唯一比对的独特读长,并计算独特读长相对于总读长的百分比(ur%);(d)同时计算每个平台的数据,设置截断值,并组织ur(y)%值;(e)使用组织的ur(y)%将男性和女性分类;(f)使用大小与染色体x和y相似的三个或更多个常染色体计算部分参考染色体(相对fgr)值;和(g)使用所述部分参考染色体(相对fgr)值计算z分数,并基于所述z分数计算g分数(gscore)和m分数(mscore)以区分性染色体异常。附图说明图1是本发明的诊断性染色体非整倍性的整个流程图。图2显示使用hiseq平台(illumina)分析的样品的性染色体独特读长%的分析结果。左纵轴表示ur(x)%,并且右纵轴表示ur(y)%。图3显示使用hiseq平台(illumina)的两个实验组的性染色体独特读长%的分析结果之间的比较。图4显示使用ionproton平台(iontorrent)的两个实验组的性染色体独特读长%的分析结果之间的比较。图5是显示使用illumina平台在51个样品的截断值的基础上的性别分类的结果的图。图6是显示在使用ionproton平台(iontorrent)分析的两个不同实验组中在与图5中截断值相同的基础上的性别分类的结果的图。图7是显示通过仅使用染色体7(chr7)和染色体20(chr20)作为内部参考计算的z分数值的图。图8是显示通过使用四个内部参考计算的m分数的图。图9是显示通过使用四个内部参考计算的g分数的图。图10是显示从中去除pcr复制的两个平台(illumina的hiseq和iontorrent的ionproton)的图。图11是显示在两个平台(illumina的hiseq和iontorrent的ionproton)上独特读长的百分比的图。图12是显示使用一个样品在两个平台上同时进行测序的流程图。图13是显示当根据本发明的示例性实施方案将样品中的女性进行分类时,通过g分数区分的性染色体异常的结果的图。图14是显示当根据本发明的示例性实施方案将样品中的男性进行分类时,通过m分数区分的性染色体异常的结果的图。图15是显示根据本发明的示例性实施方案通过添加样品使用chrx和chry区分性染色体异常的结果的图。具体实施方式除非另有定义,否则本文所使用的技术和科学术语具有与本发明所属领域的技术人员通常理解的那些相同的含义。一般地,本说明书和下述实验方法中使用的术语是本发明
技术领域:
中公知的和通常使用的。在本发明中,证实当使从两个或更多个ngs平台生成的测序数据标准化并基于截断值比对,然后使用在性染色体上使用部分参考染色体比对的独特读长%时,可能以高准确性和灵敏度区分胎儿性别和性染色体异常。换言之,在本发明的示例性实施方案中,开发了以下方法:使用两个或更多个ngs平台测序提取自母血的基因组dna(gdna),基于gc含量和映射率(mappability)定量每个染色体的独特读长的量以设置截断值,使用chr7至chr10作为用于定量染色体x的部分参考染色体和使用chr19至chr22作为用于定量染色体y的部分参考染色体,以及在男性的情况下计算m分数(男性分数)来确定正常(xy)和克兰费尔特综合征(xxy),和在女性的情况下计算g分数(女孩分数)来诊断xxx综合征(三x,xxx)以及特纳综合征(xo)和正常(xx)(图1)。因此,在一方面,本发明涉及区分胎儿性别和胎儿性染色体异常的方法,其包括:(a)通过从母亲生物样品中提取dna而在两个或更多个平台上进行下一代测序(ngs);(b)将对每个平台测序的序列(读长)与参考基因组数据库进行比对;(c)提取每个平台在染色体上唯一比对的独特读长,并计算独特读长相对于总读长的百分比(ur%);(d)同时计算每个平台的数据,设置截断值,并组织ur(y)%值;(e)使用组织的ur(y)%将男性和女性分类;(f)使用大小与染色体x和y相似的三个或更多个常染色体计算部分参考染色体(相对fgr)值;和(g)使用相对fgr值计算z分数,并基于所述z分数计算g分数(gscore)和m分数(mscore)以区分性染色体异常。在本发明中,所述生物样品可选自下组:血液、血浆、血清、尿和唾液。在本发明中,步骤(a)包括:(a)通过离心所收集的生物样品以分离血浆;(b)从分离的血浆提取cfdna;(c)使用提取的cfdna制备文库;和(d)汇集制备的文库,然后使用ngs设备解码碱基序列。汇集本发明的文库的特征可在于在illumina平台的情况下进行珠子大小选择并在生命技术平台的情况下进行e-凝胶大小选择。在本发明中,步骤(c)可包括:(a)去除测序中的pcr复制和歧义读长,仅比对完美匹配的读长,并提取读长,其中gc含量和映射率大于或等于比对序列中的截断值;和(b)基于提取的读长,根据以下等式计算唯一匹配染色体x的读长的百分比(ur(x)%)和唯一匹配染色体y的读长的百分比(ur(y)%):等式1)ur(x)%=染色体x(chrx)读长的数量/常染色体读长的总数x100,和等式2)ur(y)%=染色体y(chry)读长的数量/常染色体读长的总数x100。本发明的复制读长的去除可通过用于去除复制的任意方法进行,但是优选通过loess标准化进行。在本发明中,分析gc含量和映射率的步骤可通过将每个区域分成100kb测序单元(bin)区域来进行,其中gc含量的截断值可为0.30至0.50,并且映射率的截断值可为20%或更大。在本发明中,ur(y)%的标准化在染色体y的基础上进行以在染色体xx和xy之间进行准确分类,其中可将单一样品的结果分成ur(y)%的截断标准,但是当在两个或更多个样品的大量样品或另一个平台样品上进行标准化时,不可能设置ur(y)%的截断(图3和4)。为了不但在一个实验室中获得多个样品的结果,而且还利用已经分析的许多结果,需要在多个平台并且一次在不同位置标准化结果,然后根据截断标准将男性和女性分类,并且因此其特征在于通过根据以下等式校正ur(y)%来进行标准化,每个平台的ur(y)%如下计算:等式3)ur(y)%_新的=(ur(y)%_原始的-(三个最小ur(y)%值的平均值)在本发明中,(d)的截断值优选在0.02至0.06的范围内,并且最优选0.037。在本发明中,(e)的性别分类的特征在于,当组织的ur(y)%值大于或等于截断值时,确定为男性,并且当组织的ur(y)%值小于截断值时,确定为女性。在本发明中,为了检测性染色体非整倍性,选择大小与染色体y相似的染色体chr19至chr22作为内部染色体参考,选择大小与染色体x相似的染色体chr7至chr10作为内部染色体参考,并且计算部分参考染色体(相对fgr)(lau,t.k.,et.al,j.matern.fetalneonatalmed.2012aug;25(8):1370-4.)。从其中仅选择chr7和chr20的论文获得不正确的结果,因为测序偏差很严重(图7),但是可证实计算了从fgr(7)到fgr(10)的各个值的平均值,并且因此误差率显著减小(图8和9)。因此,在本发明中,步骤(f)可包括:(a)选择大小与性染色体x(chrx)相似的常染色体(chr7、chr8、chr9和chr10)作为内部参考染色体,并且通过以下等式计算每个fgr:等式4)fgr(7)=(ur(x)%+ur(y)%)/ur(7)%等式5)fgr(8)=(ur(x)%+ur(y)%)/ur(8)%等式6)fgr(9)=(ur(x)%+ur(y)%)/ur(9)%等式7)fgr(10)=(ur(x)%+ur(y)%)/ur(10)%,和(b)选择大小与性染色体y(chry)相似的常染色体(chr19、chr20、chr21和chr22)作为部分参考染色体,并且通过以下等式计算每个fgr:等式8)fgr(19)=(ur(x)%+ur(y)%)/ur(19)%等式9)fgr(20)=(ur(x)%+ur(y)%)/ur(20)%等式10)fgr(21)=(ur(x)%+ur(y)%)/ur(21)%等式11)fgr(22)=(ur(x)%+ur(y)%)/ur(22)%。在本发明中,步骤(g)可包括:(a)当确定胎儿的性别为女性时,通过以下步骤区分性染色体异常;a-1)在以下等式中计算每个染色体的z分数:等式12)z分数(7)={fgr(7)(平均fgr(7))}/(stdevfgr(7))等式13)z分数(8)={fgr(8)(平均fgr(8))}/(stdevfgr(8))等式14)z分数(9)={fgr(9)(平均fgr(9))}/(stdevfgr(9))等式15)z分数(10)={fgr(10)(平均fgr(10))}/(stdevfgr(10))a-2)通过以下等式使用计算的z分数计算g分数;和等式16)g分数={z分数(7)+z分数(8)+z分数(9)+z分数(10)}/4a-3)当所述g分数大于或等于1.5时,确定性染色体为xxx,当所述g分数大于-3且小于1.5时,确定所述性染色体为xx,并且当所述g分数小于或等于-3时,确定所述性染色体为xo;(b)当确定胎儿的性别为男性时,通过以下步骤区分性染色体异常;b-1)在以下等式中计算每个染色体的z分数:等式17)z分数(19)={fgr(19)(平均fgr(19))}/(stdevfgr(19))等式18)z分数(20)={fgr(20)(平均fgr(20))}/(stdevfgr(20))等式19)z分数(21)={fgr(21)(平均fgr(21))}/(stdevfgr(21))等式20)z分数(22)={fgr(22)(平均fgr(22))}/(stdevfgr(22))b-2)通过以下等式使用计算的z分数计算m分数;和等式21)m分数={z分数(19)+z分数(20)+z分数(21)+z分数(22)}/4b-3)当所述m分数大于或等于0.7时,确定所述性染色体为xxy,当所述m分数大于-3且小于0.7时,确定所述性染色体为xy。本发明还涉及一种计算机系统,其包括计算机可读介质,其具有加密的用于控制计算机系统的多个指令,以执行使用两个或更多个ngs平台从提取自母亲生物样品的gdna区分胎儿性别和性染色体异常的方法,其中所述生物样品含有胎儿的无细胞核酸分子,和所述区分胎儿性别和性染色体异常的方法包括:将对每个ngs平台测序的序列(读长)与参考基因组数据进行比对;提取每个ngs平台的对染色体唯一的独特读长,并计算独特读长(ur%)相对于总读长的百分比;同时计算每个ngs平台的数据,设置截断值,并组织ur(y)%值;使用组织的ur(y)%值将男性和女性分类;使用大小与染色体x和y相似的三个或更多个常染色体计算部分参考染色体(相对fgr);和使用所述相对fgr值计算z分数,并且基于所述z分数计算g分数(gscore)和m分数(mscore)以区分性染色体异常。实施例以下,将参考以下实施例详细描述本发明。然而,以下实施例仅用于示例本发明,并且将对本领域技术人员显而易见的是,本发明的范围不被解释为局限于这些实施例。<实施例1>使用母亲dna进行下一代测序使用离心分离从母亲收集的血液以提取血浆,然后溶解血浆以通过提取30ng或更多的cfdna制备文库。在illumina平台的情况下,在组合适配体后,使用珠子大小选择进行汇集,接着测序。在生命技术平台的情况下,在组合适配体后,使用e-凝胶大小选择进行汇集,接着测序。<实施例2>将测序后获得的序列与参考基因组(人grch38)进行比对通过使用包括仅约250个间隔的grch38(2013年12月24日发布)的最新版本,进行序列的比对以去除混杂的序列。使用samtools查看选项以及去除pcr复制的一般picard提取最小独特读长。为了分析从两个或更多个平台生成的数据,分析了初始文件大小和读长长度的分布(表1)。因为仅所有区域中的独特读长不能通过一般pcr复制去除操作来收集(图10),在使用其他过滤选项后,为hiseq收集了约85%的独特读长,并且为proton收集了约76%的独特读长(表2)。[表1]10m读长的文件大小(单位:千兆字节)样品id77916473927276626867837486857166876365706984illumina1.61.61.61.61.61.61.61.61.61.61.61.61.61.61.61.61.61.61.61.61.61.6proton2.92.92.92.92.92.92.92.92.92.92.92.92.92.82.82.82.82.82.72.72.62.610m读长的文件大小(单位:十亿字节)样品id[表2]10m读长的映射率%(单位:%)10m读长的映射率%(单位:%)<实施例3>通过标准化多样品和多平台数据的数据保持将每个染色体分成100kb测序单元区域。然后,对于gc含量,去除除0.30<=gc含量<=0.50之外的区域,并且对于映射率,仅选择具有20%或更大映射率的区域以去除pcr复制和噪音区域。仅使用z分数的平均值来观察总体变化。为了建立从illumina和proton平台之间的差异和每个实验组和位置或环境的差异获得的实验结果的偏差(图3和4)的标准,获得ur(y)%的三个最小值的平均值,并且从原始值减去该平均值,由此证实了该值的变化程度通过某一截断值显著减小(图5和6)。<实施例4>使用保持的数据值和gm分数计算的性别分类将来自在不同实验条件下使用不同平台进行的结果的相同三个最小值的平均值设置为参考线。然后,将染色体y的独特读长ur(y)%的百分比的截断值设置在0.02至0.04的范围内以将男性和女性分类。当性别是女性(xx)时,xo和xxx通过使用超出正态分布的情况来区分。当性别是男性(xy)时,将超出正态xy分布的数据评判为xxy。当关于ur(x)%(即chrx的独特读长的百分比)时,使用具有相似大小的chr7计算部分参考染色体,即相对fgr,并且计算各个z分数(图7),并且当关于ur(y)%(即chry的独特读长的百分比)时,也使用具有相似大小的chr20计算部分参考染色体,即相对fgr,并且计算各个z分数(图7),值的分布程度高度偏差。因此,为了获得具有相似大小的染色体的平均值,证实获得大小与chrx相似的chr7、chr8、chr9和chr10的平均值以计算部分参考染色体(相对fgr)和z分数,由此获得m分数,即最接近chrx的标准值的值(图9)。如在chry中,根据与chrx相同的原则,证实获得chr19、chr20、chr21和chr22的平均值以计算相对fgr和z分数,由此获得m分数,由此最精确的检测chry的变化程度(图8)。<实施例5>通过gm分数进行的最终诊断结果通过使用实施例1至4的方法,证实可清楚地区分94个正常人,具有xxx基因型的1个人和具有xxy基因型的1个人(表3),并且可满足所有截断值。然后,通过羊膜腔穿刺术证实诊断。即使使用小样品,与同源染色体的异常相比,也较难获得样品,并且提供了可从标准比率清楚区分的分数,并且因此可证实该方法能够仅通过gm分数有效地评判染色体异常(图13和14)。可通过g分数从xxx中区分xx,并且可预测通过使用-3或更小的截断值也能够评判出xo。此外,证实了可通过m分数从xy有效地区分出xxy的分布。[表3]样品的数量女性男性正常4945xxx1xxy1总计5046<实施例6>诊断另外的样品组的性别通过使用在实施例5中公开的方法,证实分析了表4所示的样品组(具有xo基因型的3个人、具有xyy基因型的1个人和121个正常人的数据)以区分性别和染色体异常,概率为100%(图15)。[表4]女性男性正常101114xxx1xo3xxy1xyy1总计105116已经基于本发明的特定特征详细描述了本发明,并且对本领域技术人员显而易见的是,这些特定技术仅是优选的实施方案,并且因此本发明的范围不局限于这些实施方案。因此,本发明的实质范围通过所附权利要求及其等同物来定义。工业实用性根据本发明的区分胎儿性别和性染色体异常的方法不但可增加使用下一代测序(ngs)的性别分类的准确性,还可增加难以被检测出的xo、xxx、xxy等的检测准确性,并且因此可增加性染色体的商业使用连同常染色体异常的非侵入性检测。因此,本发明的方法可用于产前诊断,其能够确定由于在早期在胎儿中胎儿性染色体的异常造成畸形的发生。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1