一种基于Fasta、Fastq生物大数据的压缩方法与流程
本发明涉及高压缩算法工具领域,具体为一种基于fasta、fastq生物大数据的压缩方法。
背景技术:
1、在生物信息技术领域,随着第二代、第三代基因样本数据急剧增大,信息系统面对海量的fasta、fastq格式的大数据存储。存储技术涉及磁盘阵列、sas、san、分布式存储系统等技术,每一份数据都要保存2到3个副本。面对大量的数据长期存储或冷备时,特别是数据中心、科研院所等机构需要pb(1pb=1024tb)级的数据存储时,本算法代来的经济价值非常大。然而,现有压缩gz、rar格式压缩比率还不够低,在大数据存储使用上成本不够低,在pb级的存储节省的投入不够大,并不能把专用fasta、fastq格式的大数据做到最理想的压缩效果。
2、所以,人们急需一种基于fasta、fastq生物大数据的压缩方法来解决上述问题。
技术实现思路
1、本发明的目的在于提供一种基于fasta、fastq生物大数据的压缩方法,以解决上述背景技术中提出的问题。
2、为了解决上述技术问题,本发明提供如下技术方案:
3、一种基于fasta、fastq生物大数据的压缩方法,该方法包括以下步骤:
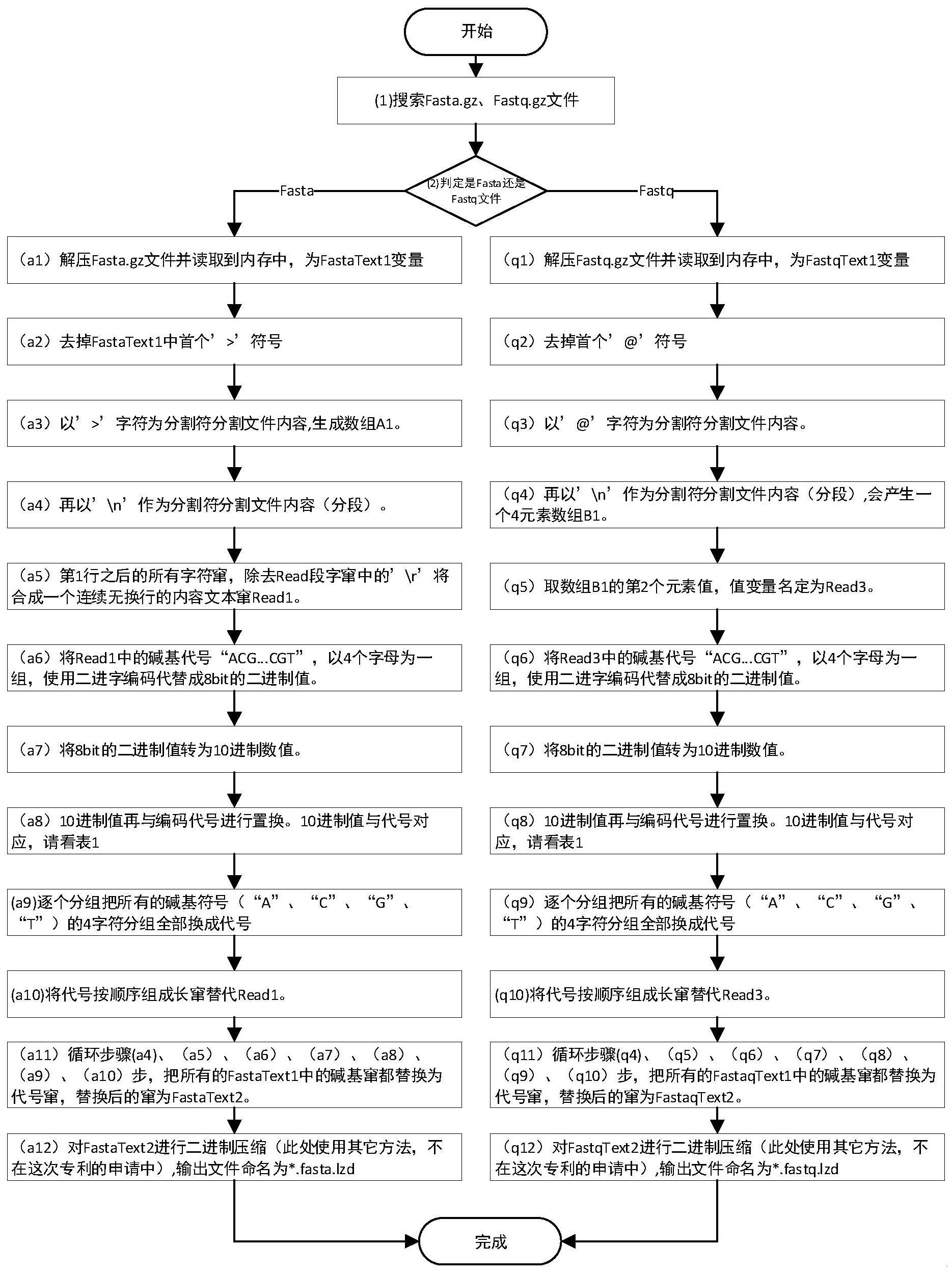
4、s1、在目录中通过文件名后缀判定fasta文件或者fastq文件类型,解压文件并读取到内存中;
5、s2、对fasta文件或者fastq文件进行分割处理,组成新的数组文件和文本文件;
6、s3、对新的数组文件进行代号字符数值分组和转换处理;
7、s4、将转换以后的代号字符重新组成新的串,并进行替代存储,从而减少字符数量提高压缩率。
8、根据上述技术方案,在步骤s1中,通过现有技术的检索程序在所述指定目录中检索后缀名为fasta或者fastq的文件类型,并对所述fasta或者fastq的文件类型进行解压并读取到所述内存中。
9、根据上述技术方案,在步骤s2中,去掉内存中后缀名为fasta的解压文件的首个“>”符号,后文以“>”字符为分割符号分割所述数组文件内容,生成新的数组文件,再以“\n”作为分隔符分割所述文本文件内容;对于后缀名为fastq的解压文件,需要去除文件中首个“@”符号,后文以“@”字符为分割符号分割所述数组文件内容,生成新的数组文件,产生一个四元素数组,再以“\n”作为分隔符分割所述文本文件内容。
10、根据上述技术方案,在步骤s3中,对于所述fasta的数组文件和文本文件的第一行之后的所有字符串,除去字符串的“\r”,重新合成一个连续无换行的内容文本串;对于所述fastq的数组文件和文本文件,取数组文件的第二个元素值,并修定值变量名。
11、根据上述技术方案,在步骤s3中,将所述的fasta或者fastq中的碱基代号以4个字母为一组,使用二进字编码代替成8bit的二进制值,以“a”、“c”、“g”、“t”转换成二进制值,分别对应“00”、“01”、“10”、“11”,4个字符,顺序不限,再将文本进行4个字母对应值二进制值,即8个bit做成分组数据,将8bit的二进制值转为10进制数值数组,10进制值与代号对应与编码代号进行转换,转换后将所有字符串连接起来转成新的文本。
12、根据上述技术方案,在步骤s4中,将所述代号字符重新组成串r2,用r2串代替所述字符串在文件中进行存储,从而减少因基序列读数文本数据内容的字符数量,从而达到压缩效果。
13、根据上述技术方案,所述的fasta或者fastq中的每个字节的值对应图3中的索引值对应固定代号字符0~255的数值,共256个值,每个值对应一个西文单字节字符或双字节字符。
14、与现有技术相比,本发明所达到的有益效果是:
15、1、本专利主要的核心原理是先将因基序列读数,即“acgt...ccgt”字符串,以“a”、“c”、“g”、“t”编排成二进制值,分别对应“00”、“01”、“10”、“11”,4个字符或8个字符一组,以4个字符为例,4个字符组成的串对应一个字节b1,每个字节的值对应十进制的0~255的数值,共256个值,每个十进制值对应一个西文单字节字符,然后将代号字符重新组成串,用重新组成的串代替原字符串在文件中进行存储,减少因基序列读数文本数据内容的50%~75%数量,从而提升压缩的效果。
16、2、针对现有压缩gz、rar格式压缩比率还不够低,在大数据存储使用上成本不够低,在pb级的存储节省的投入不够大的现状,可以在一定程度上降低存储成本,减少存储经济投入。
技术特征:
1.一种基于fasta、fastq生物大数据的压缩方法,其特征在于:该方法包括以下步骤:
2.根据权利要求1所述的一种基于fasta、fastq生物大数据的压缩方法,其特征在于:在步骤s1中,通过程序在所述指定目录中检索后缀名为fasta或者fastq的文件类型,并对所述fasta或者fastq的文件类型进行解压并读取到所述内存中。
3.根据权利要求2所述的一种基于fasta、fastq生物大数据的压缩方法,其特征在于:在步骤s2中,去掉内存中后缀名为fasta的解压文件的首个“>”符号,后文以“>”字符为分割符号分割所述数组文件内容,生成新的数组文件,再以“\n”作为分隔符分割所述文本文件内容;对于后缀名为fastq的解压文件,需要去除文件中首个“@”符号,后文以“@”字符为分割符号分割所述数组文件内容,生成新的数组文件,产生一个四元素数组,再以“\n”作为分隔符分割所述文本文件内容。
4.根据权利要求3所述的一种基于fasta、fastq生物大数据的压缩方法,其特征在于:在步骤s3中,对于所述fasta的数组文件和文本文件的第一行之后的所有字符串,除去字符串的“\r”,重新合成一个连续无换行的内容文本串;对于所述fastq的数组文件和文本文件,取数组文件的第二个元素值,并修定值变量名。
5.根据权利要求4所述的一种基于fasta、fastq生物大数据的压缩方法,其特征在于:在步骤s3中,将所述的fasta或者fastq中的碱基代号以4个字母为一组,使用二进字编码代替成8bit的二进制值,以“a”、“c”、“g”、“t”转换成二进制值,分别对应“00”、“01”、“10”、“11”,4个字符,顺序不限,再将文本进行4个字母对应值二进制值,即8个bit做成分组数据,将8bit的二进制值转为10进制数值数组,10进制值与代号对应与编码代号进行转换,转换后将所有字符串连接起来转成新的文本。
6.根据权利要求5所述的一种基于fasta、fastq生物大数据的压缩方法,其特征在于:在步骤s4中,将所述代号字符重新组成串r2,用r2串代替所述字符串在文件中进行存储。
7.根据权利要求6所述的一种基于fasta、fastq生物大数据的压缩方法,其特征在于:所述的fasta或者fastq中的每个字节的值对应索引值对应固定代号字符中0~255的数值,共256个值,每个值对应一个西文单字节字符或双字节字符。
技术总结
本发明公开了一种基于Fasta、Fastq生物大数据的压缩方法,属于高压缩算法工具领域。包括以下步骤:S1、通过文件名后缀判定Fasta文件或者Fastq文件类型,解压文件并读取到内存中;S2、对Fasta文件或者Fastq文件进行分割处理,组成新的数组文件;S3、对新的数组文件进行代号字符数值分组和转换处理;S4、将转换以后的代号字符重新组成新的串,并进行替代存储,从而减少字符数量提高压缩率。本发明充分利用Fasta、Fastq数据格式的特征与计算机底层编码原理开展数据压缩算法与工具设计,采用基因碱基符号进行字符转换,以代号字符替换原有的碱基字符后再存储数据和进一步压缩。
技术研发人员:李志达,郭涛
受保护的技术使用者:玉溪融建信息技术有限公司
技术研发日:
技术公布日:2024/1/13
- 还没有人留言评论。精彩留言会获得点赞!