对正在运行中的临床试验进行动态数据监测和实时优化的系统、方法及实施过程与流程
相关申请
本申请之要求已于2018年8月2日提交美国临时申请号no.62/713,565和2019年2月19日提交美国临时申请号no.62/807,584的优先权。此些先前申请之全部内容以引用之方式并入本申请。
本申请亦引用多个公开出版物,该等公开出版物的全部内容以引用之方式并入本申请案中以更充分地描述本发明所涉及的技术现状。
本研究发明针对进行中的临床试验研究之动态数据监测和数据优化系统,及其方法和过程之说明。
通过使用电子患者数据管理系统(如edc系统)、治疗分配系统(如iwrs系统)和客制化统计软件包,本发明是用于动态地监测并实时地优化正在进行中的临床研究试验的一个“封闭系统”。本发明的系统、方法和工序将一个或多个子系统集成为一个封闭系统,从而允许在临床研究试验中计算药物、医疗设备或其他治疗方法的治疗功效评分,而不会向任何一受试者的或参与之研究人员解盲(透露)个体治疗分配。在临床研究的各个阶段或之后的任何时间,随着新数据的累积,本发明将实施自动估计治疗效果、信赖区间(ci)、条件检定力、更新的停止界线,且根据所需的统计检定力重新估计样本数(量),并进行模拟,预测临床试验之趋势。本发明系统还可用于选择治疗方案、选择人群、识别病情预判因素、检测药物安全性信号,和在一个药物、医疗器械或治疗方案获批后,在患者治疗和医疗保健中与真实世界证据(rwe)和真实世界数据(rwd)的连接。
背景技术:
美国食品和药物管理局(fda)负责监督并保护消费者一切接触之健康相关产品(包括食品、化妆品、药物、基因疗法和医疗器械)。在fda的指导下,临床试验用于测试新的药物、医疗设备或其他治疗方法的安全性和有效性,以最终确定新的治疗方法是否适合目标患者群。本文所用术语“药物”和“药剂”可互换使用,并且包括但不限于任何药物、药剂(化学、小分子、复合物、生物制剂等)、治疗方法、医疗器械或其他需要使用临床研究、试验以获得fda批准的产品。本文所用术语“研究”和“试验”可互换使用,并且意指如本文所述的针对新药的安全性和有效性的随机临床研究。本文所用术语“研究”和“试验”包括其任何阶段或部分。
定义和缩写
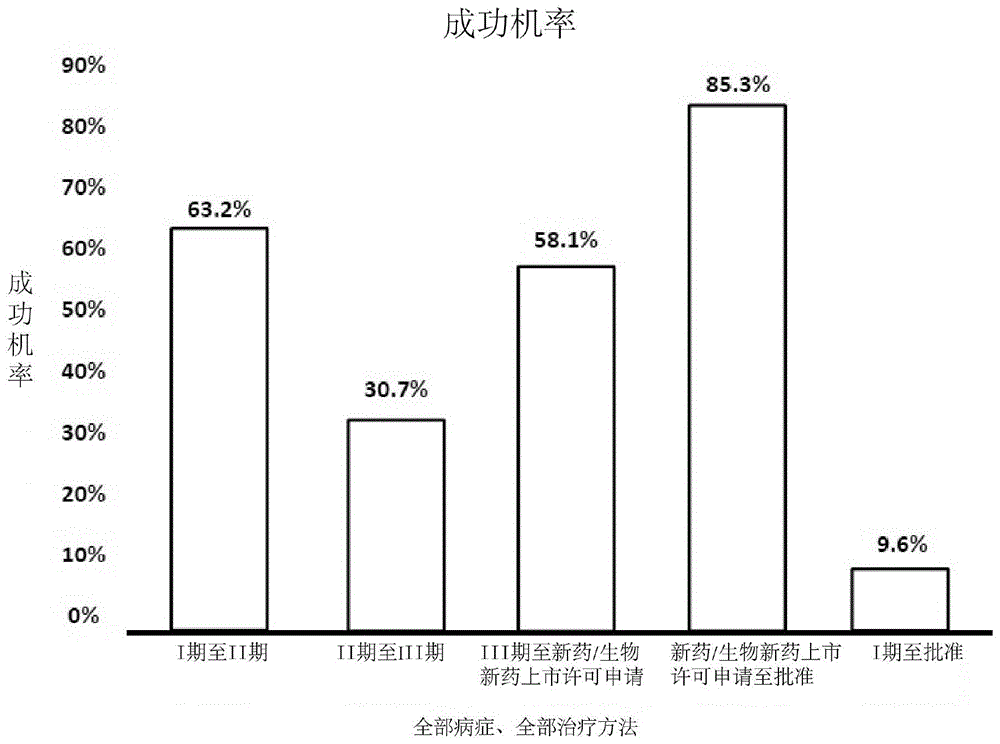
平均而言,一种新药从最初的发现到批准上市至少要花十年时间,仅临床试验平均就需要6至7年,每个成功药物的研发平均费用估计为26亿美元。如下所述,大多数临床试验皆须经过三个批准前阶段:第一阶段、第二阶段和第三阶段。大多数临床试验都在第二阶段失败,因而不能进入第三阶段。发生此失败的原因很多,但主要为安全性、功效和商业可行性相关的问题。如在2014年的报导中,完成第二阶段并进入第三阶段的试验药物,成功率仅为30.7%。请见图1。任何试验药物完成第三阶段并在fda进行新药申请(nda)成功率仅为58.1%。在初期(第一阶段)人类受试者测试的候选药物中,约只有9.6%被fda最终批准在人群中使用。因此,在寻找候选药物并最终能获得fda批准时,药厂需花费大量资金与物力,更有可能造成的人力浪费。
若在动物试验中新药物测试结果看起来令人满意,即可进行该药物的人类试验和研究。在进行人体测试之前,必须先将动物研究结果报告报与fda,以获得测试批准。提交给fda的报告被称为新药研究申请(“ind”申请,即“inda”或“ind申请”)。
候选药物在人体上的实验过程称为临床试验,其通常包括四个阶段(三个批准前阶段和一个批准后阶段)。在第一阶段,研究人类参与者(称为受试者)(大约20至50人)用以确定新药之毒性。在第二阶段,更多的人类受试者参与研究(通常为50-100人),此阶段用来确定药物的疗效并进一步确定治疗的安全性。第二阶段试验的样本量因治疗区域和人群而有异,有一些试验规模较大,可能包含数百名受试者。该药物的剂量将进行分层,以取得最佳治疗方案。一般将治疗与安慰剂或与另一种现有治疗方法进行比较。第三阶段临床试验旨在确认第二阶段临床试验结果之疗效。对于此阶段,需要更多的受试者(通常是数百到数千个)来执行更具结论性的统计结果分析。此阶段之试验设计亦是将治疗与安慰剂或与另一种现有治疗方法进行比较。在第四阶段(批准后研究),该治疗已获fda批准,但仍需进行更多测试以评估长期效果与其他可能的适应症。亦如是说,即使在fda批准之后,该药物仍会因严重不良事件而被持续监督。监督(亦称为上市后监督试验)是通过系统的报告以及样本调查和观察研究来收集不良事件。
样本量倾向于随着试验阶段而增加。第一阶段和第二阶段的试验样本量很可能在十几到一百多,而第三和第四阶段试验的样本量为一百多到一千多之间。
每个阶段的研究重点在整个过程中变化,初期测试的主要目的是确定该药物是否足够安全,是否可进行进一步的人体测试。此初期研究的重点在于确定药物的毒性特征,并寻找适当的治疗有效剂量以用于后续测试。通常,初期的试验是不设对照组的(即研究不涉及同时观察的、随机的对照组),且试验时间较短(即治疗和随访时间相对较短),并寻找合适的剂量以用于后续测试阶段。测试后期阶段的试验通常涉及传统的平行治疗设计(即,设对照组,通常涉及试验组和对照组),患者随机分组并针对所治疗疾病的典型治疗期与治疗后的追踪进行记录观察和研究。
大多数药物试验都是在药物“发起人”持有的ind下进行的。发起人通常是药品公司,但也可以是个人或是代理。
试验计划一般由研究发起人制定。试验计划书是为描述实验原因、所需受试者数量的依据、研究受试者的方法以及如何进行研究的相关指南或规则的文文件。在临床试验期间,会在医疗诊所或其他调查地点进行,并且通常由医生或其他医疗专业人员(也称为研究的“调查员”)对受试者进行评估。当参与者签署知情同意书并满足某些纳入和排除条件标准后,将成为研究对象。
参与临床研究的受试者将以随机方式分配给研究组与对照组,此是为了避免在选择试验受试者时可能出现的偏差。例如,如果病情较轻或基线风险特征较低的受试者被分配给新药组的比例高于对照组(安慰剂),那么新药组可能会出现更有利但有偏差的结果。即使是无意的,这种偏差也会使临床试验的数据和结果偏向于研究的试验药物。然而,当在只有一个研究组的情况下,将不进行随机分组。
随机临床试验(rct)设计通常用于第二阶段和第三阶段的试验,在试验中,患者会被随机分配实验药物或对照药物(或安慰剂)。通常以双盲方式随机分配,即医生和患者皆不知各是接受了何种治疗。此随机化和双盲化其目的是为减少功效评估中的偏差。而计划(或预估)的研究患者数量和试验时间,是根据研发初期对试验药物的有限了解推估而出。
通过“盲性"过程,受试者(单盲)或受试者和研究者(双盲)不知晓临床试验中受试者的研究组别分配。此盲性设计,尤其是双盲,最大程度地降低了数据的偏差风险。而在只有一个研究组的情况下,一般不进行盲性测试。
通常,在标准临床研究试验结束时(或在指定的过渡时间段,下文将进一步讨论),会将包含完整试验数据的数据库数据传输给统计学家进行分析。若看到某一特定事件,无论是不良事件还是试验药物的功效,其发生率在一组中都高于另一组,从而超过了单纯的纯随机,那么可以说已经达到统计学意义。使用众所周知的统计计算并用于此目的,组之间任何给定事件的比较发生率都可以通过被称为“p值”的数值来描述。p值<0.05表示发生事件的可能性的95%不是由于偶然的结果。在统计情况下,“p值”也称为误报率或误报概率。通常,fda接受总体假阳性率<0.05。因此,如总体p<0.05,则认为该临床试验具有“统计学意义”。
在一些临床试验中,可能不使用分组研究,甚至不使用对照组。在这种情况下,仅存在一个研究组别,则所有受试者均接受相同的治疗。此种单一组别通常同先前已有已知之临床试验数据或有相关药物治疗之历史数据进行比较,或因其他伦理原因而使用。
研究组别的设计、随机化、盲性是业内共识和fda批准的成熟技术,使得在试验过程中可以确定新药的安全性和有效性。由于这些方法需要维持盲性以保护临床试验的完整性,因此在研究进行期间,临床试验发起人无法随时取得或跟踪试验的安全性和有效性之相关关键信息。
任何临床试验的目的之一即是确定新药的安全性。然而,在两个或多个研究组别之间进行随机化的临床试验中,只有将一个研究组别与另一个研究组别的安全性参数进行分析比较后,才可确定其安全性,如果研究组别在盲性的情况下进行试验,则无法将受试者及其数据分为相应之组别进行比较。此外,如下文更详细的讨论,研究数据仅能在试验结束时或在预定的分析时点进行解盲破译和分析,使得研究对象将承受潜在的安全风险。
对于有效性,将遵循试验过程中的关键变量以得出结论。此外,研究计划中会定义某些结果或终点,以此来认定研究对像是否已完成试验计划。研究数据会随着研究的信息时间线累积,直到受试者到达各自的终点(即受试者完成研究),然而这些参数(包括关键变量和研究终点)无法随时在受试者试验进行中进行比较或分析,从而造成了在统计分析和伦理方面的不便与潜在风险。
另一个相关问题是统计检定力。定义为,当对立假设(h1)为真时,正确地拒绝虚无假设(h0)的概率,换言之,也可以是当对立假设为真时将其接受的概率。在临床研究统计设计上,旨在证明有关药物安全性和功效的对立假设,并拒绝虚无假设。为此,统计检定力是必须的,故而需要有足够大的受试者样本量和各个研究组别间的分组来获得数据。如果没有足够的受试者进入试验,则存在未达到统计学显著性水平以支持拒绝虚无假设的风险。由于随机临床试验通常是盲性的,因此直到项目结束,才可知道每个研究组别的确切受试者人数,尽管这可以保持数据收集的完整性,但是此中存在固有的低效率和对于试验的浪费。
在统计学意义的情况下,研究数据达到功效证明或无效标准界线时,应为结束临床研究的最佳时间。这一时刻可能发生在临床试验计划结论之前,但通常无法确定其发生的时间。因此,若试验已达临床统计意义而还继续进行,则是浪费许多不必要的时间、金钱、人力、物力。
而发生研究数据接近但仍未达到统计显著性的情况下,一般是由于参加研究的受试者人数不足。此这种情况下,为了获得更多支持性数据,则将需要延长临床试验的试验期,但若是仅能在试验完全结束之后方能进行统计分析,则无法及时知晓并延长试验的时间。
若是在试验药物无显著功效趋势的情况下,即使招募了更多的受试者,也几乎没有机会获得期望的结论。在这种情况下,一旦得出结论,即所研究的药物无效,并在连续的研究数据中几乎没有达到统计学意义的机会(即继续对药物进行研究),则希望可尽早结束研究。此种趋势只有在进行最终数据分析(通常在试验结束时或在预定的分析点),才能得出这样的结论。同样,由于无法及早发现,不仅浪费时间和金钱,亦使过多的受试者参与试验而浪费人力和物力。
为了克服这些问题,临床试验计划已经采取了期中分析的方法,以帮助确认研究是否具有成本效益与合乎人体试验道德,但是,即使采取了此方法也可能无法达到最佳测试的效果,因为期中分析必需要先预设时间点,而期中分析与最后的分析,两次分析之间的实验时间可能会很长,数据分析前亦须要先解盲,故需要大量时间来进行,而造成缺乏效率。
图2描绘了传统的“研究结束”随机临床试验设计,通常用于第二期和第三期试验,其中将受试者随机分配到药物(实验)组或对照(安慰剂)组。在图2中,描绘了两种不同药物的两种假设临床试验(第一种药物的名称为“试验i”,第二种药物的名称为“试验ii”)。横轴为试验时间长度(也称为“信息时间”),两个试验中的每一个点都记录了试验讯息(以p值表示的功效结果)。纵轴表示两次试验的标准分数(通常称为“z-分数”,例如标准化的均值差异)。绘制研究数据的时间t起始点为0。随着两项研究的进行,时间沿时间轴t继续,并且两项试验的研究数据(统计分析后)均随时间而累积。两项研究均在c线完成(结论线—最终分析时间)。上方的s线(“成功”线),为p<0.05的统计学显著水平的边界。当(如果有)试验结果数据超过s时,则达到统计学上的显著水平p<0.05,并且该药物被认为在研究中定义的功效为有效。下方的f线(“失败”线)是无效的边界,表明测试药物不太可能具有任何功效。s和f线均已根据试验计划书进行了预先计算和确定。图3至图7为类似的有效性/信息时间图。
图2中试验i和试验ii的假设治疗以双盲方式随机分配,其中研究者和受试者均不知道受试者是使用了药物或安慰剂。在两个试验计划书中以有限的知识估算了参与每个试验的受试者数量和试验时间。在完成各个试验后,将根据主要终点的结果,对每个试验之数据进行分析,确认是否具有统计学显著性,即p<0.05,以确定是否达到研究目标。在c线(试验结束),许多试验低于“成功”的阈值p<0.05,被认为是无效的。理想情况下,此类的无效结果试验应尽早终止,以避免对患者进行的试验测试并避免大量财务资源的支出。
图2中描述的两个试验仅有一次数据分析,即在c线处得出的试验结论。试验i在显示可能成功趋向的候选药物的同时,仍未达到(低于)s,即试验i的功效尚未达到统计学上显著的p<0.05。对于试验i,若能有更多受试者或不同剂量的研究组别,可能可使试验结束前得到p<0.05;然而试验发起者必须等到试验结束并分析结果后才能知道这一事实。另一方面,为了避免经济浪费和减少受试者进行试验,应该早些终止试验ii。图中试验ii候选药物的功效评分向下的趋势证明试验ii候选药物不具有效性。
图3为两个假想的第二期或第三期试验的随机临床试验设计,其中将受试者随机分配到测试药物(实验)组或对照(安慰剂)组中,并且利用一个或多个期中分析。图3采用了常用的群组序列(groupsequential,“gs”)设计,即试验进行中对累积的试验数据进行一个或多个期中分析。图3与图2的试验设计不同,图2为盲性测试,需在研究完成后方可进行统计分析和检查。
图3中s线和f线不是c在线的单个预定数据点,而是在试验计划书中预先建立的预定边界,反映了计划中的期中分析设计,上边界s表示药物的功效已达到统计学显著水平p<0.05(因此,该候选药物被认为在试验计划书中定义的功效评分为有效),下边界f表示该药物的功效对试验计划书中定义的功效评分为失败、无效。根据总假阳性率(α)必须小于5%的规则,图3中的gs设计的停止边界(上边界s和下边界f),由预先计算的预定点t1和t2得出(t3为完成试验终点c)。
有其他不同类型的机动型停止界线,参见flexiblestoppingboundarieswhenchangingprimaryendpointsafterunblindedinterimanalyses,chen,liddym.,etal,jbiopharmstat.2014;24(4):817-833;earlystoppingofclinicaltrials,atwww.stat.ncsu.edu/people/tsiatis/courses/st520/notes/520chapter_9.pdf。o'brien-fleming为最常使用的机动型停止界线。不似图2所示,机动型停止界线具有灵活机动性的边界,上边界s确定了药物的功效有效性(p<0.05),下边界f确定了药物的失败(无效)。
使用一个或多个期中分析的临床研究存在某些障碍。具体而言,使用一个或多个期中分析的临床研究必须是在解盲的状态,以便将关键数据提交并进行统计分析。而没有期中分析的药物试验同样会解盲研究数据,但仅当研究结束时,且须消除研究结束时才发现的偏差或侵扰的可能性。因此,使用期中分析是必要的,但同时必须保护研究的完整性(盲性和随机)。
其中一种执行期中分析研究的必要统计分析的方法,是通过独立的数据监测委员会(“dmc”或“idmc”)。该委员会通常与独立的第三方独立统计组(isg)合作。在预定的期中分析,累积的研究数据会通过dmc解盲并提供给isg,而后,isg会对实验组和对照组进行必要的统计分析比较。在对研究数据进行统计分析后,结果将返回给dmc。dmc会对结果进行审查,并根据审查结果向药物研究发起人提出建议。根据期中分析(和研究的阶段),dmc将建议是否继续进行试验;可能因为结果显示无效而建议中止试验,或者相反,研究药物已经建立了必要的统计学证据,证明该药物具有功效而建议继续试验。
dmc通常由研究发起人组织的一组临床医生和生物统计学家组成。根据fda的《临床试验发起人指南—建立和运行临床试验数据监测委员会(dmc)》,“临床试验dmc是一组具有相关专业知识的人员,他们将对一个或多个进行中的临床试验定期审查。”fda更进一步解释说:“dmc就试验受试者和尚待招募的受试者的安全性向发起人提供建议,以及评估该试验的持续有效性和科学价值。”
在极幸运的情况下,实验组无疑显示出优于控制组的结果,dmc可能建议终止试验。这将使发起人可以提早得到fda的批准,并更早的对患者群体进行治疗。然而,这种情况下,统计证据必须非常强大,但是,也可能还有其他原因需继续进行研究,例如需收集更多的长期安全性数据。dmc在向发起人提供建议时会考虑所有的相关因素。
若不幸的,研究数据显示该试验药物无效,dmc可能建议终止试验。举例来说,如果项目试验仅完成了一半,而实验组和对照组的结果几乎相同,则dmc可能建议停止研究。在此种统计证据下,如果试验继续按计划完成,极可能无法获得fda对该药的批准。发起人可以放弃该试验为其他项目节省资金,并且可以为当前和潜在的试验对象提供其他治疗方法,且将来的受试者将不用进行不必要的试验。
尽管利用期中数据的药物研究具有其优点,但也有缺点。首先,存在固有的风险,即研究数据可能被泄漏或流出。尽管无法得知是否由dmc成员泄露或利用这种机密信息,但有人怀疑isg的组成人员或为isg工作的人不当使用此类信息。其次,期中分析需要暂时停止研究并使用宝贵的时间进行后续的分析。通常,isg可能需要3到6个月的时间来执行其数据分析并准备dmc的期中结果。此外,期中数据分析只是个临时的“快照”视图,在各个相应的过渡点(tn)进行的统计分析,是无法对正在进行中的数据进行趋势分析的。
参照图3,鉴于试验i的期中信息时间点t1和t2的数据结果,dmc可能会建议试验i的药物继续研究。该结论由药物有效性评分的持续增加所支持,因此继续进行研究可增加有效性的评分并达到统计学意义p<0.05。对于试验ii,dmc可能也可能不会建议继续进行,尽管药物的有效性持续下降,但还没有越过失败的界限,但由此可推测出试验ii最终(并且很可能)是无效的;除非试验ii的药物安全性极差,dmc可能会建议继续药物研究。
总而言之,尽管gs设计利用预定的数据分析时间点来分析和审查,但是它仍然存在各种缺点。其中包括1)研究数据流向第三方(即isg),2)gs设计仅能在过渡时间点提供数据的“快照”,3)gs设计无法确定试验的具体趋势,4)gs设计无法从研究数据中“学习”以调整研究参数和优化试验,5)每个期中分析时间点需要3到6个月来进行数据分析和准备结果。
自适应群组序列(“ags”)是gs设计的改良版,通过这种方法设计试验,其分析了临时数据,并将其用于优化(调整)某些试验参数,例如重新估计样本量,且该设计试验可以属于任一阶段,从任意数量开始。换句话说,ags设计可以从期中数据中“学习”,从而调整(适应)原始试验设计并优化研究目标。参见例如2018年9月fda指南(草案指南),《药物和生物制剂临床试验的适应性设计》,www.fda.gov/downloads/drugs/guidances/ucm201790.pdf。与gs设计一样,ags设计的临时数据分析点,亦需要dmc的审查和监测,因此同样需要3到6个月的时间进行统计分析和结果的汇编。
图4描绘了ags试验设计,再次使用假设的药物研究试验i和试验ii。在预定的期中时间点t1,与图3的gs试验设计相同的方式来编译和分析每个试验数据,然而,在统计分析和审查后,可以调整研究的各种研究参数,即,使其适应优化,从而重新计算了上边界s和下边界f。
参照图4,数据进行了编译和分析并用于调整此研究的适应性,即“学习与适应”,例如,重新计算样本数(大小),并因此调整终止界线。作为这样优化的结果,研究样本大小将被修改,界线将被重新计算。在图4的期中分析时间点t1进行数据分析,并基于此分析来调整(增加)研究样本的大小,从而重新计算了停止界线、s线(成功)和f线(失败),s1和f1的初始边界不再使用,而是使用由期中分析时间点t1得出并调整之停止界线s2和f2。图4在预定的期中分析时间点t2,再次编辑和分析研究数据,并再次调整各种研究参数(即,使其适于研究优化),作为这种修改的结果,重新计算了停止界线s(成功)和f(失败)。重新计算的上边界s现标为s3,重新计算的下边界f现标为f3。
虽然图4的ags设计对图3的gs设计进行改良,但仍然存在某些不足。首先,ags的设计仍然需要dmc审查,故而需要在预定的时间点停止研究(尽管是暂时的),并且需要解盲后提交给第三方进行统计分析,从而存对数据完整性的风险。另外,ags设计不执行数据仿真来验证期中结果的有效性和可信度。与gs设计一样,ags设计期中数据分析、查看结果并提出适当的建议仍需要3到6个月才能完成。与图3的gs设计一样,在两次期中分析时间点之分析,dmc可能会建议继续进行试验i和试验ii,因为两者都在(可能经过调整的)停止范围之内;或者,dmc由数据分析中发现了试验ii可能缺乏功效而建议暂停。如果试验ii研究的药物也显示出不良安全性,那么试验ii将被建议停止。
综上所述,尽管ags设计在gs设计的基础上进行了改进,但它仍具有各种缺点。其中包括1)中断研究并解盲数据以提供给第三方,即isg;2)ags设计仍仅在期中分析点提供数据“快照”;3)ags设计无法识别试验数据累积的具体趋势;4)每一期中分析点需要3到6个月的时间进行数据分析和准备数据结果。
如上,图3和图4(gs和ags)仅能在一个或多个预定的期中分析时间点呈现数据的“快照”给dmc。即使经过统计分析,此类快照视图也可能误导dmc并干扰有关当前研究的最佳建议。然而,可期望的是,在本发明的实施例中,提供的是对试验进行的连续数据监测方法,由此对研究数据(功效和/或安全性)进行实时分析并实时记录以供后续审查。如此,在经过适当的统计分析后,将为dmc提供实时的结果和研究趋势(如所积累的数据),从而能够提出更好的建议,这对试验更有益。
图5描绘了一连续监测的设计,随着受试者数据而累积,沿着t信息时间轴记录或绘制试验i和试验ii的研究数据。每个研究数据图都针对当时累积的所有数据进行全面的统计分析。因此,统计分析并不会像在图3和4的gs和ags设计中那样等待中间的期中分析时间点tn,或如图2中须试验完成方可进行数据分析;相反,随着研究数据的累积,统计分析是实时进行的,并且沿信息时间轴t实时记录了功效和/或安全性的数据结果。在预定的期中分析时间点,给dmc显示整体的数据记录,如图5-7。
如图5所示,试验i和试验ii的研究数据实时汇总并进行统计分析,然后沿信息时间轴t记录受试者试验数据至试验终点。在期中分析时间点t1,此二试验记录研究数据将显示给dmc并进行审查。基于研究数据的当前状态,包括累积研究数据的趋势,或对于边界和/或其他研究参数的自适应重新计算,dmc能够针对此二试验研究提出更准确且最佳的建议。如图5中的试验i,dmc可能会建议继续研究该药物。至于试验ii,dmc可能会发现功效低下或缺乏功效趋势,但可能会等到下一个期中分析时间点再作进一步考虑。此外,dmc还可以基于审查的研究数据建议例如增加了样本量,并且根据样新本量重新修改计算终止界线。
图6中试验i和试验ii都持续到期中分析时间点t2。在封闭的环境中实时地统计所累积的研究数据,并且以与图5相同方式的对其进行记录。在期中分析时间点t2,试验i和试验ii所累积的研究数据进行统计分析并呈交dmc审查。在图6中,dmc可能会建议继续试验i,可能会或不会调整样本大小(因此可能会也可能不会重新计算界线s);而试验ii,在图6中的期中分析时间点t2,dmc可能会发现它有令人信服的证据,包括累积数据确定的趋势,并建议终止试验;若药物安全性较差,则尤其如此;然而,dmc仍可能会建议继续进行试验ii,因其图中显示,所累积的数据分析结果仍在停止界线内。
如图7,若不对试验i和试验ii进行连续监测,则dmc可能会建议继续进行这两试验,因为它们都在两个终止界线(s和f)之内,虽然,dmc可能会建议终止试验ii;故而,任何这样的建议都取决于dmc审查时的特定的数据统计分析方法,而本方法,在过程中,系统在死循环环境中使用,并对其所累积的数据进行实时统计分析,能够更加准确。
出于伦理、科学或经济方面的原因,大多数长期临床试验,尤其是那些病情严重的研究终点的慢性疾病,都应定期进行监测,以便在有令人信服的证据支持或反对无效试验时终止或修改试验假设。传统的群组序列设计(gsd)在固定的时间点并按预定的测试次数进行测试(pocock,1997;o'brien和fleming,1979;tsiatis,1982),通过alpha花费函数方法得到了极大的增强(lan和demets,1983;lan和wittes,1988;lan和demets,1989),且具有灵活的测试时间表和试验监测期间进行的期中分析次数。lan,rosenberger和lachin(1993)进一步提出“在临床试验中临时的或连续的监测数据”,基于连续的布朗运动过程提高gsd的灵活性。但是,由于现实原因,过去在实践中仅能执行临时的监测。进行数据收集、检索、管理,最终呈现给数据监视委员会(dmc)都是阻碍实践连续型的数据监测的因素。
当虚无假设为真时,上述gsd或连续监测方法对于通过适当控制的i型错误率来做出研究早期的决策非常有用。其最大量的信息在试验计划书中已预先固定。
临床试验设计中的另一个主要考虑因素是当虚无假设不成立时,需预估提供统计检定力所需的足够信息量。对于此任务,gsd和固定样本的设计均依靠较早的试验数据估计所需的(最大)信息量。挑战在于,由于患者人群、医疗程序或其他试验条件可能不同,这种来自外部的估计可能并不可靠。因此,一般而言,先期预估的信息或特定的样本大小可能无法提供所需的统计检定力。相比之下,在90年代初期通过利用当前试验本身的期中数据开发的样本量重新估算(ssr)程序,通过增加方案中最初指定的最大信息量来确保统计检定力(wittes和britan,1990;shih,1992;gouldandshih,1992;hersonandwittes,1993);参见shih(2001)对gsd和ssr的评论。
此二种gsd和ssr后来结合在一起,形成了过去二十年来许多人所谓的自适应gsd(agsd),包括bauer和kohne(1994),proschan和hunsberger(1995),cui,hung和wang(1999),li等(2002),chen,demets和lan(2004),posch等(2005),gao,ware和mehta(2008),mehta等(2009),mehta和gao(2011),gao,liu和mehta(2013),gao,liu和mehta(2014)等。有关最新评论,详见shih,li和wang(2016)。agsd对gsd进行了改进,使其具有使用ssr扩展最大信息的能力,并可能提早终止试验。
技术实现要素:
对于ssr,仍然存在一个关键问题,即当前的试验数据何时足够可靠,来执行有意义的重新估计。过去,由于没有有效的连续的数据监测工具可用于分析数据的趋势,因此一般建议将期中分析时间点作为准则,但是,期中分析时间点只是数据快照,并不能真正保证ssr的数据是足够的,可以通过连续监测数据来克服此点。
随着当今的计算技术和硬件的计算能力极大提高,对于实时的快速数据传输运算已不再是问题。利用ssr对累积的数据进行连续监测并根据数据进行计算,将充分发挥agsd的潜力。在本发明中,该新过程被称为动态自适应设计(dad)。
在本发明中,基于连续布朗运动过程,将lan,rosenberger和lachin(1993)中开发的连续数据监测程序扩展到dad,并使用数据指导的分析来对ssr进行计时。在试验计划书中dad可以作为一种灵活的设计方法,当dad在正在进行的试验中实施时,它可以用作有用的监测和导航工具,此称为动态数据监测系统(ddm)。在本发明中,dad和ddm的术语可以一起或互换使用。在一个实施例中,i型错误率总是受到保护,因为连续监测和ags都保护i型错误率。通过模拟,dad/ddm可以就无效性或早期效力终止做出正确的决定,或认为试验有望随着样本量的增加而到达有效性,从而大大提高了试验的效率。在一个实施例中,本发明提供了用于治疗效果的中位数不偏的点估计和精确的双向置信区间。
关于统计问题,本发明提供了一种解决方案,该解决方案涉及以下方面:如何检查数据趋势并确定是否该进行正式的临时分析、如何保护i型错误率并得到效率,以及如何在试验结束后建立治疗效果的置信区间。
本发明公开了对进行中的新药随机临床试验的动态数据监测的封闭系统、方法和过程,使得在不使用人为解盲的情况下来研究数据、连续而完整地跟踪统计参数,例如,自动计算出治疗效果、安全性、置信区间和条件检定力,并可以在信息时间轴上的所有点上进行查看,即随着试验人群累积所得到的所有数据进行查看。
附图说明
图1是柱状图,根据历史数据描绘了fda在各个阶段中批准候选药物的近似成功概率。
图2描绘了随着时间,两种候选药物的两个假设临床研究的功效评分。
图3描绘了实施群组序列(gs)设计的两个候选药物的假设临床研究的功效和期中分析。
图4描绘了实施自适应群组序列(ags)设计的两个候选药物的假设临床研究的功效和期中分析。
图5描绘了实施连续监测设计,在期中分析时间点t1的两个候选药物的假设临床研究的功效。
图6描绘了实施连续监测设计,在期中分析时间点t2的两个候选药物的假设临床研究的功效。
图7描绘了实施连续监测设计,在期中分析时间点t3的两个候选药物的假设临床研究的功效。
图8是本发明的实施例示意图。
图9是本发明的实施例示意图,描绘了其中的动态数据监测(ddm)部分/系统的工作流程。
图10是本发明的实施例示意图,描绘了其中的网络交互响应系统/部分(iwrs)和电子数据收集(edc)系统/部分。
图11是本发明的实施例示意图,描绘了其中的动态数据监测(ddm)部分/系统。
图12是本发明的实施例示意图,进一步描绘了动态数据监测(ddm)部分/系统。
图13是本发明的实施例示意图,进一步描绘了动态数据监测(ddm)部分/系统。
图14描绘了由本发明的实施例所输出的假设临床研究的统计结果。
图15描绘了通过本发明的实施例所输出的候选药物假设临床研究的功效图。
图16描绘了通过本发明的实施例所输出的候选药物假设临床研究的功效图,其中,重新估计了受试者的人数,并且重新计算了终止界线。
图17是本发明一实施例中的实施方式和步骤流程图。
图18是本发明一实施例的临床试验仿真数据。
图19是本发明一实施例的趋势比(tr)计算
图20a和20b分别显示了最大趋势比的分布,以及在试验结束时使用最大趋势比的ho的(条件)拒绝率cpmtr。
图21显示了不同表现分数区域的图形(样品大小为np;np0是具有固定样品大小设计的临床试验所需的样品大小,p0是所需的检定力。表现分数(ps)=1是最佳计分,ps=0是可接受的分数,而ps=-1是最无希望的分数)。
图22显示了试验最终失败的wald统计数据的全部纪录。
图23a至23c分别显示了试验最终成功的wald统计数据、条件检定力和样本量比率的完整纪录。
具体实施方式
药品临床试验计划书通常须包含药物剂量、测量终点、统计检定力、计划期程、显著水平、样本数估计、实验组及控制组所需之样本数等,且彼此间具有关联性。例如,以提供所需的统计显著性水平,所需的受试者(测试组,因此接受药物)人数在很大程度上取决于药物治疗的功效。如研究药品本身具有高度功效,即认为该药物将获得较高的功效评分并预计达到统计学显著水平,即在研究初期p<0.05,则相比于有益但是效果要低一些的治疗,所需患者明显要少。然而,在初期研究设计上,欲研究药品之真实效果是未知的,因此,可藉由先驱计划、文献回顾、实验室数据、动物实验数据等进行参数估计并写入试验计划书中。
在研究的执行上,依照实验设计将受试者随机分派至实验组及对照组,而随机分派的过程可藉由iwrs(interactivewebresponsesystem,网络交互响应系统)完成。iwrs是一提供随机编号或是生成随机序列列表之软件,其所包含之变量有受试者身份标示、分派组别、随机分派之日期、分层因子(如性别、年龄分组、疾病期程等)。这些数据将存放于数据库中,并针对该数据库进行加密或是设置防火墙等,使受试者及试验执行人员无从得知受试者的分派组别,如受试者是否接受药物治疗或是被给予安慰剂、替代治疗等,从而达到盲性之目的。(举例来说,为确保盲性之落实,欲试验药品及安慰剂可能会采相同包装,并以加密条形码做区别,只有iwrs能指派给予受试者该组药物,如此临床实验人员与受试者皆无法得知受试者所属组别为何。)
随着研究的进行,将定期评估治疗对于受试者所产生的影响,该评估可由临床人员或是研究人员亲自进行,也可透过合适的监测装置进行(如穿戴监测装置或是居家监测装置等)。然而,透过评估资料,临床人员及研究人员可能无法得知受试者所属组别,亦即评估数据不会呈现分组状态。可以使用适当配置的硬件和软件(例如window或linux操作系统的服务器)收集此盲性评估数据,这些服务器可以采用电子数据捕获(“edc”)系统的形式并可以存储在安全数据库中。edc数据或数据库同样可以通过例如适当的密码和/或防火墙来保护,以使数据对研究对象,包括受试者、研究者、临床医生和发起人保持盲性和不可用。
在一个实施例中,用于随机分派治疗的iwrs、用于数据库评估的edc以及ddm(dynamicdatamonitoringengine,动态数据监测引擎,一统计分析引擎)可以安全地相互链接在一起。举例来说,将数据库及ddm放置于单一服务器,该服务器本身即受到保护并与外部存取隔离,进而形成一封闭回路系统,或是透过具安全性且加密的数据网络,将安全的数据库及安全的ddm链接在一起。在适当的编程配置下ddm能从edc获取评估纪录,并从iwrs获得随机分派结果,用以进行盲性下试验药物的成效评估,如计分检定、wald检定、95%信赖区间、条件检定力以及各项统计分析等。
随着临床试验进行,即随着新增的受试者达到试验终点和研究资料完成累积,由edc、iwrs及ddm互相链接所构成的封闭系统可持续且动态地监测内部解盲数据(详细解说请参阅图17),其监测的内容可能涵盖药物疗效的点估计及其95%信赖区间、条件检定力等。可透过ddm对于已收集的数据进行以下事项:重新估计所需之样本数、预测未来趋势、修改研究分析策略、确认最佳剂量,以利研究发起人评估是否继续进行试验,并估算试验药物的有效反应之子集合,以利后续招募受试者及模拟研究以估计成功概率等。
理想情况下,由ddm所产出的分析结果及统计模拟实时地提供给dmc或研究发起人,并依照dmc所提出之建议,即早对于研究进行调整并执行。举例来说,如该试验主要目的是在评估三种不同剂量相较于安慰剂之疗效,根据ddm的分析,在试验初期如发现某一药物剂量功效显著优于其他剂量,达统计学上显著意义,即可提供给dcm,并以最有效剂量进行后续研究,如此一来,后续更进一步的研究可能仅须纳入一半人数的受试者,此将大幅减低研究成本。再者,就道德伦理层面来说,比起让受试者接受合理但疗效不佳之剂量,以更具疗效之剂量继续试验治疗,是更好的选择。
根据当前的规定,可在期中分析前将此类前导式评估结果提报给dmc;如前所述,当isg取得完整且解盲的数据数据后将进行分析,再将结果呈报给dmc,dmc将依其分析结果,对于试验是否继续及如何继续等问题给予研究发起人建议,而在某些情况下,dcm亦提供指导试验相关参数的重新估计,如样本数的重新计算、显著界线的调整。
当前执行上不足的地方包括但不限于,(1)数据解盲必然有人为参与的情况(如isg)、(2)数据数据的准备并送至isg进行期中分析须耗时约3~6个月、(3)dmc须在审查会议前约2个月,对isg所提交的期中分析进行审查(因此,dmc审查会议上所呈现的研究资料已是5~8月前的旧资料)。
而前述之不足之处可在本发明中得到解决,本发明的优势如下:(1)本发明之封闭系统不须有人为介入(如isg)来解盲;(2)预定义分析允许dmc或研究发起人能实时且持续地审阅分析结果;(3)有别于传统的dmc执行方式,本发明允许dmc随时进行追踪并监测,使安全性及疗效的监测更加完整;(4)本发明可自动执行样本数的重新估算、更新试验停止边界、预测试验的成败。
因此,本发明成功地达到期望中的效益及目的。
在一个实施例中,对于动态监测下的盲性试验,本发明提供了一封闭系统及方法,对于还在执行中的试验无须由人为的介入(如dmc、isg)解盲来进行数据分析。
在一个实施例中,本发明则提供了计分检定、wald检定、点估计及其95%信赖区间,和条件检定力等功能(即从开始研究到获得最新研究数据)。
在一个实施例中,本发明亦允许dmc和研究发起人随时审查正在执行中之试验的关键数据(安全性及功效评分),因此,无须透过isg,可避免冗长的准备过程。
在一个实施例中,本发明结合了机器学习及ai技术,可利用观察到的累积数据做出抉择,进而优化临床研究,使试验成功机率最大化。
在一个实施例中,本发明能尽早评估试验的无效性,以避免受试者承受不必要的痛苦以及减少研究成本的浪费。
相较于gsd及agsd,本发明中所描述、揭示的动态监测程序(如dad/ddm)更具优势。为求更清楚说明此情况,以下将以gps系统作为譬喻进行解说。gps导航装置通常用于提供驾驶人员目的地的路径引导,而gps一般分为汽车导航及手机导航两种。一般而言,汽车导航并未连接因特网,故无法提供实时路况数据,驾驶可能因此遇到交通壅塞的困境,而手机导航因连接因特网则可根据实时交通路况提供最快速的行车路线。简而言之,汽车导航只能提供固定且不灵活的预定路线,而手机导航则能使用最新的讯息进行动态导航。
对于期中分析数据撷取的时间点选择上,如使用传统的gsd或agsd并无法确保分析结果的稳定性,如选择的时间点过早,可能会导致不合适的试验调整决策;如选择时间点过晚,则将错失及时调整试验的机会。然而,本发明中的dad/ddm在每一位受试者进入试验后,即提供实时的连续监测功能,就如同手机导航功能,藉由实时数据的导入持续地导正试验方向。
本发明在统计问题上提供了解决方法,如对于如何检查数据趋势、是否该进行正式的期中分析、如何确保i型误差的控制、潜在的功效评估,以及如何在试验结束后建置功效的信赖区间。
本发明的实施例将更详尽的展现于附图中,附图中的说明将以相同的方式进行标示,这些实施例操作将用于本发明之阐释,但并不限于此。相关技术人员在阅读本说明书及附图后,在不违悖本发明精神之情况下,可对其适当地进行各种修改与操作的变化。
本发明的各项实施例操作之说明及图示仅能代表本发明部分功能,并不涵盖整体范围。尽管如此,在不违悖本发明之精神及范畴之下,不论是单一或是组合形式的实施例说明或图示,皆可进行细节上的修改及合并。举例来说,对于建构所使用之材料、方法、特定方位、型状、效用及应用上并无特定限制,在秉持本发明之精神及范畴下皆可进行替换,本发明对于实施例更加注重特定细节,并无意于任何形式的限制。
然而,为求达到说明之目的,附图中的图像将以简化的形式呈现,且不一定依照比例进行描绘。另外,在情况允许之下,除了在区分各项元素时给予适当的标示之外,对于图标中相同元素尽量使用相同标示,以利图示之理解。
本发明所公开的实施例仅是针对本发明之原理与应用进行阐述(特定说明、范例示范以及方法学等),在不违悖本发明之精神与范畴下,可对其进行修改及设计,甚至是将其步骤或是特色与其他实施例进行合并运用。
图17为本发明实施例主要架构之流程示意图。
步骤1701,“定义研究计划书(研究发起人)”,发起人如制药公司(不限于此),欲了解新药在某医疗情况下是否具有功效,将对此新药设计进行临床试验研究,这类研究多半采取随机分派临床试验(randomclinicaltrial,rct)之设计,如前所述,此研究设计采取双盲形式,在理想的状况下,试验之研究者、临床医师及照护人员对于药物之分派结果皆处于未知的状态。然而,有些时候基于安全进行虑,如外科手术的介入治疗,使得研究本身的条件限制而无法达到理想的双盲状态。
研究计划书应详尽说明研究内容,除定义研究目的、原理及重要性外,还可以包含受试者纳入标准、基准资料、治疗进行方式、数据收集方法、试验终点及结果(亦即已完成试验之个案功效)等。而为求最小化研究成本及降低受试者暴露于试验中,试验欲求以最少的受试者人数进行研究,同时寻求试验结果具统计学上的意义,因此,样本数估计对于试验是必要的一环,样本数估计理应纳入研究计划书中。另外,由于同时寻求最少样本数及统计上之显著结果,试验设计可能须重度仰赖复杂但已被证实效用的统计分析方法,因此,为求分析结果不受其他因子干扰,呈现其该有的临床意义,在评估单一介入因子时通常会设置严谨的控制条件。
然而,相对于安慰剂、标准治疗及替代疗法等对照组,欲于统计上求得显著意义(如具有优势、劣势),试验所需样本数大小取决于某些参数,而这类参数将定义于试验计划书中。举例来说,试验所需之样本数通常与介入效果、药物治疗成效成反比,但是在研究初期其介入效果通常是未知的,可能只能根据实验室资料、动物实验等获得近似值,而随着试验的进行,介入所造成的影响能获得更适当的定义,并对试验计划书进行适当的修改。而计划书中被定义之参数可能包含条件检定力、显著标准(通常设定为<0.05)、统计检定力、母体变异数、退出试验比率、不良事件发生率等。
步骤1702,“受试者之随机分派(iwrs)”,符合纳入试验研究之受试者可藉由iwrs生成的随机编号或随机序列表进行随机分派,在受试者完成随机分派后,iwrs亦将分配与该组别相对应之药物标签序列,用以确保受试者接收到正确的分配药物。随机化的过程通常在特定的研究地点(如诊所或医院)进行,而iwrs能够使受试者在诊所、医生办公室或通过移动设备在家中进行注册。
步骤1703,“存储分配”,iwrs可以储存相关的数据报含(不仅限于):受试者身分标示、治疗组别(候选药、安慰剂)、分层因子以及受试者之描述性数据等。这些资料将受到加密保护,受试者、调查人员、临床护理人员以及研究发起人等皆无法取得与受试者身份有关的资料。
步骤1704,“受试者之治疗与评估”,在受试者完成随机分派后,根据受试者所属组别给予试验药物、或安慰剂或替代治疗等,受试者需依照访视计划定期回访进行评估,访视次数及频率应明确定义于计划书中,依据计划书要求评估的内容可能包含生命征象、实验室检验、安全性及功效评估等。
步骤1705,“数据管理收集系统(edc)”,研究人员或临床医护人员可根据计划书中所规定之指南对受试者进行评估,并将评估数据输入edc系统中,而评估数据的收集亦可藉由行动装置获得(如穿戴监测装置)。
步骤1706,“储存装置评估”,由edc系统所收集之评估数据可存储于评估数据库,该edc系统则必须符合联邦法规,例如联邦法规的21篇第11节关于临床试验受试者及其资料之规范。
步骤1707,“解盲资料之分析(ddm)”,ddm可与edc、iwrs相互链接构成一封闭系统。而ddm可检视盲性数据库及盲性下之评估数据库,并在信息收集期间计算功效及其95%信赖区间、条件检定力等,并将结果显示于ddm仪版上。另外,在研究执行期间,ddm还可以利用解盲资料进行趋势分析与仿真。
在ddm系统中拥有类似于r程序语言之统计模块编程,使ddm可执行类似自动更新信息并进行实时运算,计算出试验当前功效、其信赖区间、条件检定力等参数,而这类参数在信息时间轴上任一时间点皆可获得。ddm将保留连续且完整的参数估计过程。
步骤1708,“机器学习与人工智能(ddm-ai)”,此步骤为ddm进一步利用机器学习和人工智能技术优化试验,最大化试验成功率,详请参看[0088]。
步骤1709,“ddm界面仪版”,ddm仪版是一edc用户接口,其可提供dmc、研究发起人或是具权限之相关人员查阅试验动态监测结果。
步骤1710,dmc可随时查看动态监测结果,如存有任何安全疑虑或试验趋近功效界线的情况下,dmc可要求召开正式的审查会议。dmc可提出关于试验是否继续进行的相关建议,而dmc做出的任何建议都将与研究发起人进行讨论;在相关规定下,研究发起人亦有权审阅动态监测结果。
图18为本发明中ddm之实施例图示。
如图所示,本发明将多个子系统整合为一封闭回路系统,其分析过程无须有任何人为的介入,数据无需进行解盲,不论任何时候,新的试验数据会不断累积。同时,此系统将自动且连续地计算出试验功效、信赖区间、条件检定力、停止边界值、再估算所需样本量并预测试验之趋势。而对于病患治疗与健康照护部分,此系统亦与真实世界数据(real-worlddata;rwd)及真实世界证据(real-worldevidence;rwe)连接,由此提供治疗方案选择、人群的选择及病情预判因子的识别等。
在一些实施例中,edc系统、iwrs及ddm将整合成一单一封闭回路系统。在一个实施例中,这种至关重要的整合确保使用治疗分配计算治疗功效(如实验组与对照组间之平均数差异)可保存于系统内。其对于不同类型之试验终点的计分功能可构建于edc系统或ddm引擎中。
图9为ddm系统之原理与工作流程之示意图,第一部分:资料抓取;第二部分:ddm规划和配置;第三部分:推导;第四部分:参数估计;第五部分:调整及修改;第六部分:数据监测;第七部分:dmc审查;第八部分:给予研究发起人建议。
如图9所示,ddm运行方式如下:
■在edc系统或ddm中,在任何时间点t(指试验期间的信息时间)皆可获得功效估计值z(t)。
■藉由时间点t之功效估计值z(t)进行条件检定力的估算。
■ddm可利用观察到的功效估计值z(t)进行n次(如n>1000)模拟,以预测后续试验的趋势走向。举例来说,观察试验中初期之100位病患所得之功效估计值z(t)及趋势,可利用其建立之统计模型推估1000多位病患之未来趋势。
■此过程可以在试验进行中动态执行。
■此方法可用于多种目的,如试验人群的选择、预后因子的判别等。
图10为图9中第一部分之实施例图示。
图10说明了如何将病患数据数据导入edc系统。edc的数据来源包括但不限于,如现场调查数据、医院电子病历纪录(electronicmedicalrecords;emr)、穿戴装置等,可将数据数据直接传输至edc系统。而真实世界数据数据,如政府数据数据、保险理赔数据、社交媒体或其他相关数据等,皆可由edc系统相互连接来获取。
参与研究的受试者可以被随机分配至治疗组。基于双盲及临床随机分派试验设计,试验执行过程中,不应向试验相关的任何人员透露受试者所属组别,iwrs将确保分派结果之独立性及安全性。在dmc常规监测中,dmc仅能得到预定义之时间点数据,其后isg通常需要大约3-6个月的时间来进行期中结果分析。这种需要大量人力参与之方法可能导致非本意的”解盲”等潜在风险产生,此为目前dmc监测的主要缺点。与目前dmc监测模式相比,如前述本发明对进行中的试验提供了更好的数据分析模式。
图11为图9中第二部分之实施例图示。
如图11所示,使用者(如研究发起人)需规范其试验终点,试验终点通常是一可定义及可量测之结果。在实际应用上,可同时指定多个试验终点,如一个或多个功效评估之主要试验终点、一个或多个试验安全终点或其任意组合等。
在一个实施例中,在选择欲监测之试验终点时,可以指定端点的类型,即是否使用特定类型的统计数据,包括但不限于于正态分布、二进制事件、事件发生时间、泊松分布或它们的任意组合。
在一个实施例中,亦可以指定试验终点的来源,如试验终点该如何量测、由何人进行、如何确认已达试验终点等。
在一个实施例中,透过参数的设定,亦可以定义ddm的统计目标,如统计显著水平、统计检定力、监测的模式(连续型监测、频率型监测)等。
在一个实施例中,在信息期间或是病患累积到一定百分比时,一次或多次的期中分析可能决定试验是否被停止,而试验被停止时数据可呈现解盲状态并进行分析。用户还可以指定要使用的停止界线的类型,例如基于pocock类型分析的边界、基于o'brien-fleming类型分析的边界,或基于alpha花费函数或其他的某种组合。
用户也可指定动态监测之模式,所要采取的行动如执行仿真、调整样本数、执行无缝设计第二/三期临床试验、选择多重比较下的剂量、选择及调整试验终点、挑选受试族群、比较安全性、评估无效性等。
图12为图9中第三、第四部份之实施例操作示意图。
在这些部分(图9第三第四部份),可以对于研究中之治疗终点数据进行分析,如无法直接从数据库中获得监测终点,系统将要求用户利用现有之数据数据(如血压、实验室检验数值等),于封闭回路系统中编写程序建立一个或多个公式,以获得终点数据相关数据。
一旦得出终点数据数据,系统便可以利用此数据自动计算各项统计数值,如在信息时间点t的估计值及其95%信赖区间、取决于患者累积的条件检定力,或其某种组合等。
图13为图9中第六部分,其显示预定之监测模式可于此部分执行。
如图13所示,ddm可以执行一种或多种预定的监测模式,且将其结果显示在ddm监测显示器上或是或视频屏幕上。其任务包括执行仿真、调整样本数、执行无缝设计第二/三期临床试验、选择多重比较下之剂量、选择及调整试验终点、挑选受试族群、比较安全性、评估无效性等。
在ddm中这些结果可能是以图形或表格的形式输出。
图14及图15为具前景之试验ddm分析结果输出范例图。
在图14及图15中所显示的项目包含功效评估、95%信赖区间、条件检定力、基于o'brien-fleming分析所得之试验停止边界值等。由图14及图15可看出,在个案人数累积至总人数75%时,其良好的功效在统计上已获得验证,故试验可提早结束。
图16呈现ddm试验调整设计之统计分析结果。
如图16所示,自适应群组序列设计初始样本量为每组100名受试者,并预计于在30%和75%的患者累积点上解盲并进行期中分析。如图所示,在累积人数达到75%时(解盲),样本数进行重新估计至每组227人,另外两次的期中分析则预计于累积人数达120及180人时进行。而当累积至180位受试者的终点数据数据时,该试验已跨过了重新计算的停止边界值,显示其候选疗法具有功效。若此试验仅以未调整之最初设定的每组100人进行试验,结果可能相去甚远,且其最初设定之结果可能无法达到统计学上的显著意义。因此,未经调整的试验可能呈现失败的结果,然而在系统连续监测并调整样本数量后,使得试验得到成功。
在一个实施例中,本发明提供了个动态监测和评估进行中的与一种疾病相关的临床试验的方法,该方法包括:
(1)由数据收集系统实时收集临床试验的盲性数据,
(2)由与所述数据收集系统协同操作的一个解盲系统自动将所述盲性数据解盲,
(3)依据所述解盲数据,通过一个引擎连续计算统计量、临界值以及成败界线,
(4)输出其一项评估估计结果,该结果表明如下情形之一:
■所述临床试验具有良好的前景,和
■所述临床试验不具效益,应终止,
所述统计量包括但不限于计分检定、点估计值
在一个实施例中,当满足以下一项或是多项条件时,该临床试验前景将被看好:
(1)最大趋势比率落于0.2~0.4之间,
(2)平均趋势比率不低于0.2,
(3)计分统计数值呈现不断上升之趋势,又或者于信息时间的期间保持正数,
(4)计分统计对于信息时间作图的斜率为正,和
(5)新样本数不超过原计划样本数的3倍,
在一个实施例中,当符合以下一项或是多项条件时,该临床试验不具效益:
(1)最大趋势比小于-0.3,且点估计值
(2)观察到的点估计值
(3)计分统计数值呈现不断下降之趋势,又或者于信息时间的期间保持负数,
(4)计分统计对于信息时间作图的斜率为0或是趋近于0,且只有极小的机会跨越成功之边界,和
(5)新样本数超过原计划样本数的3倍。
在一个实施例中,当该临床试验前景被看好的时候,该方法进而评估所述临床试验,并输出一项额外结果,该额外结果表明是否需要样本数调整。样本数比值如稳定地落于0.6-1.2区间,则样本数不需进行调整;反之落于此区间外则需样本数调整,且新的样本数通过满足以下条件来计算,其中(1-β)为期望的条件检定力:
在一个实施例中,所述方法中的数据收集系统是一个电子数据收集(edc)系统。在另一实例中,所述方法中的数据收集系统则是一个网络交互响应系统(iwrs)。又另一实例中,所述方法中的引擎为一个动态数据监测(ddm)。一实例中,所述方法中的期望的条件检定力至少为90%。
在一实际应用中,本发明提供了一种动态监测和评估进行中的与一种疾病相关的临床试验的系统,该系统包括:
(1)一个由数据收集系统,所述系统实时的从所述该临床试验中收集盲性数据,
(2)一个解盲系统,所述解盲系统与所述数据收集系统协作,自动将所述盲性数据解盲,
(3)一个引擎,所述引擎依据所述解盲资料,连续计算统计量、阈值以及成败界线
(4)一个输出模块或接口,所述输出模块或接口输出一项评估结果,该结果表明如下情形之一
■此临床试验具有良好的前景,和
■此临床试验不具效益,应终止,
其统计量包括但不限于计分检定、点估计值
在一个实施例中,当满足以下一项或是多项条件时,该临床试验前景将被看好:
(1)最大趋势比率落于0.2~0.4之间,
(2)平均趋势比率不低于0.2,
(3)计分统计数值呈现不断上升之趋势,又或者于信息时间的期间保持正数,
(4)计分统计对于信息时间作图的斜率为正,和
(5)新样本数不超过原计划样本数的3倍。
在一个实施例中,当符合以下一项或是多项条件时,该临床试验不具效益:
(1)最大趋势比小于-0.3且点估计值
(2)观察到的点估计值
(3)计分统计数值呈现不断下降之趋势,又或者于信息时间的期间保持负数,
(4)计分统计对于信息时间作图的斜率为0或是趋近于0,且只有极小的机会跨越成功之边界,
(5)新样本数超过原计划样本数的3倍。
在一个实施例中,当该临床试验前景被看好的时候,所述系统由其中引擎进一步评估所述临床试验,并输出一项额外结果,该额外结果表明是否需要样本数调整。样本数比值如稳定地落于0.6-1.2区间,则不需样本数调整;反之,落于此区间外则需样本数调整,且新的样本数通过满足以下条件来计算,其中(1-β)为期望的条件检定力:
在一个实施例中,所述系统中的数据收集系统是一个电子数据收集(edc)系统。在另一实例中,所述系统中的数据收集系统则是一个交互式网络响应系统(iwrs)。又另一实例中,所述系统中的引擎为一个动态数据监测(ddm)。一实例中,所述系统中期望的条件检定力至少为90%。
尽管对于本发明的特殊性已有一定程度的描述,但本发明的公开是藉由示范案例的模式进行,在不违悖本发明精神之情况下,可对其细节进行各种修改与操作变化。
透过提供后续的实验性细节,将能更清楚理解本发明,其实验性细节仅为说明所用,本发明并非局限于此。
在整个申请过程中,引用了各式各样的文献资料或出版物,为了更全面地叙述本发明的相关技术,这些公开的文献数据或出版物信息将结合到本发明中。而引用术语中的包括、包含等,其意思具有开放性,并不排除其他未引用的部分或是方法。
实施例
实施例一
初始设计
假定θ值为试验治疗效果,依照研究数据类型,其值可能为平均数之差异、胜算比、危险对比值等。在试验最初始设计为每组样本数为n0、显著水平为α以及其所期望的统计检定力下,进行假说检定,其虚无假说为治疗无效,对立假说为治疗有效(h0:θ=0versusha:θ>0)。考虑试验经随机分派,其主要指针服从常态分布之假设,令实验组之功效xe服从平均值为μe、变异数为
间歇监测与连续监测
此处将说明统计上的关键讯息部分。一般来说,目前的agsd仅能提供间歇的监测数据,而dad/ddm在每位受试者进入研究后,则可动态地监测试验及检验数据。数据监测的可能行为包括:试验数据的积累、发出进行正式的期中分析(可能无效或有早期效力)的信号、或调整样本量。两者(agsd与dad/ddm)的基本设定大致相似,而本发明将展示如何透过dad/ddm找到适当的时间点并进行实时且正式的期中分析,在此时间点之前,试验将持续进行且无需任何调整。而lan,rosenberger(1993)等人提出的alpha花费函数方法对于两者在信息时间中任何时间点之检定提供了高度的灵活性。然而,要找出调整样本数的时机点(尤其是增加样本数)并非易事,在增加样本数前需对功效有稳健的评估,整个试验期间可能只有一次机会调整样本数。表1显示了样本数再估计(ssr)时机点对于试验之影响,如表1中的第一种情况,该试验预期效益为
表1.进行样本数再估计之时机(令统计检定力为0.9,标准偏差为1)
在任意时间点下,令实验组之样本数为ne,其样本平均为
依上述定义,在试验最后,每组的费雪信息估计为
在期间分析score检验统计量
(1)中的条件检定力预期的个案数n与阈值c,可藉由预期治疗效果θ及目前所观测到的统计检定量
令
再者,虽然使用条件检定力进行样本再估计十分合理,但其并非是在调整样本大小时的唯一考虑,在实际执行上,可能会因预算限制的问题导致样本无法进行调整,或者为求准确的点估计值
为了控制i型错误率,临界/边界值c被认为如下。
当计划的信息时间
也就是说,在没有做任何期中分析,当在样本再估计后,在信息时间满足公式(3)将临界值调整至c1,且
如果于样本再估计前监测gs边界于早期功效,假使最终临界值为cg,则须将公式(3)中的c0替换为cg。关于在dad/ddm的连续监测中的cg,允许因有其功效而提早停止试验的部分,将于实施例3中进一步讨论。例如,进行一显著水平α=0.025、临界值c0=1.96之单尾检定(无期间分析),藉由o’brien-fleming方法得到最终临界值cg=2.24。
请注意,chen,demets和lan(2004)表明,如果在信息时间期间进行了至少50%时使用当前的点估计值
dad/ddm的数据连续累积
图18所示为治疗功效θ真值为0.25、共同变异数为1时的临床试验dad/dmm的仿真特征。此处,在显著水平为0.025(单尾)、统计检定力为90%下,每组所需样本数336人,然而预期治疗功效为θassumed=0.4,其预期样本数为每组133人(总样本数为266人),在每一位受试者进入后即开始连续监测,随着受试者(实验组及对照组)的进入,在临界值为1.96设定下,得到其点估计值
(1)所有的曲线波动皆出现在纳入总受试者的50%(n=133)及75%(n=200)的时候,这是进行中期分析的常用时间点。
(2)点估计值
(3)在每组133人的样本时,虽然wald检定量
(4)信息对比值
(5)由于wald检定量
在这个模拟的实施例中,随着试验的进行,系统对于数据行为的连续监测能提供更好的解读。而透过累积数据的分析,能检测出试验是否有继续进行的价值,如判断其不适合继续进行下去,研究发起人可决定提早终止试验,以减少成本损失及避免受试者承受不必要的痛苦。在一个实施例中,本发明关于样本数的再估计判断适合继续进行试验,最终获得了成功。此外,即使一开始使用了错误的预期功效进行试验,经由不断更新的数据分析引导设计,可将试验引导到正确的方向(如校正样本数等)。下面的实施例2将以趋势比率方法,通过使用dad/ddm评估试验是否具有前景。本文所展示的趋势比率方法及无效停止规则,可进一步协助订立决策。
实施例2
考虑ssr之dad/ddm:样本数重新估算之时机
条件检定力在计算
趋势比率和最大趋势比率
在此章节中,本研究公开使用dad/ddm的工具进行趋势分析,以评估试验是否趋向成功(即,试验是否有希望成功)。该工具使用布朗运动方法来反映轨迹的走向。为此目的,基于原先计划的讯息量
当对立假设为θ>0,s(t)函数的平均轨迹将会向上,且此曲线应会接近y(t)=θt。若检查了离散信息时间t1,t2,...上的曲线,则更多的线段s(ti+1)-s(ti)应该向上(即,sign(s(ti+1)-s(ti))=1),而非向下(即,sign(s(ti+1)-s(ti))=-1)。设l为所计算的线段总数,则长度为l的预期“趋势比”tr(l)则为
在图19中,针对每4位患者(在ti+1和ti之间)计算sign(s(ti+1)-s(ti))的趋势,并当l≥10时开始计算tr(l)。当在t12处有60位患者时,计算出l=10,11,15的tr(l)。图19中6个tr的最大值等于0.5(当l=12时)。可以预期在获取60位患者的数据趋势时,最大tr值(mtr)比平均趋势比率更为敏感。当mtr为0.5时,表示在检查的各区段中呈现正向趋势。
为了研究mtr的特性和可能的用途,针对3种情况,θ=0,0.2,0.4,分别运行了100,000次的模拟研究。在每种情况下,计划的总样本数为266,并针对在ti+1和ti之间的每4位患者,计算sign(s(ti+1)-s(ti))之趋势,并且当l≥10时开始计算tr(l)。由于通常在不超过信息分数3/4的情况下执行ssr(即,此处总共有200名患者),因此当l=10,11,12,...,50,即从t10开始到t50,根据tr(l)计算出mtr。
图20a显示了mtr在41个片段之间的经验分布。如图所示,随着θ的增加,mtr向右移动。图20b显示在不同的临界点之下使用mtr来拒绝h0:θ=0的模拟结果。特别是在a≤mtr<b下每个不同的θ的模拟,最后测试结果为
为了使用mtr来及时监测可能进行ssr的信号,图20b建议将mtr在0.2时设置为临界点。这意味着连续监测时,ssr的时机点安排很灵活;也就是说,在任何
有了tr(l),l=10,11,12,...,在
样本数比率及最小样本数比率
在此部分中,本研究公开了另一种使用dad/ddm进行趋势分析的工具,以评估试验是否趋向成功(亦即,试验是否有希望)。
使用趋势的ssr与使用单个时间点的ssr之比较
传统上,通常在t趋近于1/2但不迟于3/4的某个时间点进行ssr。如上所述,本研究中所公开的dad/ddm使用了数个时间点上的趋势分析。两者皆使用条件检定力之方法,但是在评估治疗效果时利用了不同的数据量。这两种方法通过模拟比较如下。假设一临床试验,其θ为0.25并且共同方差为1(参数与实施例1的第二部分相同),在单边i型错误率为0.025且检定力为90%的设定之下,每个治疗组所需的样本数为n=336。(两组共需672)。但是,假设在进行研究计划时使用θassumed=0.4且设定随机区块大小为4,则所需样本量为每组n=133(共266个样本)。比较两种情况:每次患者入院后使用dad/ddm程序连续监测试验,与常规ssr程序。具体而言,传统的ssr程序分别使用t趋近于1/2时间点(每组人数为66或总数为132)或t趋于3/4的时间点(每组人数为100或总数为200)计算出的
对于dad/ddm,并无预先指定执行ssr的时间点,但监测着计算mtr的时机。从tl=t10开始,每4名患者进入之后开始计算tr(l)(在t10共有40位患者)。依t10,t11,...tl进行mtr之计算,并分别在1,2,...l-9区段上找到tr(l)的最大值,直到第一次mtr≥0.2或直到t≈1/2(总共132名患者),其中tl=t33。与上述传统的t≈1/2方法比较,最大值将超过33-9=24个区段;若与传统的t≈3/4方法进行比较,当tl=t50(总共200例患者)最大值将超过50-9=41个区段。只有在第一个mtr≥0.2时,才会使用等式(2)中的
当进行ssr时,以τ表示时间分数。传统的ssr方法,是按照设计的τ=1/2或3/4进行(因此,无条件机率与条件机率在表2中是相同的)。对于dad/ddm,τ为(与第一个mtr≥0.2相关的患者数量)/266。如果τ超过1/2(第一次比较)或3/4(第二次比较),则τ=1表示未进行ssr。(因此,表2中的无条件机率和条件机率不同。)当每一组人数为133时,样本数变化的起点为n>=45,而每组的增加的数量为4。
在表1中,基于“我们是否有连续6个大于1.02或小于0.8的样本大小比率”重新估计样本大小。在每组45位患者进入后将会做出决定,但每个比率将会在每个区块中计算(即n=4、8、12、16、20、24、28、32等)。如果所有样本大小比率,在24、32、36、40、44、48处均大于1.02或全部小于0.8,则样本数将会在n=48时重新估算。然而,本研究在每个模拟试验结束后计算最大趋势比。它不会影响动态适应设计的决策。
对于这两种方法,均不允许减小样本大小(单纯ssr)。如果nnew小于最初计划的样本数,或者治疗效果为负,则试验应继续使用计划的样本量(共266)。但是,即使在这些情况下样本量保持不变,也要进行ssr。令as=(平均新样本量)/672为对立假设之下理想样本数之百分比,亦或在虚无假设之下,as=(平均新样本量)/266。两者差别如表2和表3,总结如下:
(1)当虚无假设为真时,两种方法皆将i型错误率控制在0.025。在这种情况下,样本量不应被增加。若不考虑功效无效的情况,作为保护措施,此设计之新样本总数为800(近似于266的3倍)。可以看出,对比于原本总样本为266的情况之下,以mtr方法所进行的连续监测方法(as≈183-189%)比传统的单点分析(as≈143-145%)可节省更多。如果考虑功效无效之情况(新样本量超过800,则停止),将可看到更明显的优势。无效监测的描述如下述范例。
(2)当对立假设为真时,基于高估治疗效果的情况之下,两种方法都要求增加样本量。然而,若理想样本量为672的情况下,基于mtr方法所使用的连续监测方法所求得之样本量(≈58-59%)比传统的单点分析(≈71-72%)要少,每种方法所预设的条件机率为0.8。因受试者上限为800故只能达到0.8的条件机率。
(3)相比于传统的固定时间表(t=1/2或3/4)没有执行ssr限制的条件,以mtr≥0.2为条件的连续监测方法,在何时以及决定是否进行ssr上将有条件限制。在虚无假设之下,在试验期间有50%的机会未达mtr≥0.2,因此不进行ssr。(如果不进行ssr,则τ为1)。表2呈现,在mtr≥0.2的条件限制之下的连续监测方法时τ为0.59,与之相对,不具限制条件的固定时间表t=1/2时τ为0.5。然而,在对立假设之下,在试验进行及管理中若可更早地执行可靠的ssr期中分析,将可确定是否需要增加样本量以及增加多少样本量,是更有益处的。与τ=0.5或0.75的常规单次分析相比,基于mtr方法的连续监测在τ=0.34(相对于0.5)或0.32(相对于0.75)时进行ssr的时间要早得多。dad/ddm在固定时间表上执行ssr的时间有非常明显之优势。
实施例3
考虑早期功效及i型错误率控制的dad/ddm
dad/ddm是一种基于lan,rosenbergerandlachin(1993)所提出的开创性理论的方法,针对在试验初期利用连续监测,进而看到显著功效。dad/ddm使用alpha连续花费函数
在设计中采用群组序列边界进行早期功效监测后执行ssr且最终边界值为cg时,实施例1的第二部分讨论了调整最终测试临界值之公式。对于具有连续监测的dad/ddm,cg为2.24。
另一方面,如果在执行ssr后(无论是
在一个实施例中,当使用dad/ddm的连续监测系统时,即使越过功效边界,仍可否决提前终止的建议。可基于lan,lachine和bautisa(2003)的观点推翻系统推荐的ssr信号。在这种情况下,可以收回先前花费的alpha概率,并将之重新花费或重新分配给未来的检定。lan等人(2003年)表示,使用类似o′brien-fleming的花费函数,对最终的i型错误率和研究的最终功效影响可忽略不计。其亦表示可以通过使用固定样本大小的z临界值来收回先前花费的alpha。这种简化的过程保留了i型错误率,同时将检定力之损耗降至最低。
表二:进行100000次仿真的平均结果如下。拒绝h0的总比率和条件比率(第一和第二列)#,对于目标条件概率为0.8,as=(平均样本大小)/672(第三列),ssr的拒绝时间(τ是进行ssr的信息分数)(第四和第五列)
(1)拒绝h0之机率:所有拒绝次数/模拟次数(100000)
(2)条件比率:观察到mtr≥0.2的次数/模拟次数(100000)
(3)拒绝h0之条件机率:观察到mtr≥0.2之拒绝的比率
(4)平均样本数(as)/672:仿真结果之平均样本数/672
(5)τ*:若没观察到mtr≥0.2,则视为1,平均讯息比例来自所有仿真结果
(6)τ**:只来自mtr≥0.2的平均讯息比例
#:当
+:根据公式(1),其中c1根据公式(3),c0=1.96;
++:tr(l)上的最大值,l=10,11,12,,...直到tl=t33,使用区间中与mtr相关
+++:tr(l)上的最大值,其中l=10,11,12,,...直到tl=t50,使用区间中与mtr相关
表三:拒绝虚无假设的机率:所有拒绝的次数/模拟次数(100000)
(1)条件机率:观察到minsr≥1.02的次数/sim(100,000)
(2)拒绝虚无假设的条件机率:观察到minsr(最小样本数比)≥1.02且拒绝虚无假设的机率
(3)平均样本数/672:仿真结果之平均样本数/(266or672)
(4)τ*:若没观察到minsr≥1.02,则视为1。平均讯息比例来自所有100,000次模拟结果
(5)τ**:只来自minsr≥1.02的平均讯息比例
实施例四
考虑无效性的dad/ddm
一些关于药物无效的重要因素值得一提。首先,先前所讨论的ssr程序也可能和药物无效相关。若重新估算的新样本量超出了原先计划的样本量的数倍,这将会超出试验进行之可能性,那么发起人可能会认为该试验是无效的;其次,无效性分析有时会被嵌入期中功效分析,但是,由于决定试验是否无效(据此停止试验)没有约束力,因此无效性分析计划不会影响i型错误率。相反,无效性之期中分析会增加ii型错误率,进而影响试验之检定力;第三,当无效性之期中分析和ssr以及功效分析分开进行时,应该考虑无效性分析的最佳策略,包括执行的时间和无效的条件,以最大程度地降低成本和检定力之损失。可以想象,通过在每次患者进入后利用dad/ddm连续分析当下所累积数据,可比单次的期中分析更加的可靠的、且更快速地监测试验的无效性。本节首先回顾了用于间歇数据监测之无效性分析的最佳时间,进而说明使用dad/ddm连续监测之过程,亦藉由模拟比较间歇监测和连续监测这两种方法。
间歇数据监测的无效期中分析的最佳时机
在进行ssr时,本研究藉由适当地增加样本数以确保试验之检定力,同时在虚无假设为真的情况下,也会防止不必要的增加样本数。传统的ssr通常在某个时间点进行,例如t=1/2,但不晚于t=3/4。在无效性分析中,本研究的程序可以尽早发现无效的情况,以节省成本以及因无效治疗而遭受痛苦的病患。另一方面,无效性分析会影响试验的检定力。频繁的无效分析会导致过多的检定力损失。因此,本研究可以通过在检定力损耗时找寻样本数(成本)的最小化为目标,来优化进行无效性分析的时机。这种方法已被xi,gallo和ohlssen(2017)采用。
群组序列试验中伴随可被接受边界之无效性分析
假设申办方在群组序列试验中,预计要执行k-1次的无效期中分析,其中样本数为nk,在每次执行的讯息时间为tk,而所累积的讯息量标示为ik,k=1,...,k-1。假设讯息时间
给定θ之条件下,期望之总讯息量为
期望之总讯息量可视为最大讯息量之百分率etiθ(%)=etiθ/ik。
群组序列试验检定力为
不进行无效性分析之固定样本试验设计检定力为u=p(z>zα|θ=θ*),与之相比,检定力会因为无效停止而降低为
可看出当dk越大,越容易达到无效边界并且提早停止试验,所损失的检定力也越大。因
当pl≤λ时,可找寻(tk,bk),k=1,...,k-1,以最小化eti0。这里的λ可用来防止由于无效性分析而导致的检定力降低,近而可能会错误地终止试验。xi,galloandohlssen(2017)以gamma(γ)函数为边界值,研究在各种可接受的检定力损失λ之下的最佳分析时间点。
针对一次无效性分析,执行时无须局限于无效性边界。也就是说,可以找到(t1,b1)满足eti0=[t1中(b1)+1-中(b1)]之最小化,并满足pl=p(z1≤d1,z2>zα|θ=θ*)≤λ。对于给定的λ和zα,在检测θ*时,可以在10≤t1≤.80(可每次增加0.05或0.10)之间进行搜索,藉以获取对应的边界值b1.
举例来说,当检测θ*=0.25且zα=1.96时,如果允许检定力的减少在λ=5%,则当t1=0.40处的无效边界b1=0.70为最佳执行之时间点(每次以0.10递增)。在虚无假设下,以预期总信息量衡量的成本节省(表示为固定样本量设计的比率)为eti0=54.5%。若仅允许检定力的减少为λ=1%,则通过相同的方式,则当t1=0.50处且无效边界b1=0.41为最佳执行之时间点,可节省eti0=67.0%。
针对上述无效性分析的时机点及边界,接下来所需考虑的是其稳健性。假设最佳分析时机与相关的边界值是一起设计的,但实际上在监测时,无效性分析的时机可能不在原设计的时程上。本发明想做甚么呢?通常希望保持原本的边界值(因为该边界值已记录在统计分析计划书中),因此应研究检定力耗损和eti0的变化。xi,gallo和ohlssen(2017)报告了以下内容:在试验设计中,当检定力耗损为λ=1%时,在t1=0.50且b1=0.41为最佳分析时机,可节省成本eti0=67.0%(如上所述)。假设在进行无效性分析期间进行监测的实际时点t在[0.45,0.55]之间,边界b1亦如计划书所定义的为0.41,当实际时间t从0.5偏离到0.45时,检定力的耗损会从1%增加到1.6%,且eti0会从67%些微降低至64%。当实际时间t从0.5变更成0.55时,检定力的耗损会从1%降低至0.6%,且eti0会从67%增加至70%。因此,t1=0.50,b1=0.41是最佳的无效性分析条件。
此外,在考虑最佳无效性分析条件的稳健度,还需考虑试验的治疗效果θ*。假设当θ*为0.25时,xi,galloandohlssen(2017)所使用的最佳无效性规则产生的检定力耗损介于0.1%到5%。分别比较当θ=0.2、0.225、0.275和0.25所计算出的检定力耗损,结果表明,检定力耗损的幅度非常接近。例如,对于假设最大检定力耗损为5%的情况下(假设θ*=0.25),如果实际θ=0.2,则实际检定力耗损为5.03%,如果实际θ=0.275,则实际检定力耗损为5.02。
考虑条件检定力之无效性分析
另一个在群组序列试验的无效性分析研究是使用公式(1)中的条件检定力
类似地,若根据ssr中,n=nnew所得之
在连续监测过程中最佳执行无效期中分析之时机
在公式(1)中,当n=n0或nnew时,条件检定力所得之趋势比率为
使用群组序列和使用趋势的无效性分析之比较
根据实施例2相同的设定,通常会在t≈1/2进行ssr。如前所述,dad/ddm是在多个时间点上使用趋势分析。两者都使用条件检定力方法,只是在估计治疗效果时选用的讯息量不同。比较两方法的模拟结果如下:假设试验之θ=0.25且共同变异数为1(此假设与第3.2节及第4节相同),在检定力为90%,单尾i型错误率为0.025之下,每组所需要的样本为336人(两组共672人)。然而,试验计划假设θassumed=0.4,每组计划纳入133人(两组共266人),随机区组大小为4。两种情形相比较:在每个受试者进入试验后使用dad/ddm程序进行连续监测,与考虑无效性的常规ssr。对于常规ssr,ssr与无效性分析可在t≈1/2时进行,所需每组样本为66人,两组共132人。若在θassumed=0.4假设之下的条件检定力低于40%或是所需要的新样本数会超过800,则最后因无效性停止试验。此外,若
使用dad/ddm时,没有预先设定进行ssr的时间点,但需要监测mtr的时间,当tl=t10开始,计算每四位受试者所对应的tr(l)。随着mtr,依据t10,t11,...tl,在不同的区段1,2,...l-9,分别计算并找到最大的tr(l),直到第一次出现mtr≥0.2的时间点或是t≈1/2(共132位受试者),其中tl=t33且最大区段为33-9=24。只有在第一个mtr≥0.2时,才会使用公式(2)在与mtr相关的区间中使用
在虚无假设下,计分函数s(t)~n(0,t),这代表s(t)会呈水平趋势,并在经过一半的时间之后小于0。当每一段间隔在i0,1,i0,2,...,且s(t)<0时,可表示为|i0,1|,|i0,2|,...,则(∑i|i0,i|/t)=0.5。因此当∑i|i0,i|/t接近0.5时,则试验很有可能是无效的。此外,wald统计量
表四中观察到的负值的次数具有区分θ=0与θ>0之高度特异性。例如,进行s(t)或z(t)小于零之无效性评估,当θ=0.2时,正确决策的机率是77.7%,而错误决策的机率是8%。通过更多的仿真显示,dad/ddm的评估结果优于间歇监测的无效性评估。
表四:当s(t)小于零时进行无效性分析之模拟结果(100,000次模拟)
由于每当抽取新的随机样本时会计算分数,因此可以按如下公式在时间t计算无效率fr(t):fr(t)=(s(t)小于零的次数)/(计算的s(t)总数)。
实施例五
使用带ssr的dad/ddm进行推断
dad/ddm假设初始样本数为n=n0并且具有相应的fisher信息t0,并且计分函数s(t)≈b(t)+θt~n(θt,t)随着纳入的数据不断地进行计算。假设没有任何期中分析,如果试验在计划的信息时间t0结束,且
适应设计可允许在任何时间修改样本数,当时间为t0时,观测到的计分
chen,demets和lan(2004)证明,如果在t0处的点估计值的条件检定力至少为50%,则增加样本量不会增加i型错误率,在最后检定时无需将c0更改为c1。
最后观测到的计分为
表五:点估计及信赖区间估计(最多修改两次样本数)
令
表5显示,从常态分布n(θ,1)中抽取随机样本,重复100,000次模拟结果,在不同θ之下,其点估计量及双尾信赖区间。
实施例六
比较agsd及dad/ddm
本发明首先描述进行有意义比较agsd和dad/ddm的性能度量,其后描述仿真研究及其结果。
设计的性能度量
理想的设计将能够提供足够的检定力(p),而无需在有功效(θ)之情况下使用过多的样本量(n)。此概念在图3中说明得更具体:
■一般来说,设计一个试验的检定力为p0=0.9,其p0-δ≤p(δ=0.1)是可被接受的,但p<p0-δ是不可被接受的。举例来说,预设的检定力为0.9,而0.8是可被接受的。
■在一个固定样本且检定力为p的试验中,np是所需的样本数。检定力p0>0.9的设计不常见,因为np会远大于n0.9(即,需要增加的样本数大于n0.9,但相对获得的检定力却不大。这样的样本数在罕见疾病或试验中是不可行的,因为每位患者的费用很高)。样本数n大于(1+r1)n0.9(r1=0.5)时将视为样本过大而无法接受,即便所对应之检定力微大于0.9。举例来说,为提供检定力p=0.999而要求样本大小为n0.999之设计不是理想的设计。另一方面,若样本数n<(1+r1)n0.9可以提供至少0.9的检定力,是可以被接受的。
■另一个不可接受的情况是,尽管在0.8≤p<0.9时,检定力(虽非理想)是可以接受的,但样本量并不“经济”。例如,当n>(1+r2)n0.9时(r2=0.2)。如图所示,a3为不可接受的区域。
可接受的功效大小范围为θ∈(θlow,θhigh),其中θlow是临床上最小的功效。
临界值取决很多因素,如成本、弹性度、未满足的医疗需求等等。以上讨论建议试验设计(固定样本设计或非固定样本设计)之性能由三个参数度量,即(θ,pd,nd),其中θ∈(θlow,θhigh),pd为检定力,nd是对应pd所需样本大小。因此,评估一个试验设计是需要考虑三个维度的。试验的设计评估分数如下
先前,liu等人(2008)和fang等人(2018)都使用一个维度来评估不同的设计。两种评估表都难以解释,因为它们都将三维评估简化为一维指标。本发明的评估分数保留了设计性能的三维特质,并且易于解释。
agsd与dad/ddm的模拟结果如下。如果假设θassumed=0.4,检定力为90%(单尾i型错误率为0.025),则计划的样本数为每组133。从n(θ,1)中随机抽取样本,其中θ真值分别为0,0.2,0.3,0.4,0.5,0.6,则每组的样本数上限为600。在100,000次模拟之下计算每个方案的评估分数,i型错误率不会因无效分析而减少,因为无效停止是被认为是无约束性的。
agsd之仿真规则
仿真需要自动化的规则,通常是简化的和机械化的。在agsd的模拟中,使用实践中常用的规则。这些规则是:(i)两次检视,在0.75的信息分数时进行期中分析。(ii)在期中分析中进行ssr(cui,hung,wang,1999;gao,ware,mehta,2008)。(iii)无效停止的标准:在期中分析时,
dad/ddm之仿真规则
在dad/ddm的模拟中,可利用一些简化的规则自动做出决定。这些条件(与agsd平行并与之相反):(i)在信息时间t内连续监测,0<t≤1。(ii)使用r的值对ssr计时。执行ssr时,可达到90%检定力之时机。(iii)无效停止标准:在任何信息时间t,在时间间隔(0,t)内
模拟结果
表六:比较asd及ddm之结果
注:as-ss为平均仿真之样本大小;sp为模拟之检定力;fs为无效停止(%).
表六之100,000次模拟结果比较了asd及ddm在h0下的无效性停止率、平均样本数及检定力。可清楚地显示,ddm具有更高的无效停止率(74.8%),用较少的样本数可获得所需要且可被接收的检定力。
■对于虚无假设θ=0,i型错误率在agsd及dad/ddm皆可被控制。相比agsd所使用的单点分析,dad/ddm根据趋势倾向做出的无效停止规则更加具体和可靠。因此,dad/ddm的无效停止率高于agsd,且样本数小于agsd。
■对于θ=0.2,agsd无法提供可接受的检定力。当θ=0.6,agsd会导致样本量过大。在这两种极端情况下,agsd的计分皆为ps=-1,而dad/ddm的计分是可以接受的(ps=0)。对于其他的情况,θ=0.3、0.4和0.5,agsd和dad/ddm可通过合理的样本量达到预期的条件检定力。
总之,仿真结果显示,如果功效的假设错误,则:
i)dad/ddm可以将试验引导至适当的样本量,在各种可能的情况下提供足够的检定力。
ii)如果真实功效远小于或大于默认值,则agsd将调整不良。在前一种情况下,agsd所提供的检定力会小于可接受的检定力,而在后一种情况下,会需要更多样本数。
使用后向图像进行概率计算的证明
中位数无偏点估计
假设在w(·)中调整样本数,其中给定观察值
对于给定
当
因此,(θ0.5≤θ)=0.5,
后向图像计算
单次样本数调整之估计
令
且
两次样本数调整之估计
在最后推断时,
因此,
实施例七
进行期中分析是试验中的一个重要的成本,需要时间、人力、物力来准备数据以供数据监测委员会(dmc)审议。这亦是只能偶尔进行监测的主要原因。由前面的说明可知,此种偶然进行期中分析的数据监测,仅能得到数据的“快照”,因此仍具有极大的不确定性。相反,本发明的连续数据监测系统,利用每个患者进入时的最新数据,得到的不仅仅是单点时间的“快照”,更可以揭示试验的趋势。同时,dmc通过使用dad/ddm工具,可以大大减少成本。
ddm的可行性
ddm过程需要通过连续监测正在进行的数据,这涉及连续的解盲并计算监测统计信息。如此,由独立统计小组(isg)处理是不可行的。如今随着技术的发展,几乎所有的试验都可由电子数据收集(edc)系统管理,并且使用交互式响应技术(irt)或网络交互响应系统(iwrs)处理治疗的任务。许多现成的系统都包含了edc和iwrs,而解盲和计算任务可以在此集成的系统中执行。这将避免由人去解盲并保护了数据的完整性,尽管机器辅助ddm的技术细节不是本文的重点,但值得注意的是通过利用现有技术,进行连续数据监测的ddm是可行的。
数据指导性分析
使用ddm,在实际情况下应尽早开始数据指导性的分析,可以将其内置到ddm中,自动执行分析。自动化机制实际上是利用“机器学习(m.l)”的想法。数据指导性的适应方案,例如样本量重新估计、剂量选择、人群富集等,可以被视为将人工智能(a.i)技术应用于正在进行的临床试验。显然,具有m.l和a.i的ddm可以应用于更广泛的领域,例如用于真实世界证据(rwe)和药物警戒(pv)信号监测。
实施动态自适应设计
dad程序增加了灵活性,提高了临床试验的效率。如果使用得当,它可以帮助推进临床研究,特别是在罕见疾病和试验中,毕竟每位患者的治疗费用相当昂贵。但是,该程序的执行需要仔细讨论。控制和减少潜在的操作偏差的措施是至关重要的。这样的措施可以更加有效,并确保是否可以识别和确定潜在偏差的具体内容。而在过程中置入自适应群组序列设计的程序,是可行且极具实用性的。在计划的期中分析中,数据监测委员会(dmc)将收到由独立的统计学家们所得出的汇总结果,并举行会议进行讨论。尽管在理论上可以多次修改样本大小(例如,参见cui,hung,wang,1999;gao,ware,mehta,2008),但通常仅进行一次。通常会因应dmc的建议对试验计划书进行修订,但是,dmc可以举行不定期的安全评估会议(在某些疾病中,试验功效终点也是安全终点)。dmc的当前设置(稍作修改)可用于实现动态自适应设计。主要区别在于,采用动态自适应设计时,dmc可能不会定期举行审查会议。独立的统计人员可以在数据积累时随时进行趋势分析(可以通过可不断下载数据的电子数据捕获(edc)系统来简化此过程),但结果不必经常与dmc成员共享(但是,如果必要且监管机构允许,可以通过一些安全的网站将趋势分析结果传给dmc,但无需正式的dmc会议);可以在正式dmc审查前,并认为趋势分析结果有决定性时告知dmc。因为大多数试验确实会对试验计划书进行多次修改,其中可能对样本量进行不止一次的修改,考虑到试验效率的提高,这不算是额外增加负担。当然,此类决定应由发起人做出。
dad和dmc
本发明引入了动态数据监测概念,并展示了其在提高试验效率方面的优点,其先进的技术使其能在未来的临床试验中实施。
ddm可直接服务于数据监测委员会(dmc),而大多数dmc监测试验为ii-iii期。dmc通常每3或6个月开会一次,具体时间取决于试验。例如,与没有生命威胁性的疾病试验相比,对于采用新方案的肿瘤学试验,dmc可能希望更频繁地举行会议,在试验的早期阶段更快地了解安全情况。当前的dmc做法涉及三个方面:发起人、独立统计小组(isg)和dmc。发起人的责任是执行和管理正在进行的研究。isg根据计划时间点(通常在dmc会议召开前一个月)准备盲性和解盲数据包,包括:表格、列表和图形(tlf),准备工作通常需要3到6个月的时间。dmc成员在dmc会议前一周收到数据包,并将在会议上进行审查。
当前的dmc在实践中存有一些问题。首先,显示的数据分析结果只是对于数据的一个快照,dmc看不到治疗效果(有效性或安全性)的趋势。基于数据快照的建议和能看到连续的数据追踪的建议可能会不同。如下图所示,在a部分中,dmc会建议两个试验i和ii都继续,而在b部分中,dmc可能建议终止试验ii,因其有负向的趋势。
当前的dmc进程也存在后勤问题。isg大约需要3到6个月来准备dmc的数据包。而解盲通常由isg处理。尽管假定isg将保留数据完整性,但是人工的操作过程并不能100%的保证。借助ddm的edc/iwrs系统具有安全性和有效性数据的优点,这些数据将由dmc直接进行实时监测。
减少样本量以提高效率
理论上,减小样本对于动态自适应设计和自适应群组序列设计都是有效的(例如,cui,hung,wang,1999,gao,ware,mehta,2008)。我们在asd和dad的仿真上发现,减少样本数量可以提高效率,但由于担心“操作偏差”,在目前试验中,修改样本大小通常意味着增加样本。
非固定样本设计的比较
除了asd,还有其他非固定样本的设计。lanetal(1993)提出了一种对数据进行连续监测的程序。如果实际效果大于假定效果,则可以尽早停止该试验,但是该过程不包括ssr。fisher“自我设计临床试验”(fisher(1998),shen,fisher(1999))是一种灵活的设计,它不会在初始设计中固定样本量,而是让“期中观察”的结果来确定最终的样本量,亦允许通过“方差支出”进行多个样本大小的校正。群组序列设计、asd、lan等人(1993年)的设计均为多重测试程序,其中,在每个期中分析都要进行假设检验,因此每次都必须花费一些alpha来控制i型错误率(例如lan,demets,1983,proschanetal(1993))。另一方面,fisher的自我设计试验并非多重测试程序,因为无需在“期中观察”上进行假设检验,因此不必花费任何alpha来控制i型错误率。正如shen,fisher(1999年)所阐释的:“我们的方法与经典的群组序列方法之间的显著区别是,我们不会在期中观察中测试其治疗效果。”i型错误率控制是通过加权实现的。因此,自行设计的试验确实具有上述“增加灵活性”的大部分,但是,它不是基于多点时间点分析的,也不提供无偏差点估计或信赖区间。下表总结了这些方法之间的异同。
实施例八
一项随机、双盲、安慰剂对照之iia期研究被用于评估口服候选药物的安全性和有效性。该研究未能证明功效。将ddm应用于研究数据,显示了整个研究的趋势。
图22包括具有95%信赖区间的主要试验终点估计、wald统计、计分统计、条件功效和样本量比率(新样本量/计划的样本量)。计分统计量、条件功效和样本数量是稳定的,并且接近零(图中未显示)。由于图中显示不同剂量(所有剂量、低剂量和高剂量)与安慰剂的关系有相似的趋势和规律,因此图22中仅显示所有剂量与安慰剂的关系。因标准偏差估计的原因,每组至少从两名患者开始绘制。x轴为患者完成研究的时间。示意图在每个患者完成研究后更新。
1):所有剂量对比安慰剂
2):低剂量(1000毫克)对比安慰剂
3):高剂量(2000毫克)对比安慰剂
实施例九
一项多中心、双盲、安慰剂对照、4个组别的ii期试验被用于证明治疗夜尿症的候选药物的安全性和其有效性。将ddm应用于研究数据,显示了整个研究的趋势。
相关图中包括具有95%信赖区间的主要试验终点估计、wald统计(图23a)、分数统计、条件功效(图23b)和样本量比率(新样本量/计划的样本量)(图23c)。由于图显示不同剂量(所有剂量、低剂量、中剂量和高剂量)与安慰剂的关系有相似的趋势和规律,图中仅显示所有剂量与安慰剂的关系。
由于标准偏差估计的原因,每图从组中的至少两个患者开始。x轴为患者完成研究的时间。示意图在每个患者完成研究后更新。
1:所有剂量vs安慰剂
2:低剂量vs安慰剂
3:中剂量vs安慰剂
4:高剂量vs安慰剂
参考文献
1.chandler,r.e.,scott,e.m.,(2011).statisticalmethodsfortrenddetectionandanalysisintheenvironmentalsciences.johnwiley&sons,2011
2.chenyh,demetsdl,lankk.increasingthesamplesizewhentheunblindedinterimresultispromising.statisticsinmedicine2004;23:1023-1038.
3.cui,l.,hung,h.m.,wang,s.j.(1999).modificationofsamplesizeingroupsequentialclinicaltrials.biometrics55:853-857.
4.fisher,l.d.(1998).self-designingclinicaltrials.stat.med.17:1551-1562.
5.gaop,warejh,mehtac.(2008),samplesizere-estimationforadaptivesequentialdesigns.journalofbiopharmaceuticalstatistics,18:1184-1196,2008
6.gaop,liul.y,andmehtac.(2013).exactinferenceforadaptivegroupsequentialdesigns.statisticsinmedicine.32,3991-4005
7.gaop,liul.y.,andmehtac.(2014)adaptivesequentialtestingformultiplecomparisons,journalofbiopharmaceuticalstatistics,24:5,1035-1058
8.herson,j.andwittes,j.theuseofinterimanalysisforsamplesizeadjustment,druginformationjournal,27,753d760(1993).
9.jennisonc,andturnbullbw.(1997).groupsequentialanalysisincorporatingcovarianceinformation.j.amer.statist.assoc.,92,1330-1441.
10.lai,t.l.,xing,h.(2008).statisticalmodelsandmethodsforfinancialmarkets.springer.
11.lan,k.k.g.,demets,d.l.(1983).discretesequentialboundariesforclinicaltrials.biometrika70:659-663.
12.lan,k.k.g.andwittes,j.(1988).theb-value:atoolformonitoringdata.biometrics44,579-585.
13.lan,k.k.g.andwittes,j.‘theb-value:atoolformonitoringdata’,biometrics,44,579-585(1988).
14.lan,k.k.g.anddemets,d.l.‘changingfrequencyofinterimanalysisinsequentialmonitoring’,biometrics,45,1017-1020(1989).
15.lan,k.k.g.andzucker,d.m.‘sequentialmonitoringofclinicaltrials:theroleofinformationandbrownianmotion’,statisticsinmedicine,12,753-765(1993).
16.lan,k.k.g.,rosenberger,w.f.andlachin,j.m.useofspendingfunctionsforoccasionalorcontinuousmonitoringofdatainclinicaltrials,statisticsinmedicine,12,2219-2231(1993).
17.tsiatis,a.‘repeatedsignificancetestingforageneralclassofstatisticsusedincensoredsurvivalanalysis’,journaloftheamericanstatisticalassociation,77,855-861(1982).
18.lan,k.k.g.anddemets,d.l.‘groupsequentialprocedures:calendartimeversusinformationtime’,statisticsinmedicine,8,1191-1198(1989).
19.lan,k.k.g.anddemets,d.l.changingfrequencyofinterimanalysisinsequentialmonitoring,biometrics,45,1017-1020(1989).
20.lan,k.k.g.andlachin,j.m.‘implementationofgroupsequentiallogranktestsinamaximumdurationtrial’,biometrics.46,657-671(1990).
21.mehta,c.,gao,p.,bhatt,d.l.,harrington,r.a.,skerjanec,s.,andwarej.h.,(2009)optimizingtrialdesign:sequential,adaptive,andenrichmentstrategies,circulation,journaloftheamericanheartassociation,119;597-605(includingonlinesupplementmadeapartthereof).
22.mehta,c.r.,andpinggao,p.(2011)populationenrichmentdesigns:casestudyofalargemultinationaltrial,journalofbiopharmaceuticalstatistics,21:4831-845.
23.müller,h.h.and
24.nasastandardtrendanalysistechniques(1988).https://elibrary.gsfc.nasa.gov/_assets/doclibbidder/tech_docs/29.%20nasa_std_8070.5%20-%20copy.pdf
25.o’brien,p.c.andfleming,t.r.(1979).amultipletestingprocedureforclinicaltrials.biometrics35,549-556.
26.pocock,s.j.,(1977),groupsequentialmethodsinthedesignandanalysisofclinicaltrials.biometrika,64,191-199.
27.pocock,s.j.(1982).interimanalysesforrandomizedclinicaltrials:thegroupsequentialapproach.biometrics38,(1):153-62.
28.proschan,m.a.andhunsberger,s.a.(1995).designedextensionofstudiesbasedonconditionalpower.biometrics,51(4):1315-24.
29.shih,w.j.(1992).samplesizereestimationinclinicaltrials.inbiopharmaceuticalsequentialstatisticalapplications,k.peace(ed),285-301.newyork:marceldekker.
30.shih,w.j.commentary:samplesizere-estimation-journeyforadecade.statisticsinmedicine2001;20:515-518.
31.shih,w.j.commentary:groupsequential,samplesizere-estimationandtwo-stageadaptivedesignsinclinicaltrials:acomparison.statisticsinmedicine2006;25:933-941.
32.shihwj.plantobeflexible:acommentaryonadaptivedesigns.biomj;2006;48(4):656-9;discussion660-2.
33.shih,w.j."samplesizereestimationinclinicaltrials"inbiopharmaceuticalsequentialstatisticalanalysis.editor:k.peace.marcel-dekkerinc.,newyork,1992,pp.285-301.
34.k.k.gordonlanjohnm.lachinoliverbautistaover-rulingagroupsequentialboundary—astoppingruleversusaguideline.statisticsinmedicine,volume22,issue21
35.wittes,j.andbrittain,e.(1990).theroleofinternalpilotstudiesinincreasingtheefficiencyofclinicaltrials.statisticsinmedicine9,65-72.
36.xid,gallopandohlssend.(2017).ontheoptimaltimingoffutilityinterimanalyses.statisticsinbiopharmaceuticalresearch,9:3,293-301.
权利要求书(按照条约第19条的修改)
1.1-20.删除。
一个动态监测和评估进行中的与一种疾病相关的临床试验的方法,所述方法包括:
(1)由数据收集系统实时从所述临床试验中收集盲性数据,
(2)由与所述数据收集系统协同操作的一个解盲系统自动将所述盲性数据解盲,
(3)依据所述解盲数据,通过一个引擎连续计算统计量、临界值以及成败界线,和
(4)输出一项评估结果,该结果表明如下情形之一:
·所述临床试验具有良好的前景,和
·所述临床试验不具效益,应终止,
所述统计量选自最大趋势比(maximumtrendratio;mtr)、样本数值比(samplesizeratio;ssr)及平均趋势比中的一项或多项,其中
所述mtr=max1tr(l),其中
所述ssr=a,其中新的样本数通过满足以下条件来计算:
所述平均趋势比由下式计算:
2.根据权利要求21的方法,其特征在于,所述统计量进一步包含计分检定、点估计值
3.根据权利要求21的方法,其特征在于,当满足以下一项或是多项条件时,所述临床试验前景将被看好:
(1)最大趋势(mtr)比率介于0.2~0.4之间,
(2)平均趋势比率不低于0.2,
(3)计分统计数值呈现不断上升之趋势,又或者于信息时间的期间保持正数,
(4)计分统计对于信息时间作图的斜率为正,和
(5)新样本数不超过原计划样本数的3倍。
4.根据权利要求21的方法,其特征在于,当符合以下一项或是多项条件时,所述临床试验不具效益:
(1)所述最大趋势比小于-0.3且所述点估计值
(2)观察到的点估计值
(3)计分统计数值呈现不断下降之趋势,又或者于信息时间的期间保持负数,
(4)计分统计对于信息时间作图的斜率为0或是趋近于0,且只有极小的机会跨越所述成功边界,和
(5)新样本数超过原计划样本数的3倍。
5.根据权利要求21的方法,其特征在于,当所述临床试验是有前景时,所述方法包括评估所述临床试验,并输出一项额外结果,该额外结果表明是否需要样本数调整。
6.根据权利要求25的方法,其特征在于,当ssr稳定在[0.6-1.2]之内时,不需要样本数调整。
7.根据权利要求25的方法,其特征在于,当ssr稳定并且小于0.6或大于1.2时,需要所述样本数调整。
8.根据权利要求21的方法,其特征在于,所述数据收集系统是一个电子数据收集(edc)系统。
9.根据权利要求21的方法,其特征在于,所述数据收集系统是一个网络交互响应系统(iwrs)。
10.根据权利要求21的方法,其特征在于,所述引擎是一个动态数据监测(ddm)引擎。
11.根据权利要求21的方法,其特征在于,所述期望的条件检定力为至少90%。
12.一个动态监测和评估进行中的与一种疾病相关的临床试验的系统,所述系统包括:
(1)一个数据收集系统,所述系统实时从所述临床试验中收集盲性数据,
(2)一个解盲系统,所述解盲系统与所述数据收集系统协作,自动将所述盲性数据解盲,
(3)一个引擎,所述引擎依据所述解盲数据,系统连续计算统计量、阈值以及成败界线,
(4)一个输出模块或界面,所述输出模块或界面输出一项评估结果,该结果表明如下情形之一:
·所述临床试验具有良好的前景,和
·所述临床试验不具效益,应终止,
其所述统计量选自最大趋势比(maximumtrendratio;mtr)、样本数值比(samplesizeratio;ssr)及平均趋势比中的一项或多项,其中
所述mtr=maxltr(l),其中
所述ssr=a,其中新的样本数通过满足以下条件来计算:
所述平均趋势比由下式计算:
13.根据权利要求32的系统,其特征在于,所述统计量进一步包含计分检定、点估计值
14.根据权利要求33的系统,其特征在于,当满足以下一项或是多项条件时,所述临床试验前景将被看好:
(1)最大趋势比率落于0.2~0.4之间,
(2)平均趋势比率不低于0.2,
(3)计分统计数值呈现不断上升趋势,又或者于信息时间的期间保持正数,
(4)计分统计与信息时间间斜率为正,和
(5)新样本数不超过原计划样本数的3倍。
15.根据权利要求33的系统,其特征在于,当符合以下一项或是多项条件时,所述临床试验不具效益:
(1)所述最大趋势比小于-0.3且所述点估计值
(2)观察到呈现负值的点估计值
(3)计分统计数值呈现不断下降趋势,又或者于信息时间的期间保持负数,
(4)计分统计对于信息时间作图的斜率为0或是趋近于0,且只有极小的机会可跨越所述成功边界,和
(5)新样本数超过原计划样本数的3倍。
16.根据权利要求34的系统,其特征在于,所述临床试验有前景时,所述引擎评估所述临床试验,并输出一项额外结果,该额外结果表明是否需要样本数调整。
17.根据权利要求36的系统,其特征在于,当ssr稳定在[0.6-1.2]之内时,不需要样本数调整。
18.根据权利要求36的系统,其特征在于,当ssr稳定并且小于0.6或大于1.2时,需要所述样本数调整。
19.根据权利要求32的系统,其特征在于,所述数据收集系统是一个电子数据收集(edc)系统。
20.根据权利要求32的系统,其特征在于,所述数据收集系统是一个交互式网络响应系统(iwrs)。
21.根据权利要求32的系统,其特征在于,所述引擎是一个动态数据监测(ddm)引擎。
22.根据权利要求32的系统,其特征在于,所述期望的条件检定力为至少90%。
23.一个动态监测和评估进行中的与一种疾病相关的临床试验的方法,所述方法包括:
(1)由数据收集系统实时从所述临床试验中收集盲性数据,
(2)由与所述数据收集系统协同操作的一个解盲系统自动将所述盲性数据解盲,
(3)依据所述解盲数据,通过一个引擎连续计算统计量、临界值以及成败界线,其中所述临界值以及所述成败界线通过模拟由所述统计量中连续获得,和
(4)输出一项评估结果,该结果表明如下情形之一:
·所述临床试验具有良好的前景,和
·所述临床试验不具效益,应终止,
所述统计量选自计分检定、点估计值
所述mtr=max1tr(l),其中
所述ssr=a,其中新的样本数通过满足以下条件来计算:
所述平均趋势比由下式计算:
24.根据权利要求43的方法,其特征在于,步聚(4)中的所述评估结果根据临界边界获得,并据此控制第一型错误,所述临界边界由如下公式调整并计算:
- 还没有人留言评论。精彩留言会获得点赞!