一种基于概率密度估计的数据差异分析方法及系统与流程
本发明涉及数据分析领域,更具体地,涉及一种基于概率密度估计的数据差异分析方法及系统。
背景技术:
显著变化的数据往往具有关键性。如通过蛋白质质谱技术,我们可以得到各个蛋白质在实验组与对照组的表达量,表达显著差异的蛋白质可能在该过程中起关键的调控作用。人们往往根据差异倍数来找差异蛋白质,认为变化倍数越大的蛋白质差异越显著。然而,在多数情况下,这种假设不能成立,比如1变为2与10变为20都是变化2倍,但是不代表其差异显著性相同。又例如影响蛋白质修饰状态的氨基酸突变,突变前后使蛋白质修饰状态显著变化的突变往往更重要,omar等人开发了一种预测突变对磷酸化影响的方法(mimp)。然而,对于独立的二维变量,mimp中的联合概率的计算公式是不成立的。并且其方法不能计算突变对磷酸化的影响的统计显著性水平。目前,面对此类问题,人们没有很好的解决方法,因而发展新方法解决诸如此上的问题非常关键。本发明发展了一种基于概率密度估计的数据差异分析方法,本方法具有统计学意义并且无论数据是什么分布,该方法均可适用。
技术实现要素:
本发明解决了现有技术中的数据差异分析方法不仅受数据分布限制,而且缺乏统计学意义的技术问题。本发明根据概率密度估计方法求得变化前后数据联合概率密度分布,然后根据假设检验判断数据变化的显著性。本方法可以不受数据分布的限制求得每个数据的显著程度,用于发现显著变化的数据。
根据本发明的第一方面,提供了一种基于概率密度估计的数据差异分析方法,含有以下步骤:
(1)将数据集中数据的组数记为n组,所述n为正整数;任意一组数据中含有变化前的数值和变化后对应的数值,记变化前的值为x,变化后的值为y,以变化前的数据为横坐标,以变化后的数据为纵坐标建立坐标系u,所述任意一组数据对应的坐标点为(xi,yi),所述i的取值范围为1≤i≤n;
(2)利用基于高斯核的概率密度估计法去估计变化前后数据联合概率密度分布,运用的公式为:
(3)固定变化前数据大小x,求得在该固定x的情况下,变化后的数据y在步骤(2)所述最佳联合概率分布中的概率密度分布;首先,在固定x的情况下,以y的分布作为x’轴,以该固定x在所述最佳h条件下的概率密度为y’轴建立坐标系u’;然后,对于数据集中的任意一组数据(xi,yi),求得在该xi的情况下,变化后的数据大小y的概率密度分布,根据yi在所述坐标系u’的x’轴上的位置,求得该组数据(xi,yi)的变化趋势和变化程度,具体方法为:在所述坐标系u’的x’轴上取一点,经过该点作垂直于坐标系u’的x’轴的直线,该直线将密度曲线与x轴所围成的面积平均分成左右两部分,记该点为y0,如果yi大于y0,则数据点(xi,yi)的变化是上调的,上调的显著程度p为y>yi时分布中的面积比上密度曲线与x’轴所围成的面积,如果yi小于y0,则数据点(xi,yi)的变化是下调的,下调的显著程度p为y<yi时分布中的面积比上密度曲线与x’轴所围成的面积,如果yi等于y0,则数据点(xi,yi)没有发生变化。
优选地,步骤(1)所述数据集中任意一组数据为氨基酸位点周围的至少一个氨基酸发生突变前和突变后,该氨基酸位点发生修饰的概率值。
优选地,步骤(1)所述数据集中任意一组数据为赖氨酸位点前后各n个氨基酸中至少一个氨基酸发生错义突变前和错义突变后,该赖氨酸位点发生琥珀酰化的概率值;所述n为整数,n的取值范围为0<n≤50。
优选地,所述n的取值范围为5≤n≤15。
优选地,步骤(1)所述数据集为药物处理细胞前和处理细胞后,该细胞产生rna或表达蛋白质水平的数据。
优选地,所述n大于等于1000。
根据本发明的另一方面,提供了一种基于概率密度估计的数据差异分析系统,包括:
数据集建立模块:所述数据集建立模块用于建立待分析差异的数据集;将数据集中数据的组数记为n组,所述n为正整数;任意一组数据中含有变化前的数值和变化后对应的数值,记变化前的值为x,变化后的值为y,以变化前的数据为横坐标,以变化后的数据为纵坐标建立坐标系u,所述任意一组数据对应的坐标点为(xi,yi),所述i的取值范围为1≤i≤n;
最佳窗口宽度计算模块:所述最佳窗口宽度计算模块用于计算最佳窗口宽度h,并得到最佳联合概率分布;利用基于高斯核的概率密度估计法去估计变化前后数据联合概率密度分布,运用的公式为:
数据集中数据差异分析模块:所述数据集中数据差异分析模块用于分析数据集中数据变化前后的差异;固定变化前数据大小x,求得在该固定x的情况下,变化后的数据y在步骤(2)所述最佳联合概率分布中的概率密度分布;首先,在固定x的情况下,以y的分布作为x’轴,以该固定x在所述最佳h条件下的概率密度为y’轴建立坐标系u’;然后,对于数据集中的任意一组数据(xi,yi),求得在该xi的情况下,变化后的数据大小y的概率密度分布,根据yi在所述坐标系u’的x’轴上的位置,求得该组数据(xi,yi)的变化趋势和变化程度,具体方法为:在所述坐标系u’的x’轴上取一点,经过该点作垂直于坐标系u’的x’轴的直线,该直线将密度曲线与x轴所围成的面积平均分成左右两部分,记该点为y0,如果yi大于y0,则数据点(xi,yi)的变化是上调的,上调的显著程度p为y>yi时分布中的面积比上密度曲线与x’轴所围成的面积,如果yi小于y0,则数据点(xi,yi)的变化是下调的,下调的显著程度p为y<yi时分布中的面积比上密度曲线与x’轴所围成的面积,如果yi等于y0,则数据点(xi,yi)没有发生变化。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,主要具备以下的技术优点:
(1)本发明公开了一种基于概率密度估计的数据差异分析方法,本方法具有统计学意义并且无论数据是什么分布,该方法均可适用,没有条件限制,有助于人们从差异变化的数据中发现关键的事物。
(2)本发明是这样实现的:1变为2与10变为20都是变化2倍,但是不代表其差异显著性相同。然而,1变为3相较于1变为2的差异更显著。我们基于该原理,通过估计变化前每个数据对应的突变后数据的概率密度分布来评估突变前后差异的显著性。
(3)本发明中的联合概率密度分布公式中的h值,影响数据联合概率密度分布估计的好坏,为了获取联合概率密度分布的最佳估计,本发明运用最大似然法去选择最佳的h,对不同的h(0<h<1),每次取数据集中的任意一个点,用数据集中剩下的n-1个点去构建联合概率分布,计算所述任意一个点在该联合概率分布上的联合概率密度值,得到n个联合概率密度值;似然值为n个联合概率密度值之积,使似然值最大的h为最佳h,因为该h下的概率密度分布最可能符合实际分布。
(4)本发明中固定每个变化前数据大小x,求得在该x的情况下,变化后数据大小y的概率密度分布;利用该分布进行假设检验,通常认为,p-value<0.05的数据为显著变化的数据,相较于变化前数值增加,我们认为是上调;反之,则是下调。
附图说明
图1本发明中方法的流程图。
图2是218个包含ksums的基因在癌症基因与药靶基因数据集中的富集情况。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
实施例1
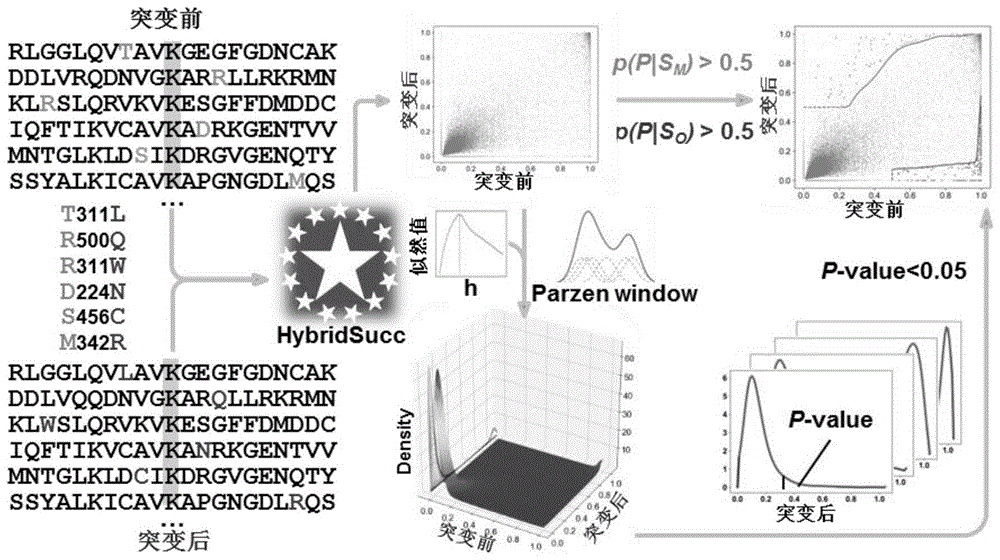
我们将发明方法用于预测显著影响现有的琥珀酰化位点的突变。这有助于发现通过改变琥珀酰化网络影响癌症的基因,并提供对疾病生物学和治疗发展的深入了解。在突变对琥珀酰化的影响分析中,我们从癌症基因数据库thecancergenomeatlas(tcga)中整合了33种主要癌症类型/亚型的11,659个肿瘤样本中的1,779,214个错义突变。其中有63693个发生在赖氨酸位点周边(左右各10个氨基酸)的错意突变(ksums)。如图1所示,我们对63693个包含ksums的肽段用琥珀酰化位点预测平台求得概率分,概率分反映了该位点琥珀酰化程度。然后,利用基于高斯核的parzenwindow法去估计突变前后的贝叶斯后验概率的联合概率密度:
其中h为windowwidth,n为ksums的数量,这里,n=63693。h的选取决定着概率密度估计的好坏,我们根据最大似然法去选择最佳的h,对不同的h,每次取1个点,用n-1个点去估计联合概率密度,求1个点的概率密度值,最后得到n个概率密度值。似然值为n个概率密度值之积f((x1,y1),(x2,y2),...,(xn,yn)|h)=f((x1,y1)|h)×f((x2,y2)|h)×…×f((xn,yn)|h)。使似然值最大的h为最佳h,最佳h=0.018。
最后,概率密度分布如图2所示,固定x,求得在该x的情况下,y的概率密度分布,我们使用p-value<0.05作为阈值进行假设检验,得到突变前后使琥珀酰化显著增强和减弱的ksums。我们设置上调后的后验概率大于0.5,以保证突变后为该位点发生琥珀酰化,下调前的后验概率大于0.5作为阈值,以保证突变前为该位点发生琥珀酰化。最终得到306个使琥珀酰化显著减弱的ksums和64个使琥珀酰化显著增强的ksums,其存在于218个基因上。
如图2所示,将218个基因分别映射到数据库cancergenecensus(cgc)中719个癌症基因和药靶数据库drugbank的2921个药靶基因数据集上,通过超几何分析发现在2个数据集中均显著富集,富集程度分别为2.62倍(p-value=3.03e-04)和4.15倍(p-value=1.20e-44),暗示该218琥珀酰化基因与癌症的相关程度较高,也说明了我们结果的可靠程度较高。
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!