一种光催化生产过氧化氢反应机理的分析方法
1.本发明属于光催化领域,涉及一种光催化生产过氧化氢反应机理的分析方法,具体涉及一种结合实验与理论计算分析光催化生产过氧化氢反应机理的方法。
背景技术:
2.光催化技术得益于其低成本、低能耗、可持续等特点,在生产过氧化氢的方向受到人们的广泛关注。然而,目前大多数研究者对于光催化产过氧化氢的反应机理不够重视且提出的机理也是非常简单,即光生空穴氧化醇产生醛和氢离子,而光生电子促进氧气的一步两电子或两步单电子还原生成过氧化氢,这些机理没有考虑到在光反应系统中生成的、也具有氧化能力的活性氧物种也可能直接氧化醇而直接生成过氧化氢,因而分析光催化生产过氧化氢的机理有助于发现过氧化氢生成过程中的限制和促进因素,对进一步提高过氧化氢的产量极其重要。因此,基于目前所提出的机理的局限性和揭示完整反应机理的重要性,开发准确、高效的机理分析方法是十分有必要的。
技术实现要素:
3.本发明要解决的技术问题是克服现有技术的不足,提供一种普适性强、效率高、成本低、准确的光催化生产过氧化氢反应机理的分析方法。
4.为解决上述技术问题,本发明采用以下技术方案:
5.一种光催化生产过氧化氢反应机理的分析方法,包括以下步骤:
6.s1、构建待研究光催化剂的模型;
7.s2、分析待研究光催化剂对应模型的电子和空穴分布;
8.s3、根据待研究光催化剂在光催化生产过氧化氢中产生的活性物种及各活性物种的转化过程,以及步骤s2中分析得到的待研究光催化剂对应模型的电子和空穴分布结果,模拟待研究光催化剂对应模型中包含的反应路径,分析待研究光催化剂生产过氧化氢的反应机理。
9.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,步骤s1中,所述待研究光催化剂为改进型光催化剂时,构建待研究光催化剂的模型,包括以下步骤:
10.s1
‑
1、根据待研究光催化剂和所述待研究光催化剂对应的原始光催化剂的x射线光电子能谱表征结果、元素分析表征结果和傅里叶红外光谱表征结果,分析待研究光催化剂的原子变化情况、各个官能团变化情况;
11.s1
‑
2、根据待研究光催化剂的原子变化情况、各个官能团变化情况,在原始光催化剂的模型的基础上,构建待研究光催化剂的模型。
12.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,步骤s1中,所述待研究光催化剂为新合成光催化剂时,构建待研究光催化剂的模型,包括以下步骤:
13.s1
‑
1、根据待研究光催化剂的x射线光电子能谱表征结果、元素分析表征结果和傅里叶红外光谱表征结果,分析待研究光催化剂的原子情况、各个官能团情况;
14.s1
‑
2、根据待研究光催化剂的原子情况、各个官能团情况,构建待研究光催化剂的模型。
15.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,采用计算机软件构建待研究光催化剂的模型;所述计算机软件为vesta。
16.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,步骤s2中,采用计算机软件对步骤s1中待研究光催化剂的模型进行理论计算,分析待研究光催化剂对应模型的电子和空穴分布情况,得到待研究光催化剂对应模型的电子空穴等值面图;所述计算机软件为gaussian 16c01,所用的泛函为m06
‑
2x,基组为6
‑
311g**。
17.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,步骤s3中,采用计算机软件模拟待研究光催化剂对应模型中包含的反应路径,根据各反应路径对应的限速步的能垒结果,确定待研究光催化剂生产过氧化氢的反应机理;所述计算机软件为cp2k,所用的模块为quickstep,所用的方法是dimer和ci
‑
neb。
18.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,以限速步的能垒最低值对应的反应路径作为待研究光催化剂生产过氧化氢的反应机理。
19.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,步骤s3中,所述待研究光催化剂在光催化生产过氧化氢中产生的活性物种及各活性物种的转化过程,由待研究光催化剂的牺牲剂实验结果和自由基捕获实验结果分析得到,包括以下步骤:
20.(1)考察不同牺牲剂对待研究光催化剂光催化产过氧化氢量的影响,根据不同牺牲剂条件下过氧化氢的产量结果,分析待研究光催化剂在光催化生产过氧化氢中产生的活性物种;
21.(2)根据步骤(1)中得到的待研究光催化剂在光催化生产过氧化氢中产生的活性物种类型,选择相应的捕获剂;
22.(3)考察相应的捕获剂对步骤(1)中分析得到的活性物种之间转化关系的影响,根据活性物种之间的转化关系,分析待研究光催化剂在光催化生产过氧化氢中产生的活性物种的转化过程。
23.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,所述待研究光催化剂的牺牲剂实验过程中采用的牺牲剂为对苯醌或色氨酸;所述牺牲剂实验的反应体系中牺牲剂的浓度控制在0.1m~10m;所述待研究光催化剂的牺牲剂实验过程中采用的捕获剂为temp或dmpo;所述自由基捕获实验的反应体系中捕获剂的浓度控制在0.1m~10m。
24.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,所述待研究光催化剂为改进型光催化剂时,包括元素掺杂型光催化剂、负载型光催化剂或缺陷型光催化剂;所述元素掺杂型光催化剂包括各种元素掺杂的氮化碳、氮化硼和共价有机骨架材料中的其中一种;所述负载型光催化剂包括单原子负载的氮化碳、氮化硼和共价有机骨架材料中的其中一种;所述缺陷型光催化剂为氮缺陷的氮化碳、氮化硼和共价有机骨架材料中的其中一种。
25.上述的光催化生产过氧化氢反应机理的分析方法,进一步改进的,所述待研究光催化剂为氮缺陷的氮化碳时,在光催化生产过氧化氢中产生的活性物种为超氧阴离子自由基和单线态氧;所述活性物种的转化过程是先由超氧阴离子自由基转化成单线态氧,再由单线态氧转化成过氧化氢;
26.所述待研究光催化剂为氮缺陷的氮化碳时,模拟得到的氮缺陷的氮化碳的各反应路径如下:
27.第一条:异丙醇上的亚甲基氢先转移到氮缺陷附近的一个氮原子上,而氧气得到一个电子被还原为超氧阴离子自由基,然后异丙醇上的羟基氢自发转移到超氧阴离子自由基上变为ooh物种,最后,ooh与氮原子上的氢结合为过氧化氢;
28.第二条:异丙醇上的亚甲基氢先转移到氰基附近的一个氮原子上,而氧气得到一个光生电子被还原为超氧阴离子自由基,然后异丙醇上的羟基氢自发转移到超氧阴离子自由基上变为ooh物种,最后,ooh与氮原子上的氢结合为过氧化氢;
29.第三条:异丙醇上的亚甲基氢先转移到氮缺陷的一个碳原子上,而氧气得到一个光生电子被还原为超氧阴离子自由基,然后异丙醇上的羟基氢自发转移到超氧阴离子自由基上变为ooh物种,最后,ooh与氮原子上的氢结合为过氧化氢;
30.第四条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,异丙醇的亚甲基氢和羟基氢直接转移到超氧阴离子自由基上形成过氧化氢;
31.第五条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,随后被空穴氧化为单线态氧,异丙醇的亚甲基氢和羟基氢直接转移到单线态氧上形成过氧化氢;
32.第六条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,随后被空穴氧化为单线态氧,异丙醇的亚甲基氢先转移到相邻的一个水分子上形成h3o
+
,这个h3o
+
又将一个氢转移到另一个水分子上形成h3o
+
,同时自身恢复为水分子,生成的h3o
+
将一个氢转移到单线态氧上,同时异丙醇的羟基氢也转移到单线态氧上形成过氧化氢;
33.第七条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,表面的超氧阴离子自由基脱附到溶液中形成自由的超氧阴离子自由基,异丙醇的亚甲基氢和羟基氢直接转移到自由的超氧阴离子自由基上形成过氧化氢;
34.第八条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,随后被空穴氧化为单线态氧,随后表面的单线态氧脱附到溶液中形成自由的单线态氧,异丙醇的亚甲基氢和羟基氢直接转移到自由的单线态氧上形成过氧化氢;
35.其中,第一至第八条反应路径对应的限速步的能垒依次为0.82ev、2.37ev、1.75ev、0.79ev、0.80ev、0.66ev、0.77ev、0.71ev;
36.对比各反应路径的限速步的能垒,以限速步的能垒最低值对应的第六条反应路径作为氮缺陷的氮化碳光催化生产过氧化氢的反应机理。
37.与现有技术相比,本发明的优点在于:
38.本发明提供了一种光催化生产过氧化氢反应机理的分析方法,根据待研究光催化剂在光催化生产过氧化氢中产生的活性物种及各活性物种的转化过程,以及待研究光催化剂对应模型的电子和空穴分布结果,模拟待研究光催化剂对应模型中包含的反应路径,分析待研究光催化剂生产过氧化氢的反应机理。本发明中,采用实验手段确定实验结果,可以保障结果的准确性,同时理论计算能在分子甚至原子尺度上更直观和具体的分析出化学反应的具体过程,进而在实验结果与理论计算分析的相互验证下,进一步提高准确性,因而能在高准确性的前提下高效地分析光催化产过氧化氢的反应机理。另外,无论是实验手段还
是理论计算都能在短时间内得到结果,分析效率非常高,非常有利于根据最终结果指导实际的光催化剂制备,包括对光催化剂进行改良。此外,相比于使用限制大且昂贵的原位表征实验,本发明方法的分析成本很低。本发明光催化生产过氧化氢反应机理的分析方法具有普适性强、效率高、成本低、准确等优点,可广泛用于分析、确定待研究光催化剂光催化生产过氧化氢的反应机理,使用价值高,应用前景好。
附图说明
39.为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整的描述。
40.图1为本发明实施例1中氮化碳和氮缺陷的氮化碳的n/c原子比对比图。
41.图2为本发明实施例1中氮化碳和氮缺陷的氮化碳的傅里叶红外光谱图。
42.图3为本发明实施例1中氮化碳和氮缺陷的氮化碳的高分辨光电子能谱图。
43.图4为本发明实施例1中氮缺陷的氮化碳模型的电子空穴等值面图。
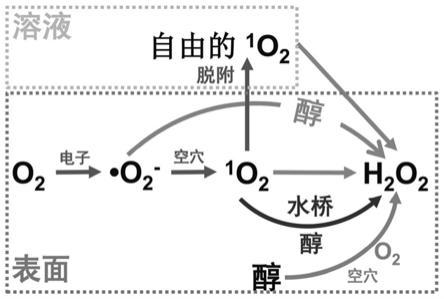
44.图5为本发明实施例1中氮缺陷的氮化碳在不同条件下光催化产过氧化氢的产量对比图。
45.图6为本发明实施例1中氮缺陷的氮化碳在不同条件下的电子顺磁共振图。
46.图7为本发明实施例1中氮缺陷的氮化碳对应模型中包含的反应路径图。
47.图8为本发明实施例1中氮缺陷的氮化碳对应模型中包含的各个反应路径的限速步能垒图。
具体实施方式
48.以下结合说明书附图和具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。
49.以下实施例中所采用的原料和仪器均为市售;其中光源系统为pls
‑
sxe 300c氙灯,购于北京泊菲莱科技有限公司。所使用的软件为gaussian 16c01、cp2k、vesta。
50.实施例1
51.一种光催化生产过氧化氢反应机理的分析方法,具体为:分析氮缺陷的氮化碳在光催化产过氧化氢中的反应机理,包括以下步骤:
52.s1、构建氮缺陷的氮化碳的模型:
53.s1
‑
1、分别对氮化碳(原始光催化剂)和氮缺陷的氮化碳(待研究光催化剂)进行x射线光电子能谱表征、元素分析表征和傅里叶红外光谱表征,根据氮化碳(原始光催化剂)和氮缺陷的氮化碳(待研究光催化剂)的x射线光电子能谱表征结果、元素分析表征结果和傅里叶红外光谱表征结果,分析待研究光催化剂的原子变化情况、各个官能团变化情况,具体如下:
54.对氮化碳和氮缺陷的氮化碳进行有机元素分析和x射线光电子能谱分析,同时由有机元素分析和光电子能谱确定氮化碳和氮缺陷的氮化碳的n/c原子比,结果如图1所示。图1为本发明实施例1中氮化碳和氮缺陷的氮化碳的n/c原子比对比图。由图1可知,相比于氮化碳,氮缺陷的氮化碳的n/c原子比都减少了,这说明氮缺陷的氮化碳中的n元素含量减少了,其中由光电子能谱确定的n/c原子比减少得更显著,进一步说明主要是表面的n元素
减少了。
55.对氮化碳和氮缺陷的氮化碳进行傅里叶红外光谱分析,结果如图2所示。图2为本发明实施例1中氮化碳和氮缺陷的氮化碳的傅里叶红外光谱图。由图2可知,相比于氮化碳,氮缺陷的氮化碳产生了氰基基团(c≡n),且氨基基团(n
‑
h)减少。
56.对氮化碳和氮缺陷的氮化碳进行高分辨的x射线光电子能谱分析,结果如图3所示。图3为本发明实施例1中氮化碳和氮缺陷的氮化碳的高分辨光电子能谱图。由图3可知,氮缺陷的氮化碳的制备方法中,naoh的添加导致了氰基基团的产生、氨基基团的减少,同时二配位的n原子(n
2c
)也减少了。由于氰基基团的产生、氨基基团的减少不会导致n元素的减少,这说明n缺陷主要是n
2c
减少所导致的。
57.s1
‑
2、根据步骤s1
‑
1中氮缺陷的氮化碳的原子变化情况、各个官能团变化情况,在氮化碳的模型的基础上,采用计算机软件(vesta,版本为3.5.7)构建氮缺陷的氮化碳的模型,具体为:根据氮缺陷的氮化碳的氮原子的变化情况以及官能团的变化情况,在计算机软件中对氮化碳的模型进行调整,具体步骤为添加氰基基团、减少氨基基团、添加氮缺陷,构建得到氮缺陷的氮化碳的模型。
58.s2、采用计算机软件(gaussian 16c01,所用的泛函为m06
‑
2x,基组为6
‑
311g**)对步骤s1中氮缺陷的氮化碳的模型进行理论计算,分析氮缺陷的氮化碳对应模型的电子和空穴分布情况,得到氮缺陷的氮化碳对应模型的电子空穴等值面图,结果如图4所示。
59.图4为本发明实施例1中氮缺陷的氮化碳模型的电子空穴等值面图。由图6可知,d0→
d2是亮激发且相应的空穴和电子分别分布在氮缺陷位点和氮缺陷附近的melon的1,4位点(nv1,4),d0→
d3也是亮激发且相应的空穴和电子分别分布在氮缺陷附近的n原子上和氮缺陷位点,而d0→
d1和d0→
d4是暗激发。
60.s3、根据氮缺陷的氮化碳在光催化生产过氧化氢中产生的活性物种及各活性物种的转化过程,以及步骤s2中分析得到的氮缺陷的氮化碳对应模型的电子和空穴分布结果,模拟氮缺陷的氮化碳对应模型中包含的反应路径,分析氮缺陷的氮化碳生产过氧化氢的反应机理,具体如下:
61.s3
‑
1、考察氮缺陷的氮化碳在不同条件下对过氧化氢产量的影响,分析氮缺陷的氮化碳在光催化生产过氧化氢中产生的活性物种及各活性物种的转化过程。
62.对照组:将氮缺陷的氮化碳、异丙醇和水在避光环境下混合,所得混合液在可见光(模拟太阳光光源)下进行光催化反应1h,以此制备过氧化氢,反应结束后,检测过氧化氢的产量;所得混合液中氮缺陷的氮化碳的浓度为1.0g/l,异丙醇和水的体积比为1/9。
63.牺牲实验组:在进行光催化反应之前,分别往混合液中添加不同的牺牲剂(对苯醌、对苯醌),其他反应条件与对照组相同,反应结束后,检测在不同牺牲剂条件下的过氧化氢的产量。牺牲实验组中,混合液中牺牲剂的浓度均为0.14m。
64.基于对照组(无牺牲剂)的过氧化氢的产量,通过对比不同牺牲剂下对应的过氧化氢产量数据,分析确定氮缺陷的氮化碳在光催化生产过氧化氢中产生的活性物种,结果如图5所示。图5为本发明实施例1中氮缺陷的氮化碳在不同条件下光催化产过氧化氢的产量对比图。由图5可知,在常规条件下,氮缺陷的氮化碳在1个小时能通过光催化产过氧化氢将近500μm。加入超氧阴离子自由基的牺牲剂,对苯醌后,过氧化氢的产量为零。加入单线态氧的牺牲剂,色氨酸后,过氧化氢的产量大大降低。由此可以推测超氧阴离子自由基和单线态
氧可能是光催化反应中的中间产物。
65.基于牺牲剂实验结果,以temp为捕获剂,继续进行自由基捕获实验。
66.自由基捕获实验组:在进行光催化反应之前,分别往混合液中添加对苯醌或对苯醌/temp,其中自由基捕获实验组中,控制混合液中对苯醌和temp的浓度均为0.14m,其他反应条件与对照组相同。在反应过程中,检测氮缺陷的氮化碳在光催化生产过氧化氢中产生的不同活性物种的信号强度,具体为:
67.对不同捕获剂条件下的氮缺陷的氮化碳进行电子顺磁共振实验,结果如图6所示。图6为本发明实施例1中氮缺陷的氮化碳在不同条件下的电子顺磁共振图。由图6a、6b可知,氮缺陷的氮化碳在光照下分别表现出明显的超氧阴离子自由基和单线态氧信号,这证明了超氧阴离子自由基和单线态氧的确存在于光催化反应中。为了进一步验证超氧阴离子自由基和单线态氧之间的关系,在使用对苯醌捕获超氧阴离子自由基后进行了单线态氧的电子顺磁共振信号检测。图6c中的结果表明单线态氧的信号完全消失,仅检测到对苯醌和temp相互作用产生的信号,因为在对苯醌/temp系统中观察到的信号与氮缺陷的氮化碳/对苯醌/temp相同。因此,部分超氧阴离子自由基可能不会直接转化为过氧化氢,而是首先转化为单线态氧。
68.基于上述结果可知,氮缺陷的氮化碳在光催化生产过氧化氢中产生的活性物种为超氧阴离子自由基和单线态氧,且活性物种的转化过程是先由超氧阴离子自由基转化成单线态氧,再由单线态氧转化成过氧化氢。
69.s3
‑
2、采用计算机软件(cp2k,版本7.1,所用的模块为quickstep,所用的方法是dimer和ci
‑
neb)对步骤s3
‑
1中氮缺陷的氮化碳在光催化生产过氧化氢中产生的活性物种及转化过程、步骤s2中分析得到的氮缺陷的氮化碳对应模型的电子空穴等值面图进行过渡态理论计算,模拟氮缺陷的氮化碳对应模型中包含的反应路径,结果如图7所示,同时得到各反应路径对应的限速步的能垒结果,如图8所示,根据各反应路径对应的限速步的能垒结果,确定待研究光催化剂生产过氧化氢的反应机理。
70.图7为本发明实施例1中氮缺陷的氮化碳对应模型中包含的反应路径图。由图7可知,模拟得到的氮缺陷的氮化碳的各反应路径如下:
71.第一条:异丙醇上的亚甲基氢先转移到氮缺陷附近的一个氮原子上,而氧气得到一个电子被还原为超氧阴离子自由基,然后异丙醇上的羟基氢自发转移到超氧阴离子自由基上变为ooh物种,最后,ooh与氮原子上的氢结合为过氧化氢;
72.第二条:异丙醇上的亚甲基氢先转移到氰基附近的一个氮原子上,而氧气得到一个光生电子被还原为超氧阴离子自由基,然后异丙醇上的羟基氢自发转移到超氧阴离子自由基上变为ooh物种,最后,ooh与氮原子上的氢结合为过氧化氢;
73.第三条:异丙醇上的亚甲基氢先转移到氮缺陷的一个碳原子上,而氧气得到一个光生电子被还原为超氧阴离子自由基,然后异丙醇上的羟基氢自发转移到超氧阴离子自由基上变为ooh物种,最后,ooh与氮原子上的氢结合为过氧化氢;
74.第四条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,异丙醇的亚甲基氢和羟基氢直接转移到超氧阴离子自由基上形成过氧化氢;
75.第五条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,随后被空穴氧化为单线态氧,异丙醇的亚甲基氢和羟基氢直接转移到单线态氧上
形成过氧化氢;
76.第六条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,随后被空穴氧化为单线态氧,异丙醇的亚甲基氢先转移到相邻的一个水分子上形成h3o
+
,这个h3o
+
又将一个氢转移到另一个水分子上形成h3o
+
,同时自身恢复为水分子,生成的h3o
+
将一个氢转移到单线态氧上,同时异丙醇的羟基氢也转移到单线态氧上形成过氧化氢;
77.第七条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,表面的超氧阴离子自由基脱附到溶液中形成自由的超氧阴离子自由基,异丙醇的亚甲基氢和羟基氢直接转移到自由的超氧阴离子自由基上形成过氧化氢;
78.第八条:吸附在氮缺陷的氮化碳表面的氧气得到一个光生电子转化为超氧阴离子自由基,随后被空穴氧化为单线态氧,随后表面的单线态氧脱附到溶液中形成自由的单线态氧,异丙醇的亚甲基氢和羟基氢直接转移到自由的单线态氧上形成过氧化氢。
79.图8为本发明实施例1中氮缺陷的氮化碳对应模型中包含的各个反应路径的限速步能垒图。由图8可知,第一至第八条反应路径对应的限速步的能垒依次为0.82ev、2.37ev、1.75ev、0.79ev、0.80ev、0.66ev、0.77ev、0.71ev。
80.由图7和图8可知,通过对比各反应路径的限速步的能垒,结果表明:以限速步的能垒最低值对应的第六条反应路径作为氮缺陷的氮化碳光催化生产过氧化氢的反应机理,即以超氧阴离子自由基或单线态氧为氧化驱动力的路径都比以空穴为氧化驱动力的路径更有利,其中反应路径六是最有利的,这表明表面的单线态氧和水桥的协同作用对过氧化氢的生成起着主要的作用。
81.本实施例中,采用的氮化碳的制备方法,包括以下步骤:
82.(1)15克三聚氰胺在马弗炉中以10℃min
‑1的速率在550℃下煅烧4小时即可得到氮化碳样品。
83.本实施例中,采用的氮缺陷的氮化碳的制备方法,包括以下步骤:
84.(1)15克三聚氰胺溶解在含有30毫升水和1.5克naoh的naoh水溶液中搅拌均匀。
85.(2)将步骤(1)中得到的混合液中的水蒸发至干。
86.(3)将步骤(2)中得到的所得固体混合物在马弗炉中以10℃min
‑1的速率在550℃下煅烧4小时。
87.(4)将步骤(3)中得到的样品用乙醇和水清洗多次后烘干,得到氮缺陷的氮化碳样品。
88.以上实施例仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例。凡属于本发明思路下的技术方案均属于本发明的保护范围。应该指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下的改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1