一种生物数据的索引方法、系统及电子设备与流程
本申请属于计算机科学与生物信息学的交叉领域,特别涉及一种生物数据的索引方法、系统及电子设备。
背景技术:
生物数据索引的建立,实质就是基于一些字符串进行哈希的过程。生物数据,特别是基因数据,数据索引的建立、基于索引的统计分析,两者都是生物数据分析的基础。如生物数据的聚类分类、生物数据的特征提取、信息挖掘都需要基于索引信息进行统计分析。
以基因数据为代表的测序技术的飞速发展、第二代以及第三代测序技术的普及,生命科学领域进入了以爆发式的海量基因数据为特征的大数据时代。基因数据正在以12-18个月10倍以上超过摩尔定律的速度指数增长。早在2008年中英美德等国家就发起了“国际千人基因组计划”,该计划的数据总量高达200tb;在宏基因组研究领域,1克土壤样品测序后便可产生50tb的原始基因数据。然而当前传统的串行基因组数据分析方法或者单节点的并行分析以及索引方法已然不能满足以上各种应用对tb乃至pb级别的基因组数据分析需求,这就对生物大数据的索引及分析提出了巨大挑战。
近年来出现了不少值得关注的单节点高效基于k-mer(长度为k的基因序列)频率计数基因组统计分析工具,其中也涉及了生物数据的索引。例如马里兰大学gmarcais设计的无锁哈希压缩存储的多线程并行k-mer频率计数工具jellyfish[m
随着基因测序技术的快速发展和测序成本的不断降低,生物信息分析研究的数据越来越多,计算模型也越来越复杂。普通个人计算机以及传统串行计算方式已经无力解决生物信息分析中的大规模数据处理问题。为了解决生物信息分析中对计算能力和计算时间的高要求,必须借助高性能计算技术和超级计算机集群系统。
然而,上述的现有基于生物大数据的索引及统计分析工具大都是单节点并行,扩展性方面的表现都较差,在处理tb-pb级数据时存在诸多问题,并且由于这些工具在设计之初并没有考虑到超大规模的生物数据的处理,所以耗时较长,而且当数据量过大时易出现运行出错、异常、终止等问题。
技术实现要素:
本申请提供了一种生物数据的索引方法、系统及电子设备,旨在至少在一定程度上解决现有技术中的上述技术问题之一。
为了解决上述问题,本申请提供了如下技术方案:
一种生物数据的索引方法,包括以下步骤:
步骤a:对生物数据进行并行化输入,并对输入的生物数据进行质量筛选;
步骤b:对质量筛选后的生物数据进行k-mer提取;
步骤c:k-mer提取完成后,通过两级哈希分别将每个k-mer散列到对应的处理器上,得到生物数据的分布式哈希索引。
本申请实施例采取的技术方案还包括:在所述步骤a中,所述对生物数据进行并行化输入具体为:获取集群中处理器的个数,设进程的个数为p,生物数据的大小为s,每个进程所需输入的生物数据大小为s/p,根据每个进程所需输入的生物数据的大小得到每个进程的数据起始地址。
本申请实施例采取的技术方案还包括:在所述步骤b中,所述对质量筛选后的生物数据进行k-mer提取具体为:每个进程分别对并行化i/o模块输入的数据进行reads到k-mer的并行提取,用一个长度为k的窗口对长度为m的reads从第一个位置开始扫描,直到窗口末端与reads的末端重合。
本申请实施例采取的技术方案还包括:在所述步骤c中,所述通过两级哈希分别将每个k-mer散列到对应的处理器上具体为:k-mer提取完成后,根据k-mer的值调用一个卷积哈希函数hash_func进行第一级哈希散列,分别找到每个k-mer映射到对应处理器的procid;完成第一级哈希后,每个进程分别对各自的k-mer进行第二级哈希,形成生物数据的分布式哈希索引。
本申请实施例采取的技术方案还包括:所述步骤c后还包括:根据分布式哈希索引统计k-mer的出现频率进行生物数据的统计分析。
本申请实施例采取的另一技术方案为:一种生物数据的索引系统,包括:
数据输入模块:用于对生物数据进行并行化输入;
质量控制模块:用于对输入的生物数据进行质量筛选;
k-mer提取模块:用于对质量筛选后的生物数据进行k-mer提取;
哈希模块:用于在k-mer提取完成后,通过两级哈希分别将每个k-mer散列到对应的处理器上,得到生物数据的分布式哈希索引。
本申请实施例采取的技术方案还包括:所述数据输入模块对生物数据进行并行化输入具体为:获取集群中处理器的个数,设进程的个数为p,生物数据的大小为s,每个进程所需输入的生物数据大小为s/p,根据每个进程所需输入的生物数据的大小得到每个进程的数据起始地址。
本申请实施例采取的技术方案还包括:所述k-mer提取模块对质量筛选后的生物数据进行k-mer提取具体为:每个进程分别对并行化i/o模块输入的数据进行reads到k-mer的并行提取,用一个长度为k的窗口对长度为m的reads从第一个位置开始扫描,直到窗口末端与reads的末端重合。
本申请实施例采取的技术方案还包括:所述哈希模块包括第一哈希单元和第二哈希单元;所述第一哈希单元用于在k-mer提取完成后,根据k-mer的值调用一个卷积哈希函数hash_func进行第一级哈希散列,分别找到每个k-mer映射到对应处理器的procid;所述第二哈希单元用于在完成第一级哈希后,每个进程分别对各自的k-mer进行第二级哈希,形成生物数据的分布式哈希索引。
本申请实施例采取的技术方案还包括统计分析模块,所述统计分析模块用于根据分布式哈希索引统计k-mer的出现频率进行生物数据的统计分析。
本申请实施例采取的又一技术方案为:一种电子设备,包括:
至少一个处理器;以及
与所述至少一个处理器通信连接的存储器;其中,
所述存储器存储有可被所述一个处理器执行的指令,所述指令被所述至少一个处理器执行,以使所述至少一个处理器能够执行上述的生物数据的索引方法的以下操作:
步骤a:对生物数据进行并行化输入,并对输入的生物数据进行质量筛选;
步骤b:对质量筛选后的生物数据进行k-mer提取;
步骤c:k-mer提取完成后,通过两级哈希分别将每个k-mer散列到对应的处理器上,得到生物数据的分布式哈希索引。
相对于现有技术,本申请实施例产生的有益效果在于:本申请实施例的生物数据的索引方法、系统及电子设备通过利用高性能集群,设计了一个高可伸缩高可扩展的框架,充分利用计算集群,通过多节点共享内存以及两级哈希的方式,实现大规模生物数据的分布式哈希,解决了单节点所能进行索引及统计分析的哈希规模的限制,实现生物数据的高效索引和极速分析。
附图说明
图1是本申请实施例的生物数据的索引方法的流程图;
图2是本申请实施例的哈希索引建立过程示意图;
图3是本申请实施例的生物数据的索引系统的结构示意图;
图4是本申请实施例提供的生物数据的索引方法的硬件设备结构示意图。
具体实施方式
为了使本申请的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本申请进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本申请,并不用于限定本申请。
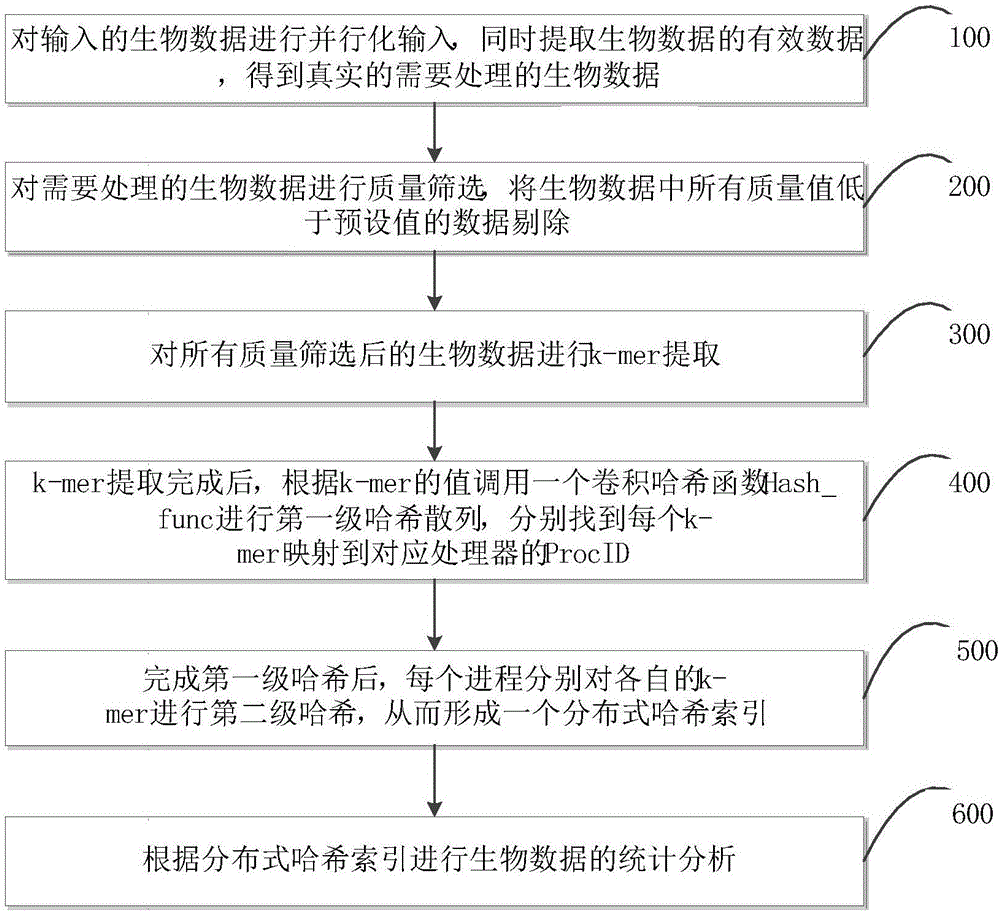
请参阅图1,是本申请实施例的生物数据的索引方法的流程图。本申请实施例的生物数据的索引方法包括以下步骤:
步骤100:对输入的生物数据进行并行化输入,同时提取生物数据的有效数据,得到真实的需要处理的生物数据;
在步骤100中,对输入的生物数据进行并行化输入具体为:首先获取集群中处理器的个数,同时设进程的个数p,假设输入的生物数据的大小为s,每个进程所需输入的生物数据大小即为s/p,然后根据每个进程所需输入的生物数据大小得到每个进程的数据起始地址start(p)、end(p)。
步骤200:对需要处理的生物数据进行质量筛选,将生物数据中所有质量值低于预设值的数据剔除;
在步骤200中,对需要处理的生物数据进行质量筛选具体为:由步骤100可知,生物数据被分为p块,对每一块加载的生物数据分别进行预处理,即进行生物数据质量的控制,利用软件包fastx_toolkit中的质量过滤工具fastq_quality_filter将生物数据中质量低于预设值的序列过滤掉。本申请实施例中,预设值优选为20,具体可根据实际应用进行设定。
步骤300:对所有质量筛选后的生物数据进行k-mer提取;
在步骤300中,k-mer提取过程可以独立并行,k-mer提取过程具体为:各进程分别对并行化i/o模块不断输入的数据进行reads到k-mer的并行提取,设一行生物数据reads的长度为m,k-mer的长度为k,整个k-mer的提取过程即用一个长度为k的窗口对长度为m的reads从第一个位置开始扫描,直到窗口末端与reads的末端重合。
步骤400:k-mer提取完成后,根据k-mer的值调用一个卷积哈希函数hash_func进行第一级哈希散列,分别找到每个k-mer映射到对应处理器的procid(处理器的编号);
在步骤400中,本申请调用一个卷积哈希函数hash_func(x)进行哈希散列,从而通过k-mer值找到对应处理器的procid,完成k-mer在p个处理器上的哈希划分和存储。
步骤500:完成第一级哈希后,每个进程分别对各自的k-mer进行第二级哈希,从而形成生物数据的分布式哈希索引;
在步骤500中,请一并参阅图2,是本申请实施例的哈希索引建立过程示意图。每一个k-mer都需要经过两级哈希才能散列到最终正确的位置(对应处理器)上,当k-mer被哈希散列到对应处理器后,待插入k-mer可直接根据其哈希值进行插入。本申请利用高性能集群,通过多节点共享内存以及两级哈希的方式,实现大规模生物数据的分布式哈希,任何数据的极速索引都可通过两级哈希完成,从而实现生物数据的高效索引。
步骤600:根据分布式哈希索引进行生物数据的统计分析;
在步骤600中,生物数据的统计分析都是基于分布式哈希索引进行,通过对每个k-mer的出现频率或特定k-mer的出现频率进行统计,数据频谱分布服从何种分布也可以从中得到,统计分析的核心是对于hash_map及分布式哈希索引进行合并分析(reduce)的过程,实现生物数据的极速统计分析。
请参阅图3,是本申请实施例的生物数据的索引系统的结构示意图。本申请实施例的生物数据的索引结构包括数据输入模块、质量控制模块、k-mer提取模块、哈希模块和统计分析模块。
数据输入模块:用于对输入的生物数据进行并行化输入,同时提取生物数据的有效数据,得到真实的需要处理的生物数据;其中,对输入的生物数据进行并行化输入具体为:首先获取集群中处理器的个数,同时设进程的个数p,假设输入的生物数据的大小为s,每个进程所需输入的生物数据大小即为s/p,然后根据每个进程所需输入的生物数据大小得到每个进程的数据起始地址start(p)、end(p)。
质量控制模块:用于对需要处理的生物数据进行质量筛选,将生物数据中所有质量值低于预设值的数据剔除;其中,对需要处理的生物数据进行质量筛选具体为:生物数据被分为p块,对每一块加载的生物数据分别进行预处理,即进行生物数据质量的控制,利用软件包fastx_toolkit中的质量过滤工具fastq_quality_filter将生物数据中质量低于预设值的序列过滤掉。本申请实施例中,预设值优选为20,具体可根据实际应用进行设定。
k-mer提取模块:用于对所有质量筛选后的生物数据进行k-mer提取;其中,k-mer提取过程可以独立并行,k-mer提取过程具体为:各进程分别对并行化i/o模块不断输入的数据进行reads到k-mer的并行提取,设一行生物数据reads的长度为m,k-mer的长度为k,整个k-mer的提取过程即用一个长度为k的窗口对长度为m的reads从第一个位置开始扫描,直到窗口末端与reads的末端重合。
哈希模块:用于在k-mer提取完成后,通过两级哈希分别将每个k-mer散列到对应的处理器上,得到生物数据的分布式哈希索引;具体地,哈希模块包括第一哈希单元和第二哈希单元:
第一哈希单元:用于在k-mer提取完成后,根据k-mer的值调用一个卷积哈希函数hash_func进行第一级哈希散列,分别找到每个k-mer映射到对应处理器的procid(处理器的编号);其中,本申请调用一个卷积哈希函数hash_func(x)进行哈希散列,从而通过k-mer值找到对应处理器的procid,完成k-mer在p个处理器上的哈希划分和存储。
第二哈希单元:用于在完成第一级哈希后,通过每个进程分别对各自的k-mer进行第二级哈希,形成生物数据的分布式哈希索引;本申请实施例中,每一个k-mer都需要经过两级哈希才能散列到最终正确的位置(对应处理器)上,当k-mer被哈希散列到对应处理器后,待插入k-mer可直接根据其哈希值进行插入。本申请利用高性能集群,通过多节点共享内存以及两级哈希的方式,实现大规模生物数据的分布式哈希,任何数据的极速索引都可通过两级哈希完成,从而实现生物数据的高效索引。
统计分析模块:用于根据分布式哈希索引进行生物数据的统计分析;本申请实施例中,生物数据的统计分析都是基于分布式哈希索引进行,通过对每个k-mer的出现频率或特定k-mer的出现频率进行统计,数据频谱分布服从何种分布也可以从中得到,统计分析的核心是对于hash_map及分布式哈希索引进行合并分析(reduce)的过程,实现生物数据的极速统计分析。
图4是本申请实施例提供的生物数据的索引方法的硬件设备结构示意图。如图4所示,该设备包括一个或多个处理器以及存储器。以一个处理器为例,该设备还可以包括:输入系统和输出系统。
处理器、存储器、输入系统和输出系统可以通过总线或者其他方式连接,图4中以通过总线连接为例。
存储器作为一种非暂态计算机可读存储介质,可用于存储非暂态软件程序、非暂态计算机可执行程序以及模块。处理器通过运行存储在存储器中的非暂态软件程序、指令以及模块,从而执行电子设备的各种功能应用以及数据处理,即实现上述方法实施例的处理方法。
存储器可以包括存储程序区和存储数据区,其中,存储程序区可存储操作系统、至少一个功能所需要的应用程序;存储数据区可存储数据等。此外,存储器可以包括高速随机存取存储器,还可以包括非暂态存储器,例如至少一个磁盘存储器件、闪存器件、或其他非暂态固态存储器件。在一些实施例中,存储器可选包括相对于处理器远程设置的存储器,这些远程存储器可以通过网络连接至处理系统。上述网络的实例包括但不限于互联网、企业内部网、局域网、移动通信网及其组合。
输入系统可接收输入的数字或字符信息,以及产生信号输入。输出系统可包括显示屏等显示设备。
所述一个或者多个模块存储在所述存储器中,当被所述一个或者多个处理器执行时,执行上述任一方法实施例的以下操作:
步骤a:对生物数据进行并行化输入,并对输入的生物数据进行质量筛选;
步骤b:对质量筛选后的生物数据进行k-mer提取;
步骤c:k-mer提取完成后,通过两级哈希分别将每个k-mer散列到对应的处理器上,得到生物数据的分布式哈希索引。
上述产品可执行本申请实施例所提供的方法,具备执行方法相应的功能模块和有益效果。未在本实施例中详尽描述的技术细节,可参见本申请实施例提供的方法。
本申请实施例提供了一种非暂态(非易失性)计算机存储介质,所述计算机存储介质存储有计算机可执行指令,该计算机可执行指令可执行以下操作:
步骤a:对生物数据进行并行化输入,并对输入的生物数据进行质量筛选;
步骤b:对质量筛选后的生物数据进行k-mer提取;
步骤c:k-mer提取完成后,通过两级哈希分别将每个k-mer散列到对应的处理器上,得到生物数据的分布式哈希索引。
本申请实施例提供了一种计算机程序产品,所述计算机程序产品包括存储在非暂态计算机可读存储介质上的计算机程序,所述计算机程序包括程序指令,当所述程序指令被计算机执行时,使所述计算机执行以下操作:
步骤a:对生物数据进行并行化输入,并对输入的生物数据进行质量筛选;
步骤b:对质量筛选后的生物数据进行k-mer提取;
步骤c:k-mer提取完成后,通过两级哈希分别将每个k-mer散列到对应的处理器上,得到生物数据的分布式哈希索引。
本申请实施例的生物数据的索引方法、系统及电子设备通过利用高性能集群,设计了一个高可伸缩高可扩展的框架,充分利用计算集群,通过多节点共享内存以及两级哈希的方式,实现大规模生物数据的分布式哈希,解决了单节点所能进行索引及统计分析的哈希规模的限制,实现生物数据的高效索引和极速分析。
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本申请。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本申请的精神或范围的情况下,在其它实施例中实现。因此,本申请将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
- 还没有人留言评论。精彩留言会获得点赞!