用于治疗HBV感染的组合物的制作方法
本发明涉及具有乙型肝炎病毒(HBV)组分的免疫原性组合物,其可以是基于核酸或多肽。所述免疫原性组合物可用于刺激或增强对HBV的免疫应答,目标是提供针对HBV感染和由HBV感染引起或与其相关的任何疾病或病症的预防性或治疗性效果。本发明还涉及用于表达这样的HBV组合物的表达载体和它们的治疗性或预防性用途。本发明特别关注免疫治疗,更具体是用于治疗感染HBV(尤其那些慢性感染)的患者的免疫治疗领域。
背景
乙型肝炎是主要的公共健康问题,全世界有超过3.5亿人口慢性感染,他们中的20到40%处于发展慢性肝疾病、肝硬化和肝癌的风险中。尽管存在有效的预防疫苗,乙型肝炎病毒(HBV)感染在许多国家,甚至发达国家中仍然猖獗,估计全世界每年有4百五十万新感染病例。与WHO实现世界性疫苗接种的建议不同,全程预防接种的覆盖率从在亚洲25%至在欧洲75-90%不等。目前乙型肝炎是导致死亡率(1百万死亡/年左右)的第10名,并且HBV相关肝癌为第5常见癌症。HBV感染的地理分布是不均匀的,在西方国家普遍低于1%,在东南国家、非洲和赤道南美洲大部分超过10%。在高HBV慢性携带者的流行区域,从感染的母亲传染给新生儿的垂直传播是最常见的污染模式并且几乎总是造成慢性肝炎(90%的病例)。通过出生后立即对感染的婴儿进行预防性接种,该比率可被降低至15%。在西方国家,感染最有可能通过水平传播发生于成年期,其通过体液如血液、精液、唾液,在85%的患者中造成急性和自我恢复感染,但仍然在15%的例子中造成慢性感染。
乙型肝炎病毒(HBV)是嗜肝DNA病毒科(hepadnaviridae)的成员并且主要感染肝脏,在肝细胞中复制。感染性颗粒是所谓的42-45nm的“Dane颗粒”,其由外部脂蛋白包膜(含有3种不同的表面蛋白质(HBs))和内部核衣壳组成,该核衣壳的主要结构蛋白为核心(core)蛋白(HBcAg)。在核衣壳中是与病毒聚合酶蛋白(P)连接的单拷贝的HBV基因组。除了42-45nm的病毒体,感染HBV的患者的血液还含有20nm的球颗粒(sphere),其由HBsAg和宿主来源的脂质(由被感染细胞释放)组成。这些球颗粒数量超过病毒体的104-106倍。
HBV基因组为约3,200核苷酸的松散环状的部分双链DNA,由全长负链和较短的正链所组成。其含有4个重叠的开放读码框(ORF),C、S、P和X。C ORF编码了核心蛋白(或HBcAg),长183个氨基酸的蛋白质,其构成了HBV核衣壳,以及在病毒复制期间发现于患者血清中的第二种蛋白质,名为HBeAG,其包含前核心(precore)N末端的延伸和HBcAg的一部分。核心蛋白的C末端是非常碱性的并包含4个富含Arg的结构域,预测其可结合核酸和大量磷酸化位点(核心的磷酸化状态与衣壳颗粒的构象变化相关,如在Yu和Sommers,1994,J.Virol.68:29(55)中描述的)。S ORF编码3种表面蛋白质,它们都拥有相同的C末端,但在N末端是不同的,由于存在3个在同一读码框的ATG起始密码子,其将S ORF分为3个区域,分别为S(226个氨基酸)、前S2(55个氨基酸)和前S1(108个氨基酸)。大表面抗原蛋白(L)通过在第一个ATG起始密码子起始翻译而产生,并包含389个氨基酸残基(前S1-前S2-S)。中表面抗原蛋白质(M)由S区域和前S2区域翻译产生,起始于第二个起始ATG,而226个氨基酸的小表面抗原蛋白质(S,也叫做HBsAg)由S区域翻译产生,起始于第三个起始ATG密码子。HBV表面蛋白为糖蛋白,带有由N糖苷键连接的碳水化合物侧链(聚糖)。P ORF编码病毒聚合酶而X ORF包含称为X蛋白的蛋白质,其被认为是转录激活剂。
病毒体进入肝细胞后,通过尚未发现的受体,核衣壳将HBV基因组DNA转运至核,在其中松散环状DNA转换为共价闭合环状DNA(cccDNA)。该cccDNA起四种病毒RNA转录模板的功能,所述RNA输出至细胞质并用作翻译HBV蛋白质的mRNA。最长(前基因组)的RNA还起HBV复制模板的功能,其发生于细胞质内的核衣壳中。然后一些含HBV DNA和聚合酶的衣壳被转运回细胞核,在其中它们释放新生成的松散环状DNA以形成另外的cccDNA。具有比肝细胞更长半衰期的cccDNA负责HBV的持续性。其它衣壳通过出芽进入内质网而包膜,并在通过高尔基复合体后被分泌。
许多临床前和临床研究已强调了CD4+和CD8+T细胞免疫应答对有效抗病毒应答的重要性(Ferrari等人,1990,J Immul 145:3442;Penna等人,1996,J Clin Invest,98:1185;Penna等人,1997,Hepatology,25:1022)。也就是说,已天然地从乙型肝炎恢复的患者获得了由辅助性T(TH)和细胞毒性T淋巴细胞(CTL)介导的多特异性和持续的应答,其可容易地在外周血中检测到。识别病毒肽后,CTL获得了通过非细胞病变的、细胞因子介导的对HBV复制的抑制来治愈HBV感染的细胞和/或通过穿孔素-Fas配体和TNFα-介导的死亡通路杀死它们的能力。在分辨急性乙型肝炎和持续至临床痊愈后的I型T细胞应答期间,两种效应子功能都已被观察到。其经常伴随着血清丙氨酸氨基转移酶(ALT)水平升高和HBcAg特异的IgM和IgG的出现。抗HBe和抗HBs抗体较晚出现并表明感染的有利的结果。HBsAg特异性抗体是中和抗体,介导保护性免疫并在临床痊愈后终生保持。然而,慢性HBV感染极少被免疫系统解决。当这发生时,病毒的清理与提高的CTL活性和提高的ALT水平相关,其由免疫系统破坏感染的肝细胞所引起。然而,绝大多数慢性感染的患者显示了微弱的和暂时的CD4和CD8T细胞免疫应答,其对病毒感染的清除是抗原限制性的和无效的,尽管个别的HBV特异性T细胞克隆已从肝组织分离并扩增。此乙型肝炎细胞免疫应答效应子功能改变的原因目前是未知的。然而,已显示在一些患者中,当病毒载量低于106IU/mL的阈值时,功能性T细胞应答可被部分修复(Webster等人,2004,J.Virol.78:5707)。这些数据清楚地鼓励并强调了对能够诱导有效T细胞应答的免疫调节策略的需求。
理想的是,慢性乙型病毒性肝炎的治疗首先应允许在不可逆的肝损伤之前抑制HBV复制,从而消灭病毒,预防疾病发展为肝硬化或肝癌并改善患者存活率。慢性乙型肝炎的常规治疗包括PEG化的干扰素α(IFNα)和核苷/核苷酸类似物(NUCs)如拉米夫定,和最近的恩替卡韦、替比夫定、阿德福韦和替诺福韦(EASL Clinical Practice Guidelines:management of chronic hepatitis B,2009)。IFNα是强效的抗病毒分子,由此抑制病毒复制,然而其在25-30%的患者中引起严重的副作用。NUCs作为HBV聚合酶的竞争性抑制剂,旨在抑制前基因组RNA反转录成负链DNA然后为双链病毒DNA中。它们限制了新病毒体的形成,但对消灭隐藏于已感染肝细胞的细胞核中的超螺旋cccDNA是无效的,这些细胞构成了新子代病毒的来源。这可解释为什么NUC疗效是暂时的,并且在停止治疗后会立即发生病毒反弹,需要患者保持终生接受治疗。此外,由于耐受性HBV突变体的出现(一年后超过24%而拉米夫定治疗四年后大约66%,如Leung等人,2001,Hepatology 33:1527中所描述的),长期疗效也是受到限制的,尽管更新的NUC(恩替卡韦、替比夫定和替诺福韦)在提高HBV DNA抑制的同时显示出少得多的HBV耐药性突变的出现。然而,这些新药物的长期治疗数据是有限的,并且该更高的疗效尚未与显著更高的HBs血清转化相关联。
除了抗病毒治疗,目前正在努力开发旨在改善宿主免疫应答的(特别是那些由细胞毒性T淋巴细胞和辅助性T淋巴细胞所介导的)辅助治疗。大部分现有免疫治疗方法都重点在HBV表面蛋白质S、前S1和/或前S2的用途(Smith等人,1983,Nature 302:490;Lubeck等人,1989,Proc.Natl Acad.Sci.USA 86:6763;Adkins等人,1998,BioDrugs 10:137;Loirai等人,2000,J.Immunol.165:4748;Funuy-Ren et al.2003,J.Med.Virol,71:376;Kasaks等人,2004,J.Gen,Virol,85:2665;Xiangming Li等人,2005,Intern.Immunol.17:1293;Mancini-Bourguine等人,2006,Vaccine 24:4482;Vandepapeliere等人,2007,Vaccine 25:8585)。至少在刺激免疫应答方面取得了令人鼓舞的成果。例如,Mancini-Bourguine等人(2006,Vaccine,24:4482)报告了在HBV慢性感染患者中注射前S2-S-编码的DNA疫苗可诱导和/或唤起T细胞应答,其是免疫系统在这些患者中仍可操作的良好的指征。
HBcAg(Yi-Ping Xing等人,2005,World J,Gastro.11:4583)以及在其表面携带外来表位的嵌合HbcAg衣壳(WO92/11368;WO00/32625;Koletzki等人,1997,J.Gen,Virol.78:2049)还用作免疫原。从免疫原性的角度,插入表位最有前途的位置似乎是外环位点,其被预测位于HBcAg的表面位置80的附近(Argos等人,1988,FMBO J.7:819)。Schodel等人(1992,J.Virol.66:106)和Borisova等人(1993,J.Virol.67:3696)能够在此区域插入前S1和HBsAg表位,并且报告了用嵌合颗粒的成功的免疫。
也已研究旨在同时靶向多重HBV抗原的多价抗体候选物。值得注意的是,编码细胞毒性T淋巴细胞(CTL)和辅助T淋巴细胞(HTL)表位(存在于包膜、核心和聚合酶蛋白中)融合多肽的多表位DNA疫苗在临床前小鼠模型中显示出可诱导多重CTL和HTL应答(Depla等人,2008,J.Virol.82:435)。开发了基于编码HBsAg、HBcAg和HBV聚合酶的DNA质粒混合物的若干疫苗制剂(WO2005/056051;WO2008/020656),并在慢性乙型肝炎的转基因小鼠模型中显示了特异性抗HBV的细胞和体液应答(Chae Young Kim等人,2008,Exp.Mol.Medicine 40:669)。在乙肝病毒携带者中与拉米夫定治疗联合的I期临床试验已在韩国(South korea)开始(Yang等人,2006,Gene Ther.13:1110)。
因此,在有需要的个体如HBV慢性感染患者中,仍存在对可选的以更强力和有效的方式诱导免疫应答的免疫治疗方法的需要,特别是细胞介导的免疫应答。此外,存在提供能够以稳定和持续的方式表达HBV抗原的基于载体的组合物的需要。
通过提供在权利要求书中定义的实施方案,该技术难题已被解决。
通过下列本发明目前优选的实施方案,本发明其它和另外的方面、特点和优点将显而易见。给出这些实施方案是为了公开的目的。
发明概述
因此,第一方面,本发明提供了免疫原性组合物,其包含至少一种多肽或编码所述至少一种多肽的核酸分子,其中所述至少一种多肽选自:
i)聚合酶部分,其包含源自第一HBV病毒的聚合酶蛋白的至少450个氨基酸残基;
ii)核心部分,其包含源自第二HBV病毒的核心蛋白的至少100个氨基酸残基;和
iii)包膜(env)部分,其包含源自第三HBV病毒的HBsAg蛋白的一或多个15至100个连续氨基酸的残基免疫原性结构域;或
所述聚合酶部分、核心部分、包膜部分、编码所述聚合酶部分的所述核酸分子、编码所述核心部分的核酸分子和/或编码所述包膜部分的所述核酸分子的任意组合。
定义
如本文中所用,在整个申请中,术语“一(a)”和“一(an)”用于此种意义,即其意指“至少一”、“至少第一”、“一或多”或“多个”所指的化合物或步骤,除非上下文另有说明。例如,术语“细胞”包括多种细胞,包括其混合物。
本文中无论何处使用术语“和/或”包含“和”、“或”和“通过所述术语关连的元件的全部或任意其它组合”的意思。
如本文中所用,术语“约”或“近似”意指给出值或范围的10%之内,优选8%之内,并最优选5%之内。
如本文中所用,当用于定义产物、组合物和方法时,术语“包含(comprising)”(和任何包含的形式,如“包含(comprise)”和“包含(comprises)”),“具有(having)”(和任何具有的形式,如“具有(have)”和“具有(has)”),“包括(including)”(和包括的任何形式如“包括(include)”和“包括(includes)”)或“含有(containing)”(和含有的任何形式,如“含有(contain)”和“含有(contains)”)是开放式的,而不排除额外的、未记载的元件或方法步骤。“基本上由...组成”意指排除其它具任何实质意义的组合物或步骤。因此,基本上由所记载的组分组成的组合物将不排除痕量污染和药物学可接受的载体。“由...组成”意指排除多于痕量的其它组合物或步骤。例如,当多肽除了所记载的氨基酸序列之外不包含任何氨基酸时,该多肽“由该氨基酸序列组成”。当这样的氨基酸序列存在最终仅几个额外的氨基酸残基时,该多肽“基本由该氨基酸序列组成”。当氨基酸序列至少为多肽的最终氨基酸序列的一部分时,该多肽“包含”该氨基酸序列。这样的多肽可拥有一些多至几百个额外的氨基酸残基。
术语“氨基酸”、“残基”和“氨基酸残基”为同义词并涵盖天然氨基酸和氨基酸类似物(如,非天然的、合成的和修饰的氨基酸,包括D或L光学异构体)。
在本文中术语“多肽”、“肽”和“蛋白质”可互换地使用,是指氨基酸残基的聚合物,其包含九或更多个通过肽键结合的氨基酸。聚合物可为线性的、分支的或环状的,并可包含天然存在的和/或氨基酸类似物,并且其可被非氨基酸打断。作为一般的表示法,如果氨基酸聚合物很长(如,多于50个氨基酸残基),其优选称为多肽或蛋白质,而如果其为50个氨基酸或更少,其优选称为“肽”。
就本发明而言,术语“核酸”、“核酸分子”、“多核苷酸”和“核苷酸序列”可互换地使用,并定义了任何长度的多聚脱氧核糖核苷酸(DNA)(如,cDNA、基因组DNA、质粒、载体、病毒基因组、分离的DNA、探针、引物和其任意的混合物)或多聚核糖核苷酸(RNA)分子(如,mRNA、反义RNA)或混合的多聚核糖-多聚脱氧核糖核苷酸。其涵盖了单或双链、线性或环状、天然或合成的多核苷酸。此外,多核苷酸可包含非天然存在的核苷酸,如甲基化的核苷酸和核苷酸类似物(见US 5,525,711、US 4,711,955或EPA 302 175,作为修饰的实例)并且可被非核苷酸组分打断。如果存在,对核苷酸的修饰可在聚合之前或之后加入。
如本文中所用,术语“免疫原性组合物”是指包含以下描述的1、2、3、4种或更多组分(如,聚合酶部分、核心部分、包膜部分、编码聚合酶部分的核酸分子、编码核心部分的核酸分子和/或编码包膜部分的核酸分子)以及任选的其它组分(如,佐剂、载体、稀释剂等)的制剂。本发明的免疫原性组合物通常为这样的形式,即其能够向宿主生物体施用并诱导足够诱导或刺激抗HBV免疫力的保护性或治疗性免疫应答,获得治疗性的益处,如预防HBV感染、减少和/或减缓至少一种由HBV感染引起或与之相关的病症(如,降低病毒载量、减少或延缓肝脏病变如肝硬化或肝癌的风险、改善肝历史等),和/或减少血清HBeAg或HBsAg或HbeAg和HBsAg两者的水平,和/或诱导HBe血清转换、HBs血清转换或HBe血清转换和HBs血清转换,和/或提高另一种抗HBV治疗或预防的效果。引入宿主生物体后,本发明的免疫原性组合物可以引起免疫应答,包括但不限于,抗体和/或细胞因子的产生和/或细胞毒性T细胞、B、T淋巴细胞、抗原呈递细胞、辅助T细胞、树突状细胞、NK细胞的活化,导致产生针对至少一种HBV抗原/表位的固有免疫应答和/或特异性体液和/或细胞免疫应答。
“免疫原性结构域”是指能够被抗体或T细胞受体结合的HBV蛋白的结构部分。通常,这样的免疫原性结构域包含一或多个B和/或T表位,尤其CTL或TH表位或CTL和TH表位两者,并涉及特定的抗体或T细胞受体(就MHC(主要组织相容性复合体)来说)的识别。“表位”对应共同形成由抗体、T细胞受体或HLA分子识别的位点的最小的肽基序(通常一组9-11个氨基酸残基)。这些残基可为连续的(线性表位)或不连续的(包含彼此不直接相邻的残基的构象表位)。一般相信T细胞对T细胞表位的识别是通过这样的的机制,即其中T细胞识别抗原肽片段,所述抗原肽片段结合在抗原呈递细胞上表达的I类或II类MHC分子。
如本文中所用,“HBV”和“乙型肝炎病毒”可互换地使用,是指嗜肝DNA病毒科的任何成员(见如Ganem and Schneider in Hepadnaviridae(2001)“The viruses and their replication”(2923-2969页),Knipe DM等人,eds.Fields Virology,第4版或其后的版本,Philadelphia,Lippincott Williams和Wilkins)。大规模系统发育分析已将乙型肝炎病毒分为8个主要基因型(A至H),其显示出至少8%的序列差异。不同的HBV基因型显示出截然不同的地理分布并且可显露出多样的病症和/或临床表现(outcome)。不同的HBV被分成九个不同的亚型(aywl,ayw2,ayw3,ayw4,ayr,adw2,adw4,adrq+和adqr-),根据HBsAg-相关的血清学(见综述,Mamum-Al Mahtab等人,2008,Hepatobiliary Pancrease Dis Int 5:457;Schaeffer,2007,World Gastroenterol.7:14;Norder等人,1993,J.Gen Virol.74:1341)。每种基因型和血清型涵盖不同的HBV株系和分离株。分离株对应分离自特定HBV来源(如,患者样本或其它生物学HBV储源)的特定病毒,而株系涵盖不同的、就基因组序列而言彼此非常近似的分离株。
本领域已表述许多适用于本发明的HBV,尤其是在GenBank中。示例性的A基因型HBV包括但不限于分离株HB-JI444AF和株系HB-JI444A(登录号AP007263)。示例性的B基因型HBV包括但不限于克隆pJDW233(登录号D00329)、分离株HBV/14611(登录号AF121243)、由Hou等人,2001鉴定的HBV-B1(GenBank登录号AF282917.1)、由Zhang等人(2005,Arch.Virol.150,721-741)鉴定的HBV株系Whutj-37(GenBank登录号AY2933309.1)、由He等人鉴定的中国HBV株系GDH1(GenBank登录号AY766463.1)和由Jiang等人鉴定的HBV亚型adw的分离株57-1(GenBank登录号AY518556.1)。示例性的C基因型HBV包括但不限于分离株AH-1-ON980424(登录号AB113877)、由Fang等人鉴定的HBV分离株SWT3.3(GenBank登录号EU916241.1)、由Zhu等人鉴定的HBV分离株H85(GenBank登录号AY306136.1)、由Tu等人鉴定的HBV株系C1248(GenBank登录号DQ975272.1)、由Wang等人(2007,J.Viral Hepat 14,426-434)鉴定的HBV分离株CHN-H155(GenBank登录号DQ478901.1)和由Zhou等人鉴定的HBV分离株GZ28-1(GenBank登录号EF688062)。示例性的D基因型HBV包括但不限于分离株KAMCHATKA27(登录号AB188243)、ALTAY136(登录号AB188245)和Stoll-Becker等人(1997,J.Virol.71:5399)描述的并在GenBank登录号Y07587下可得到的Y07587,以及在登录号AB267090下描述的HBV分离株。示例性的E基因型HBV包括但不限于分离株HB-JI411F和株系HB-JI411(登录号AP007262)。典型的F基因型HBV包括但不限于分离株HBV-BL597(登录号AB214516)和HBV-BL592(登录号AB166850)。示例性的G基因型HBV包括但不限于分离株HB-JI444GF和株系HB-JI444G(登录号AP007264)。示例性的H基因型HBV包括但不限于分离株HBV ST0404(登录号AB298362)和分离株HB-JI260F和株系HB-JI260(登录号AP007261)。然而,本发明并不受限于这些示例性的HBV。事实上在不同的HBv分离株和基因型间核苷酸和氨基酸序列可变化,并且天然遗传差异和如下描述的非天然的修饰都包含在本发明的范围内。
如本文所用,“天然HBV蛋白”是指可发现、分离、获取自天然HBV来源的蛋白质、多肽或肽(如,聚合酶蛋白、核心蛋白或HBsAg等),其区别于那些在实验室中由人人工修饰或改变的那些。因此,该术语将包括天然存在的HBV蛋白、多肽或肽,除非另有规定。这样的天然来源包括收集自感染或暴露于HBV的对象的生物学样本(如,血液、血浆、血清、精液、唾液、组织切片、活检标本等)、培养的细胞(如,HepG2.2.15、HuH6-C15(Sureau等人,1986,Cell 47:37;Sells等人,1987,Proc.Natl.Acad.Sci.84(4):1005);HuH7.TA61或HuH7.TA62(Sun等人,2006,J Hepatol.45(5):636))、组织培养物和重组材料。重组材料包括但不限于HBV分离株(如,可于保藏机构获取)、HBV基因组、基因组RNA或cDNA文库、包含HBV基因组或其片段的质粒,或任何已知包含这样的元件的现有技术载体。
编码不同HBV蛋白的核苷酸序列可发现于特定的数据库(如,上述提及的)和文献(见如,Valenzuela等人,1980,The nucleotide sequence of the hepatitis B viral genome and the identification of the major viral genes(57-70页),“Animal Virus Genetics”;编辑B.Fields等人;Academic Press Inc.,New York和Vaudin等人,1988,J.Gen.Virol.69:1383)。天然聚合酶、核心和HBsAg多肽的代表性的例子分别在示于SEQ ID NO:1-3(SEQ ID NO:1提供HBV分离株Y07587的天然聚合酶蛋白氨基酸序列,SEQ ID NO:2提供HBV分离株Y07587的天然核心蛋白氨基酸序列而SEQ ID NO:3提供HBV分离株Y07587的天然包膜(HBsAg)氨基酸序列)。为了说明性的目的,编码所述Y07587天然HBV聚合酶、核心和HBsAg的核苷酸分别示于SEQ ID NO:4、5和6。然而,如上述所讨论,HBV蛋白不限于这些示范性的序列,并且遗传差异也包含在本发明的范围内。
如本文所用,术语“部分”(如,聚合酶、核心和/或包膜部分)是指源自天然HBV蛋白、多肽或肽,其如本文所描述由实验室人员人工修饰或改变的蛋白质、多肽或肽。术语“修饰”涵盖缺失、取代或添加一或多个核苷酸/氨基酸残基、这些可能的任意组合(如,简并天然核苷酸序列以降低由本发明组合物编码的HBV序列间的同源性、引入合适的限制性酶切位点)和非天然排列(如,融合两种或多种HBV蛋白、多肽或肽或部分)。当预期若干种修饰时,可考虑连续的残基和/或不连续的残基。可通过大量本领域技术人员已知的方法生成修饰,如定点诱变(如,使用Amersham的SculptorTM体外诱变系统,Les Ullis,France)、PCR诱变、DNA改组和化学合成技术(如,得到合成的核酸分子)。本发明预期的修饰涵盖不改变所编码的HBV多肽氨基酸序列的沉默修饰,和翻译成所编码多肽后导致其与对应的天然多肽相比有修饰的氨基酸序列的修饰。术语“源自”(或源自)用于确认分子的来源,但不意味着限制制造该分子的方法,所述方法可为,例如,化学合成或重组手段。
优选地,本发明使用的每种HBV部分都与对应的天然HBV蛋白在全长蛋白质或其部分上保持高度的氨基酸序列相同性。两种多肽间的相同性百分比是所述序列共有的相同位置的数目的函数,其考虑缺口(需要被引入以得到最优的比对)的数目和每个缺口的长度。在本领域可得到多种计算机程序和数学算法以确定氨基酸序列间相同性的百分比,例如在NCBI可得到的Blast程序(如,Altschul等人,1997,Nucleic Acid Res.25:3389;Altschul等人,2005,FEBS J.272:5101)。同样可应用于核苷酸序列。用于确定核苷酸序列同源性的程序也可得自专门的数据库(Genebank或Wisconsin Sequence Analysis Package)中,例如BESTFIT、FASTA和GAP程序。
例如,除了以下描述的修饰(如,降低的酶活性等)外,任何或所有包含本发明组合物或由本发明组合物编码的HBV部分可被修饰以对特定的基因型是具代表性的,并因此包含对应于共有或接近共有序列的氨基酸序列,所述共有序列或接近共有序列通常在对特定基因型的不同HBV多肽序列比对之后确定。
如本文所用,术语“组合”是指至少两种包含本发明的免疫原性组合物或由本发明的免疫原性组合物编码的组分之间的任意类型的组合,以及多种组分(特别优选2或3种所述组分)任意可能的排列。这涵盖了两或多种多肽的混合物、两或多种核酸分子/载体的混合物、一或多种多肽混合物和一或多种核酸分子/载体,以及以提供具有两或多种HBV部分(如,非天然排列)的单一多肽链的两或多种核酸分子的融合。
在优选的实施方案中,本发明的免疫原性组合物包含所述聚合酶部分、核心部分、包膜部分中至少两种的组合,或包含编码所述聚合酶部分的所述核酸分子、编码所述核心部分的所述核酸分子和/或编码所述包膜部分的所述核酸分子中至少两种的组合。本发明特别优选的组合物选自:(i)组合物,其包含本文中定义的聚合酶部分和核心部分的组合,或包含编码所述聚合酶部分和所述核心部分的核酸分子的组合;(ii)组合物,其包含本文中定义的核心部分和包膜部分的组合,或包含编码所述核心部分和所述包膜部分的核酸分子的组合;和(iii)组合物,其包含本文中定义的聚合酶部分、核心部分和包膜部分的组合,或包含编码所述聚合酶部分、所述核心部分和所述包膜部分的核酸分子的组合。
如本文所用,术语“融合”或“融合蛋白”是指至少两种多肽(或其片段)相互组合于单一的多肽链。优选地,不同多肽间的融合通过遗传手段实行,如通过符合读码框地融合编码每种所述多肽的核苷酸序列。“符合读码框地融合”意指融合的编码序列的表达导致单一蛋白质,在每个融合的多肽之间没有任何翻译终止子。融合可以是直接的(如,在中间不含有任何额外的氨基酸残基)或通过接头。接头的存在可能有利于融合蛋白正确形成、折叠和/或发挥功能。本发明不受所采用的接头序列的形式、大小或数目限制,并且多拷贝的接头序列可插入至融合多肽间的连接处。根据本发明,合适的接头为长3至30个氨基酸并由如甘氨酸、丝氨酸、苏氨酸、天冬氨酸、丙氨酸和/或脯氨酸的氨基酸残基重复组成(见例如Wiederrecht等人,1988,Cell 54 841;Aumailly等人,1990,FEBS Lett.262,82;和Dekker等人,1993,Nature 362,852),如Ser-Gly-Ser或Gly-Ser-Gly-Ser-Gly接头。
如本文中所用,术语“异源疏水性序列”是指疏水性质的肽(包含高数目的疏水性氨基酸残基如Val、Leu、Ile、Met、Tyr和Trp残基)。“异源”是指对选定的HBV部分所源自的天然HBV蛋白、多肽或肽来说为外来的序列。其可为对HBV病毒为外来的肽(如,来自麻疹或狂犬病毒的肽),或为来自HBV病毒的肽但其位于在病毒基因组中通常不发现该肽的位置。异源疏水序列能符合读码框地融合于HBV部分的N末端、于C末端或内部,并可在多肽运输、促进多肽产生或纯化、延长半衰期和其它过程中起作用。根据本发明,合适的异源疏水序列长15至100个氨基酸并含有高度疏水性结构域。
如本文所用,术语“载体”是指表达载体和非表达载体,并包括病毒和非病毒载体,其包括染色体外载体(如,多拷贝质粒)和整合载体(其设计为可并入宿主染色体)。就本发明来说特别重要的是用于将核酸分子转移进病毒基因组的载体(所谓的转移载体)、用于免疫治疗的载体(如,能够将核酸分子递送至宿主生物体的载体)和在多种表达系统或在宿主生物体中使用的表达载体。
如本文所用,术语“病毒载体”涵盖载体DNA和其产生的病毒颗粒。病毒载体可为可复制的或可被遗传失活(disabled)而使得其为复制缺陷或复制受损的。如本文所用,术语“可复制的”涵盖选择性复制和条件型复制的病毒载体,其被工程化以更好地或选择性地在特定宿主细胞(如,肿瘤细胞)中复制。
如本文所用,术语“调控序列”是指在给定的宿主细胞或生物体中的任何允许、有助于或调节核酸分子表达的序列,其包括复制、重复、转录、剪切、翻译、稳定性和/或将所述核酸或其衍生物(mRNA)的一种运输进宿主细胞或生物体中。
如本文中所用,术语“宿主细胞”应广义地,没有任何限制地理解为考虑组织、器官或分离的细胞中的特定有机体。这样的细胞可能为独特类型的细胞或一组不同类型的细胞,并涵盖细菌、低等和高等真核细胞,以及培养的细胞系、原代细胞和增殖细胞。本术语包括可接受或已接受本发明的组合物、核酸分子、载体或感染性病毒颗粒的细胞以及这样的细胞的后代。
术语“宿主生物体”是指脊椎动物,特别是哺乳动物物种的成员,尤其是家养动物、农场动物、运动动物以及包括人类的灵长类动物。优选地,宿主生物体为患有慢性HBV感染的患者。感染的HBV可来自同样的基因型或血清型,如本发明使用的第一、第二或第三HBV中的至少一种。
如本文所用,术语“分离的”是指从其天然环境中分离出的蛋白质、多肽、肽、核酸分子、宿主细胞或病毒(即,从至少一种其天然伴随的其它组分中分离)。
如本文所用,“治疗有效量”是足够减轻一或多种通常与HBV感染相关的症状或任何由HBV感染引起或与之相关的疾病或病症的剂量。当考虑预防性的用途时,本术语意指足够预防或延缓HBV感染确立的剂量。“治疗性”组合物被设计并施用至已被HBV感染的宿主生物体,其目的为减轻或改善至少一种由HBV感染引起或与之相关的疾病或病症,最终结合一或多种本文中所描述的常规治疗形式(如,用核苷或核苷酸类似物治疗)。例如,用于诱导免疫应答的治疗有效量可为引起免疫系统激活(如,导致抗HBV应答发展)的必须量。术语“癌症”涵盖任何癌性的病症,包括弥散性或局限性的肿瘤、转移、癌性息肉以及前癌病变(如肝硬化)。
如本文所用,“药物学可接受的载体”旨在包括任何和所有的载体、溶剂、稀释剂、赋形剂、佐剂、分散介质、包衣、抗细菌剂和抗真菌剂、和吸收延缓剂等等,其与药物学施用兼容。
就本发明来说,本发明的组合物包含或编码的所述聚合酶、核心和/或包膜部分可独立地源自目前鉴定的任何HBV基因型、株系或分离株,如上述术语“HBV”中描述的那些。此外,每种聚合酶、核心和包膜部分可源自天然的相应HBV蛋白质或源自修饰的HBV多肽(如,被修饰以对特定基因型是代表性的)。因此,本发明的组合物包含或编码的聚合酶、核心和包膜部分所源自的第一、第二和第三HBV病毒可独立地来自相同的或不同的基因型、血清型和/或分离株。例如,使用源自两或三种不同HBV基因型的HBV部分允许提供针对更宽范围的HBV基因型的保护。
在一个实施方案中,使本发明的免疫原性组合物适应于特定地理区域(通过使用至少一种来自流行于该区域的HBV基因型的HBV部分)是有意义的。为了说明性的目的,基因型A和C是在美国最流行的,而来自欧洲西部国家的患者大部分被基因型A和D感染,而地中海盆地的患者主要被基因型D感染。来自印度的有限的数据表明,在印度基因型A和D是最普遍的。另一方面,在中国基因型B和C是最流行的。例如,以欧洲国家为目的地的本发明的组合物可包含源自基因型A的聚合酶部分和源自基因型D的包膜部分,或反过来。另外,聚合酶和核心部分可来自基因型A而包膜部分来自基因型D。作为另一实例,以欧洲国家和美国为目的地的本发明的组合物可包含独立地源自基因型A、C和D的聚合酶、核心和包膜部分。另一方面,以中国为目的地的本发明的组合物可包含或编码独立地源自基因型B和/或C的聚合酶、核心和包膜部分。
也可以使本发明的免疫原性组合物适应待治疗的患者群体。例如,基因型A在美国白人、非裔美国人和性生活获得HBV感染的人群中更普遍,而另一方面,基因型B和C在亚裔美国人、生于亚洲的患者和母婴传播的HBV感染人群中更普遍。HBV基因型还与不同的临床表现相关(Schaeffer等人,2005,J.Viral.Hepatitis 12:111),相比于基因型A,基因型D和F与更严重的疾病进展和更差的预后相关(Sanchez-Tapias等人,2002,Gastroenterology 123:1848)。通过选择合适的HBV基因型、血清型、株系和/或分离株,根据待治疗的人群和/或地理区域修改本发明的组合物是技术人员可达成的。
根据有利的实施方案,第一、第二和第三HBV病毒中的至少两种,优选全部,来自同样的基因型,尤其是来自基因型D。独立地,它们可源自同样的分离株,特别优选第一、第二和第三HBV病毒来自HBV分离株Y07587。优选地,本发明使用的聚合酶部分包含氨基酸序列,其显示与SEQ ID NO:1所示氨基酸序列或其包含至少450个氨基酸残基的部分具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。可选地或组合地,本发明使用的核心部分包含氨基酸序列,其显示与SEQ ID NO:2所示氨基酸序列或其包含至少100个氨基酸残基的部分具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。可选地或组合地,本发明使用的包膜部分的一或多个免疫原性结构域包含氨基酸序列,其显示与SEQ ID NO:3所示氨基酸序列内的15-100个氨基酸残基的部分具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。
聚合酶部分
根据一个实施方案,本发明的组合物包含或编码的聚合酶部分与相应的天然HBV聚合酶相比为修饰的。
合适的修饰是截短至少20个以及至多335个通常存在于天然HBV聚合酶N末端的氨基酸残基。该修饰对本发明的还含有第二多肽的组合物特别重要,以减少或缺失聚合酶和核心部分间的重叠部分。技术人员有能力在所记载的最少20个氨基酸残基以及最多335个氨基酸残基的范围内修改本发明的组合物的截短。关于天然HBV聚合酶,在本发明上下文所用的聚合酶部分有利地被截短至少30个氨基酸残基以及至多200个氨基酸残基,理想地至少35个氨基酸残基以及至多100个氨基酸残基,优选至少40个氨基酸残基以及最多60个氨基酸残基,并且更优选至少45个氨基酸残基以及至多50个氨基酸残基,特别优选包含位于天然HBV聚合酶N末端的在起始甲硫氨酸残基后的前47或46个氨基酸残基的截短。优选地,截短从SEQ ID NO:1的1(甲硫氨酸起始子)或2位延展至47位。
优选的实施方案涉及聚合酶部分,其包含氨基酸序列,所述氨基酸序列显示与SEQ ID NO:1所示氨基酸序的从大约48位至大约832位的部分具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性;并甚至更优选涉及聚合酶部分,其包含氨基酸序列,所述氨基酸序列显示与SEQ ID NO:7中所示氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。
可选地或组合地,本发明所用的聚合酶部分被修饰从而显示相对于天然HBV聚合酶的降低的反转录酶(RTase)活性。有利的是,所述RTase活性的降低通过在负责RTase酶活性的结构域的一或多个突变来提供。
HBV聚合酶的结构和功能组织在几乎20年前已被研究(见例如Radziwill等人,1990,J.Virol.64:613)。它们为多重功能的蛋白质,具有3个催化HBV复制的主要步骤(引导(priming)、DNA合成和去除RNA模板)的功能性结构域和按如下顺序排列的非必须的间隔物:
o从1位延展至约177位的第一结构域负责HBV末端蛋白活性,
o间隔物位于从约178位至约335位,
o从约336位延展至约679位的DNA聚合酶结构域负责RTase活性,和
o从约680位至C末端(约832位)的RNase H结构域涉及RNaseH活性。
四个残基涉及RTase活性,其形成了通常存在于天然HBV聚合酶的大约538位至大约541位(如,对应于SEQ ID NO:1的第538、539、540和541位,和SEQ ID NO:7的492、493、494和495位)的“YMDD”(代表Tyr、Met、Asp和Asp残基)基序,并且本发明涵盖了该基序中或所述RTase结构域中其余地方的任何突变,所述突变在保持免疫原性特性的同时与显著降低(即至少10倍的降低)或消除的RTase活性相关。合适的RTase缺陷聚合酶突变体的代表性实例在参考文献中描述,例如Radziwill等人(1990,J.Virol.64:613)中、Bartenschlager等人(1990,J.Virol.64:5324)中和Jeong等人(1996,Biochem.Bioph.Res.Commun.223(2):264)中。优选地,本发明所用的聚合酶部分包含在YMDD基序(对应SEQ ID NO:1的位点540和SEQ ID NO:7的位点494)第一个Asp残基或位于天然HBV聚合酶等效位点的氨基酸残基被除Asp之外的任意残基取代,特别优选被His残基取代(D540H突变)。RTase活性的降低或消除可使用本领域众所周知的测定来进行(如,在Radziwill等人,1990,J.Virol.64:613中描述的内源聚合酶测定)。
可选地或组合地,本发明所用的聚合酶部分被修饰从而显示相对于天然HBV聚合酶的降低的RNase H活性。有利的是,所述RNase H活性的降低通过在负责RNase H活性的结构域的一或多个突变来提供。如上述讨论,涉及RNase H活性的功能性结构域已被定位在HBV聚合酶的C末端部分,更具体的从680位到C末端832位,并且本发明涵盖在此结构域中与RNase H活性的显著降低(即至少降低10倍)或消除有关并且不损害免疫原性特性的任何突变。合适的RNase H缺陷的聚合酶的代表性的实例在参考文献中被描述,如Radziwill等人(1990,J.Virol.64:613)中,Bartenschlager等人(1990,J.Virol.64:5324)中。优选地,本发明所用的聚合酶部分包含在对应于SEQ ID NO:1的718位和SEQ ID NO:7的672位的Glu残基或位于天然HBV聚合酶等效位点的氨基酸残基被除Glu之外的任意残基取代,特别优选被His残基取代(E718H突变)。RNase H活性的降低或消除可使用本领域众所周知的测定来进行(如,在Radziwill等人,1990,J.Virol.64:613中或Lee等人,1997,Biochem.Bioph.Res.Commun.233(2):401中描述的体外RNase H活性测定或DNA-RNA串联分子测定)。
优选地,本发明所用的聚合酶部分被突变以降低或消除RTase和RNase活性,并包含上文讨论的与这些酶功能有关的修饰,特别优选D540H和E718H突变。
本发明优选的实施方案涉及聚合酶部分,其包含这样的氨基酸序列(或者基本上由,或者由其组成),所述氨基酸序列显示与SEQ ID NO:8所示氨基酸序列或与在494位的Asp残基被His残基取代和672位的Glu残基被His残基取代的SEQ ID NO:7所示氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。
在另一个优选的实施方案中,本发明所用的聚合酶部分符合读码框地与异源疏水性序列相融合以改善合成、和/或稳定性、和/或在表达宿主细胞表面的呈递和/或呈递至宿主的I类MHC和/或II类MHC抗原。合适的异源疏水序列包括如信号肽和/或跨膜肽序列,其允许将聚合酶部分靶向于分泌通路中。这样的肽在本领域是众所周知的。简而言之,信号肽通常存在于存在于膜的或分泌的多肽的N末端并且起始其进入内质网(ER)。它们包含15至35个基本上疏水的氨基酸,其随后被特定的ER定位的内肽酶切除以获得成熟多肽。跨膜肽通常为高度疏水性质的并用于将多肽锚定在细胞膜上(见例如Branden和Tooze,1991,Introduction to Protein Structure 202-214页,NY Garland;WO99/03885)。本发明上下文可用的的跨膜肽和/或信号肽有大量的选择。它们可以从任何膜锚定的和/或分泌多肽中获取(如,细胞或病毒多肽),如免疫球蛋白、组织纤溶酶原(plasminigen)激活子、胰岛素、狂犬病糖蛋白、HIV病毒包膜糖蛋白或麻疹病毒F蛋白,或可是合成的。优选的插入信号肽的位点为N末端翻译起始密码子的下游,而插入跨膜肽的位点为C末端,例如,终止密码子的直接上游。此外,可用接头肽使信号肽和/或跨膜肽与聚合酶部分连接。
在本发明中,可采用其它的疏水序列,如通常存在于包膜或膜结合蛋白包括HBsAg中的那些。就本发明而言特别感兴趣的是将聚合酶部分与本文描述的一或多个具疏水性质的免疫原性结构域(env1、env2、env3和/或env4)相融合。所述一或多个疏水性结构域可符合读码框地融合于聚合酶部分的N末端、C末端或内部。
优选地,就本发明而言,本发明所用的聚合酶部分符合读码框地与狂犬病毒糖蛋白的信号肽和跨膜肽融合。如所附实施例部分表明,所述狂犬病毒信号序列符合读码框地融合于所述聚合酶部分的N末端,并且所述狂犬病毒跨膜序列符合读码框地融合于其C末端。最优选的实施方案涉及聚合酶部分,其包含这样的氨基酸序列(或者基本上由,或者由其组成),所述氨基酸序列显示与SEQ ID NO:9所示氨基酸序具有至少80%的相同性,有利地至少85%的相同性,尤其至少90%的相同性,优选至少95%的相同性并更优选100%的相同性。
核心部分
可选地或结合上文描述的组合物,本发明还提供了包含源自第二HBV的核心部分的组合物。如本文有关HBV病毒的描述,本发明所用的核心部分源自与聚合酶部分所源自的HBV病毒相同的或不同的HBV。优选地,核心和聚合酶部分都源自D基因型HBV,特别优选Y07587HBV分离株,或可选地源自在中国流行的HBV基因型(如,基因型B或C)。
根据一个实施方案,本发明所用的核心部分可为天然HBV核心(如,SEQ ID NO:2所示的)或与相应的天然HBV核心相比修饰的核心,所述修饰的核心保留核心蛋白的至少100个氨基酸残基,特别优选包含120至180个氨基酸残基、理想地125至148氨基残基并优选130至143氨基酸残基(如,大约140氨基酸残基)的核心部分。
合适的修饰是截短。理想地,截短涵盖至少10个以及至多40个通常存在于天然HBV核心C末端或在C末端部分(即,涵盖最后40个氨基酸残基的部分)内的氨基酸残基。该修饰对还含有聚合酶部分的本发明的组合物特别重要,以减少或缺失聚合酶和核心部分间的重叠部分。其对在核心的该区域缺失NLS(核定位信号)和/或抑制其与HBV聚合酶的相互作用也是重要的。技术人员有能力在所记载的最少10个氨基酸残基以及最多40个氨基酸残基的范围内修改本发明的组合物的截短。合适的截短包括10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39或40个通常存在于天然HBV核心C末端或在C末端部分内的连续的氨基酸残基。关于天然HBV核心,本发明所用的核心部分有利地被截短位于天然HBV的C末端或在C末端部分内的至少20个氨基酸残基以及至多40个氨基酸残基,优选至少30个氨基酸残基以及至多38个氨基酸残基,更优选至少34个氨基酸残基以及至多37个氨基酸残基,特别优选包含天然HBV核心最后35个氨基酸残基的截短(换句话说,截短从核心多肽的大约149位延伸至其C末端)。
尤其感兴趣的是核心部分,其包含氨基酸序列,所述氨基酸序列显示与SEQ ID NO:2所示氨基酸序的从1位至大约148位的部分具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性;并甚至更优选涉及核心部分,其包含氨基酸序列,所述氨基酸序列显示与SEQ ID NO:10中所示氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。
可选地或组合地,本发明所用的核心部分被修饰以显示相对于天然HBV核心的降低的对HBV包膜蛋白质的识别和/或相互作用。有利的是,所述识别/相互作用的降低通过在内部定位于残基80附近的区域中的一或多个突变提供,该突变被预测形成暴露于核心颗粒表面的外部环(Argos等人,1988,EMBO J.7:819)。对包膜蛋白的识别或相互作用的降低或消除可使用本领域众所周知的测定来进行(如电子显微镜、分析细胞中核衣壳形成、在HuH7中瞬时转染后的分泌病毒体,如Seitz等人,2007,EMBO J.,26:416或Ponsel等人,2003,J.Virol.77(1):416中所描述的)。
优选的修饰包括在核心的大约75位延伸至大约85位的区域(对应于SEQ ID NO:2和SEQ ID NO:10的残基75-85)缺失一或多个氨基酸残基,特别优选缺失核心部分从大约77位延伸至大约84位的部分(对应于SEQ ID NO:2和SEQ ID NO:10的残基77-84)。优选的核心部分包含这样的氨基酸序列(或者基本上由,或者由其组成),所述氨基酸序列显示与SEQ ID NO:11所示氨基酸序列或SEQ ID NO:2所示(缺失残基77-84)氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。
包膜部分
可选地或结合上文描述的组合物,本发明还提供了包含包膜部分的组合物。所述包膜部分包含一或多个存在于源自第三HBV病毒的HBs蛋白质中的至少15个连续氨基酸残基的免疫原性结构域。优选地,每个所述免疫原性结构域对应于天然的或修饰的(如,被修饰以对特定基因型是具代表性的)HBsAg蛋白质中的至少20个氨基酸以及至多100个氨基酸的部分。每个免疫原性结构域可源自相同的或不同的HBV病毒,其相对于核心和聚合酶部分所源自的HBV病毒可为相同的或不同的。优选地,每个包膜免疫原性结构域源自D基因型HBV,并特别优选Y07587HBV分离株,或可选地,源自在中国流行的HBV基因型(如,基因型B或C)。
有利的是,一或多个免疫原性结构域的每个都包含对限制于多种I类和/或II类MHC抗原(如,A2、A24、DR、DP等)的T辅助(TH)细胞和/或细胞毒性T(CTL)细胞特异的T细胞表位。这样的表位在本领域已被描述(WO93/03764;WO94/19011;Desombere等人,2000,Clin,Exp.Immunol.122:390;Loirat等人,2000,J.Immunol.165:4748;Schirmbeck等人,2002,J.Immunol.168:6253;Depla等人,2008,J.Virol.82:435),并且本领域技术人员有能力设计本文描述的包含来自天然包膜蛋白质(如,SEQ ID NO:3所示的)的B、TH和/或CTL表位的在所记载的15至100个氨基酸残基的范围内的合适的免疫原性结构域。优选地,本发明的组合物包含或编码的包膜部分不包含任何源自前S1和前S2区域的免疫原性结构域。
本发明涵盖了包含一个免疫原性结构域和包含两个、三个或更多免疫原性结构域的包膜部分。
理想地,本发明所用的一或多个免疫原性结构域选自:
o包膜蛋白质(HBsAg)14-51位的部分(env1结构域);
o包膜蛋白质(HBsAg)165-194位的部分(env2结构域);
o包膜蛋白质(HBsAg)81-106位的部分(env3结构域);
o包膜蛋白质(HBsAg)202-226位的部分(env4结构域);和
o其任何组合。
用于本发明的特别合适的免疫原性结构域包含这样的氨基酸序列(或者基本上由、或者由其组成),其显示与SEQ ID NO:12-15所示任一氨基酸序具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,以及甚至更优选100%的相同性。
就本发明来说,免疫原性结构域的组合可为本发明组合物中的单独的免疫原性结构域的混合物的形式或为符合读码框地按任何可能的排列的两或多个免疫原性结构域的融合物形式(如,env1-env2、env2-env1、env1-env3、env3-env1、env1-env4、env4-env1、env2-env-3、env3-env2、env2-env4、env4-env2、env3-env4、env4-env3、env1-env2-env3、env2-env1-env3、env1-env2-env4等的融合物或混合物)。此外,所述组合可包含单个拷贝或其若干拷贝(如,env1-env2-env1、env1-env4-env1等)。每个免疫原性结构域间的融合可为直接的或通过接头。
就本发明来说,尤其感兴趣的包膜部分包含SEQ ID NO:12-15中所示的两或三个免疫原性结构域的融合物,特别优选env1-env2融合物,其包含这样的氨基序列(或者基本上由,或者由其组成),所述氨基酸序列显示与SEQ ID NO:16所示氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性;或env1-env2-env4融合物,其包含这样的氨基酸序列(或者基本上由,或者由其组成),所述氨基酸序列显示与SEQ ID NO:17中所示氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,甚至更优选100%的相同性。
根据特定的实施方案,本发明的组合物包含或编码的聚合酶、核心和/或包膜部分可成对或全部一起符合读码框地融合。例如,可设想聚合酶和包膜部分在单一多肽链中的融合。另外,核心和包膜部分可符合读码框地融合于单一多肽链中。此外,编码核酸序列也可直接地或通过接头融合。有利的是,包膜部分符合读码框地融合于核心或聚合酶部分的C末端。
核心部分与包膜部分的融合多肽的优选的实例选自:
o多肽,其包含这样的氨基酸序列(或者基本上由、或者由其组成),所述氨基酸序列显示与SEQ ID NO:18(core*t-env1)所示氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,以及甚至更优选100%的相同性;
o多肽,其包含这样的氨基酸序列(或者基本上由、或者由其组成),所述氨基酸序列显示与SEQ ID NO:19(core*t-env1-env2)所示氨基酸序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,以及甚至更优选100%的相同性;和
o多肽,其包含这样的氨基酸序列(或者基本上由、或者由其组成),所述氨基酸序列显示与以下序列具有至少80%的相同性,有利地至少85%的相同性,优选至少90%的相同性,更优选至少95%的相同性,并甚至更优选100%的相同性:SEQ ID NO:20(core*t-env1-env2-env4)所示氨基酸序列或与SEQ ID NO:20所示氨基酸序列从残基1开始到残基251终止(core-env1-env2)的部分,或与SEQ ID NO:20所示氨基酸序列从残基1开始到残基221终止(core-env1)的部分;或它们的缺失版本,所述缺失版本缺少核心部分中的残基77-84。
就本发明而言,本发明的组合物包含或编码的聚合酶部分、核心部分和/或包膜部分还可包含额外的修饰。合适的修饰是那些有利于所得多肽的合成、加工、稳定性和可溶性的修饰(如,旨在如本文所述修饰潜在的裂解位点、潜在的糖基化位点和/或膜锚定)和那些有利于所得组合物的免疫原性的修饰(如并入或融合一或多个能够增强免疫原性特性的化合物)。这样的能增强免疫原性特性的化合物已在文献中描述,并包括但不限于钙网蛋白(Cheng等人,2001,J.Clin.Invest.108:669),结核杆菌(Mycobacterium tuberculosis)热休克蛋白70(HSP70)(Chen等人,2000,Cancer Res.60:1035),泛素(Rodriguez等人,1997,J.Virol.71:8497),细菌毒素如绿脓杆菌(Pseudomonas aeruginosa)外毒素A(ETA(dIII))的易位结构域(Hung等人,2001,Cancer Res.61:3698),以及T辅助细胞表位如泛Dr肽(Sidney等人,1994,Immunity 1:751)、pstS1GCG表位(Vordermeier等人,1992,Eur.J.Immunol.22:2631)、破伤风类毒素肽P2TT(Panina-Bordignon等人,1989,Eur.J.Immunol.19:2237)和P30TT(Demotz等人,1993,Eur,J,Immunol.23:425),流感表位(Lamb等人,1982,Nature 300:66)和血凝素表位(Rothbard等人,1989,Int.Immunol.1:479)。
核酸分子
本发明还提供了分离的核酸分子以及包含这样的核酸分子的组合物,所述核酸分子独立地或组合地编码本发明所用的聚合酶、核心和包膜部分。
尤其感兴趣的是:
o编码如本文描述的聚合酶部分的核酸分子,特别优选那些包含SEQ ID NO:7、8、9中所示任一氨基酸序列的聚合酶部分,或包含在494位的Asp残基被His残基取代以及在672位的Glu残基被His残基取代的SEQ ID NO:7所示的氨基酸序列的聚合酶部分;
o编码本文中描述的核心部分的核酸分子,特别优选那些包含SEQ ID NO:10或11中所示氨基酸序列的核心部分;
o编码本文中描述的包膜部分的核酸分子,特别优选那些包含SEQ ID NO:12-17中所示任一氨基酸序列的包膜部分;和
o编码本文中描述的融合的核心和包膜部分的核酸分子,特别优选的那些核心和包膜部分包含:SEQ ID NO:20中所示氨基酸序列或SEQ ID NO:20所示氨基酸序列从残基1开始到残基251终止(core-env1-env2)的部分或SEQ ID NO:20所示氨基酸序列从残基1开始到残基221终止(core-env1)的部分;或它们的缺失版本,所述缺失版本缺少核心部分的残基77-84。
理想地,可最优化本发明的核酸分子以在特定的宿主细胞或生物体,如哺乳动物、酵母(如,酿酒酵母(Saccharomyces cerevisiae)、裂殖酵母(Saccharomyces pombe)或毕赤酵母(Pichia pastoris))或细菌(如,大肠杆菌、枯草杆菌(Bacillus subtilis)或李斯特菌(Listeria))中提供高水平的表达。实际上已观察到,当超过一种密码子可用于编码给定氨基酸时,生物体密码子利用模式是高度非随机的(见例如Wada等人,1992,Nucleic Acids Res.20:2111),并且密码子的利用在不同宿主之间可能是显著不同的(见例如Nakamura等人,1996,Nucleic Acids Res.24:214)。由于本发明所用的核苷酸序列大多数为病毒(HBV)来源的,它们可能具有对在宿主细胞如细菌、低等或高等真核细胞中有效表达来说不合适的密码子利用模式。一般地,通过将一或多个对应于不常用于感兴趣的宿主细胞中的“天然”(如HBV)密码子替换为一或多个更常用的编码同样氨基酸的密码子来实施密码子优化。将所有的对应于不常用密码子的天然密码子都替换掉是没有必要的,因为仅通过部分替换就可获得提高的表达。此外,可作出对严格遵循优化密码子利用的一些偏差,以容纳限制性酶切位点引入至所得核酸分子。
除了密码子使用的优化,在宿主细胞或生物体中的表达可进一步通过对核苷酸序列额外的修饰改善。例如,本发明的核酸分子可被修饰以预防罕见、非优化密码子聚类出现在集中区域和/或被修饰以抑制或修饰至少部分预计将负面影响表达水平的阴性序列元件。这样的阴性序列元件包括但不限于具有非常高(>80%)或非常低(<30%)GC含量;AT富集或GC富集序列区段;不稳定的同向或反向重复序列;RNA二级结构;和/或内部隐藏调控元件如内部TATA盒、chi位点、核糖体进入位点、和/或剪切供体/受体位点。本发明另一个实施方案涉及本发明核酸分子的片段,如限制性内切酶和PCR产生的片段。这样的片段可用作探针、引物或编码第一和/或第二多肽免疫原性部分的片段。
本发明优选的核酸分子选自:
o核酸分子,其包含显示与SEQ ID NO:21所示核苷酸序列(编码SEQ ID NO:7截短的聚合酶)具有至少80%相同性的核苷酸序列;
o核酸分子,其包含显示与SEQ ID NO:22所示核苷酸序列(编码SEQ ID NO:8的突变的聚合酶)具有至少80%相同性的核苷酸序列;
o核酸分子,其包含显示与在1480位的G核苷酸被C取代,2014位的G核苷酸被C取代以及在2016位的A核苷酸被T取代的SEQ ID NO:21所示核苷酸序列(编码SEQ ID NO:7截短的聚合酶,其含有D540H和E718H取代突变)具有至少80%相同性的核苷酸序列;
o核酸分子,其包含显示与SEQ ID NO:23所示核苷酸序列(编码SEQ ID NO:9的截短的和突变的聚合酶-TMR)具有至少80%相同性的核苷酸序列;
o核酸分子,其包含显示与SEQ ID NO:24所示核苷酸序列(编码SEQ ID NO:18的core*t-env1)具有至少80%相同性的核苷酸序列;
o核酸分子,其包含显示与SEQ ID NO:25所示核苷酸序列(编码了SEQ ID NO:19的core*t-env1-env2)具至少80%相同性的的核苷酸序列;
o核酸分子,其包含显示与以下具有至少80%相同性的核苷酸序列:SEQ ID NO:26所示核苷酸序列(编码SEQ ID NO:20的core-env1-env2-env4),或SEQ ID NO:26所示核苷酸序列从核苷酸1开始到核苷酸753结束的部分(编码core-env1-env2),或SEQ ID NO:26所示核苷酸序列从核苷酸1开始到核苷酸663结束的部分(编码core-env1);或它们的缺失版本,所述缺失版本缺少SEQ ID NO:26从位点229的G开始到位点252的A的部分(对应于核心部分中残基77-84缺失)。
本发明的核苷酸分子可使用本领域可获取的序列数据和本文中所提供的序列信息而产生。编码每种HBV聚合酶的DNA序列可直接通过常规分子生物学或PCR技术从含HBV的细胞、cDNA和基因组文库、病毒基因组,或任何现有技术已知包含其的载体中分离,并可被修饰(如,如本文所描述)。另外,本发明的核酸分子还可通过自动化的化学合成方法产生(如,从重叠的合成寡核苷酸组装,例如在Edge,1981,Nature 292,756;Nambair等人,1984,Science 223:1299;Jay等人,1984,J.Biol.Chem.259:6311中所描述)。
本发明还提供的是包含一或多种本发明核酸分子的载体以及包含这样的载体的组合物。
就本发明而言,可使用多种宿主-载体系统,包括适合于在原核宿主生物体如细菌(如,大肠杆菌、枯草杆菌或李斯特菌)中表达的噬菌体、质粒或柯斯质粒载体;适合于在酵母(如,酿酒酵母、裂殖酵母或毕赤酵母)中表达的载体;适合于在昆虫细胞系统(如,Sf9细胞)中表达的病毒表达载体(如,昆虫杆状病毒);适合于在植物细胞系统中表达的病毒或质粒表达载体(如,Ti质粒、花椰菜花叶病毒CaMV;烟草花叶病毒TMV);以及适合于在高等真核细胞或生物体中表达的质粒或病毒载体。这样的载体在文献中被大量描述并为商业可得到的(如,Stratagene、Amersham Biosciences、Promega等)。合适的质粒载体的代表性的实例包括但不限于,pREP4、pCEP4(Invitrogen)、pCI(Promega),pCDM8(Seed,1987,Nature 329,840)和pMT2PC(Kaufman等人,1987,EMBO J.6:187),pVAX和pgWiz(Gene Therapy System Inc;Himoudi等人,2002,J.Virol.76:12735)。就本发明来说,还可使用许多源自多种不同病毒的病毒载体(如逆转录病毒、腺病毒、AAV、痘病毒、疱疹病毒、麻疹病毒、泡沫病毒、甲病毒、水泡性口炎病毒等等)。
尤其感兴趣的是腺病毒载体,其对基因导入或重组产生具有许多有据可查的优点(综述见“Adenoviral vectors for gene therapy”,2002,编辑,D.Curiel和J.Douglas,Academic Press)。本发明所用的腺病毒载体可来源于多种人或动物来源(如,犬、羊、猴腺病毒等)。可采用任何血清型,特别优选人腺病毒并特别优选C亚属如Ad2(Ad2)、5(Ad5)、6(Ad6),B亚属如11(Ad11)、34(Ad34)和35(Ad35),以及D亚属如19(Ad19)、24(Ad24)、48(Ad48)和49(Ad49)。使用动物Ad也是有利的,特别优选黑猩猩Ad,如黑猩猩Ad3(Peruzzi等人,2009,Vaccine 27:1293)和黑猩猩Ad63(Dudareva等人,2009,Vaccine 27:3501)。所引用的腺病毒可得自美国典型培养物保藏中心(ATCC,Rockville,Md.),或已作为大量描述了其序列、组织和产生方法的出版物的对象,使得技术人员可使用它们(见例如US 6,133,028;US 6,110,735;WO 02/40665;WO00/50573;EP 1016711;Vogels等人,2003,J.Virol.77:8263;WO00/70071;WO02/40665;WO2004/001032;WO2004/083418;WO2004/097016;WO2005/010149)。
在一个实施方案中,本发明的腺病毒为复制缺陷型。优选的复制缺陷型腺病毒载体为E1缺陷的(见例如US 6,136,594和US 6,013,638),其含有从约459位至3328位或从约459位至3510位的E1缺失(参考公开于GeneBank编号M 73260下和Chroboczek等人,1992,Virol.186:280中的5型人腺病毒的序列)。克隆容量可通过额外缺失腺病毒基因组的部分(全部或部分非必要的E3区域或其它必要的E2、E4区域,如在WO94/28152;Lusky等人,1998,J,Virol.72:2022中描述的)而进一步改善。
本发明的核酸分子可插入至腺病毒基因组的任何位置,特别优选替换E1区域的插入。它/它们可定位于相对所考虑区域的天然转录方向的有义或反义方向。
就本发明而言,其它合适的病毒载体来源自痘病毒(见例如Cox等人,“Viruses in Human Gene Therapy”,编辑,J.M.Hos,Carolina Academic Press)。就本发明来说,痘病毒载体可获得自任何痘病毒科的成员,特别是金丝雀痘病毒、鸡痘病毒和痘苗病毒,优选为后者。合适的痘苗病毒包括但不限于哥本哈根(Copenhagen)株系(Goebel等人,1990,Virol.179:247和517;Johnson等人,1993,Virol.196:381)、Wyeth株系和修饰后的Ankara株系(Antoine等人,1998,Virol.244:365)。构建重组痘病毒的一般条件是本领域众所周知的(见例如EP 206 920;Mayr等人,1975,Infection 3:6;Sutter和Moss,1992,Proc.Natl.Acad.Sci.USA 89:10847;US 6,440,422)。本发明的核酸分子优选插入到痘病毒基因组中非必须的基因座内。胸苷激酶基因尤其适合哥本哈根痘苗病毒载体的插入(Hruby等人,1983,Proc.Natl.Acad.Sci.USA 80:3411;Weir等人,1983,J.Virol.46:530)以及缺失II或III适于MVA载体的插入(Meyer等人,1991,J.Gen.Virol.72:1031;Sutter等人,1994,Vaccine 12:1032)。
本发明还涵盖与脂质或聚合物复合以形成颗粒结构如脂质体、阳离子脂质体或纳米颗粒的载体(如质粒DNA)。这样的技术可在本领域得到(见例如Arangoa等人,2003,Gene Ther.10:5;Eliaz等人,2002,Gene Ther.9:1230和Betageri等人,1993,“Liposome drug delivery systems”,Technomic Publishing Company,Inc.)。
根据优选的实施方案,本发明的载体以适合于在宿主细胞或生物体中表达的形式包含本发明的核酸分子,其意指所述核酸分子置于一或多个适合于所述载体和/或宿主细胞的调控序列的控制下。本领域技术人员应理解,调控序列可根据于如宿主细胞、所需的表达水平等因素选择。
启动子特别重要,并且就本发明来说有用的合适启动子包含指导所述核酸分子在许多类型宿主细胞表达的组成型启动子,和那些仅在某些宿主细胞中(如,肝特异性调控序列)或响应特定事件或外源因子(如,温度、营养添加剂、激素或其它配体)时指导所述核酸分子表达的启动子。
适合于在哺乳动物细胞中组成型表达的启动子包括但不限于,巨细胞病毒(CMV)即时早期启动子(Boshart等人,1985,Cell 41:521)、RSV启动子、腺病毒主要晚期启动子、磷酸甘油激酶(PGK)启动子(Adra等人,1987,Gene60:65)、和单纯疱疹病毒(HSV)-I的胸苷激酶(TK)启动子。痘苗病毒启动子特别适于在痘病毒载体中表达。具代表性的实例包括但不限于痘苗病毒7.5K、H5R、11K7.5(Erbs等人,2008,Cancer Gene Ther.15:18)、TK、p28、p11和K1L启动子,以及合成的启动子,如在Chakrabarti等人(1997,Biotechniques 23:1094,与pSE/L启动子相关)、Hammond等人(1997,J.Virological Methods 66:135)和Kumar和Boyle(1990,Virology 179:151)中描述的那些和早/晚期嵌合启动子。肝脏特异的启动子不受限制地包含以下的启动子:HMG-CoA还原酶(Luskey,1987,Mol.Cell.Biol.7:1881);类固醇调控单元1(SRE-1;Smith等人,1990,J.Biol.Chem.265:2306);白蛋白(Pinkert等人,1987,Genes Dev.1:268);磷酸烯醇式丙酮酸激酶(PEPCK)(Eisenberger等人,1992,Mol.Cell Biol.12:1396);人C反应蛋白质(CRP)(Li等人,1990,J.Biol.Chem.265:4136);人葡萄糖激酶(Tanizawa等人,1992,Mol.Endocrinology 6:1070);胆固醇7-α羟化酶(CYP-7)(Lee等人,1994,J.Biol.Chem.269:14681);α-1抗胰蛋白酶(Ciliberto等人,1985,Cell 41:531);胰岛素样生长因子结合蛋白质(IGFBP-I)(Babajko等人,1993,Biochem.Biophys.Res.Comm.196:480);人转铁蛋白质(Mendelzon等人,1990,Nucleic Acids Res.18:5717);I型胶原蛋白(Houglum等人,1994,J.Clin.Invest.94:808);和FIX(US 5,814,716)基因。
本领域技术人员应理解,控制本发明核酸分子表达的调控元件可另外包括额外的元件,其用于在宿主细胞或生物体中正确地起始、调控和/或终止转录(如,多聚A转录终止序列),mRNA转运(如,核定位信号序列)、加工(如,剪接信号)和稳定性(如,内含子和非编码的5'和3'序列),翻译(如,起始子甲硫氨酸、三联前导序列、核糖体结合位点、Shine-Dalgamo序列等)和纯化步骤(如,标签)。
根据本发明,本发明的编码所述聚合酶部分、所述核心部分和/或所述包膜部分的核酸分子可由相同的载体或至少两种载体(如,两或三种独立的载体)携带。因此本发明涵盖携带编码所述聚合酶部分、所述核心部分和所述包膜部分的核酸分子的载体,以及独立的载体,其每个仅携带一或两种编码所述聚合酶部分、所述核心部分和所述包膜部分的核酸分子。本发明也提供了这样的载体,以及包含这样的载体的组合物。当使用不同的载体时,其可来自不同的来源或相同的来源。例如,可设想由缺陷型痘病毒(如,MVA)表达一种HBV部分,并由另一痘病毒载体(如,哥本哈根载体)表达其它两种部分。作为另一个实例,可设想组合物,其包含编码所述聚合酶部分的腺病毒载体和编码所述核心和包膜部分的腺病毒载体。此外,就本发明来说,由不同的病毒载体(如,由腺病毒载体表达聚合酶部分并由MVA表达核心和/或包膜部分,反之亦然)表达和由质粒和病毒载体表达HBV部分也是合适的。
本发明优选的实施方案涉及载体,其选自:
i)MVA载体,其包含置于痘苗病毒启动子如7.5K启动子控制下并编码了聚合酶部分的核酸分子,所述聚合酶部分包含SEQ ID NO:7、8、9所示氨基酸序列,或包含在494位的Asp残基被His残基取代以及672位的Glu残基被His残基取代的SEQ ID NO:7所示氨基酸序列。优选地,所述核酸分子插入在MVA基因组的缺失III中。
ii)MVA载体,其包含置于痘苗病毒pH5r启动子控制下并编码核心部分和包膜部分的核酸分子,所述核心部分和包膜部分包含SEQ ID NO:18或19所示的氨基酸序列。优选地,所述核酸分子插入在MVA基因组的缺失III中。
iii)E1缺陷型Ad载体,其包含被插入以取代E1区域的置于CMV启动子控制下并编码聚合酶部分的核酸分子,所述聚合酶部分包含SEQ ID NO:7、8、9所示氨基酸序列,或包含在494位的Asp残基被His残基取代以及672位的Glu残基被His残基取代的SEQ ID NO:7所示氨基酸序列。
iv)E1缺陷型Ad载体,其包含被插入以取代E1区域的置于CMV启动子控制下并编码核心部分和包膜部分的核酸分子,所述核心部分和包膜部分包含SEQ ID NO:18、19、20所示氨基酸序列,或包含SEQ ID NO:20从残基1开始并在残基251结束的部分(core-env1-env2)或SEQ ID NO:20从残基1开始并在残基221结束的部分(core-env1)。
以及涉及包含载体(i)和(ii),或(iii)和(iv)的组合物。
如果需要,本发明的载体或组合物可另外包含一或多种转基因,如感兴趣的、待与本发明的核酸分子共同表达于宿主细胞或生物体中的基因。理想地,所述转基因的表达对HBV感染或由HBV感染引起或与之相关的疾病或病症具有治疗性或保护性活性,或能够增强本发明的组合物的免疫原性。合适的转基因不受限制地包含一或多种额外的HBV多肽/肽或编码核酸分子,如X蛋白或其片段,免疫调节剂如细胞因子(如,IL-2、IL-7、IL-12、IL-15、IL-18、IL-21、IFNg)、细胞因子融合物(如WO2005/14642中描述的)或其编码核酸分子,以及尤其对治疗肝脏肿瘤有用的自杀基因产物或编码核酸分子(如胞嘧啶脱氨酶(CDase),尿嘧啶磷酸核糖转移酶(UPRTase),FCU-1基因产物(在WO99/54481中描述)和其衍生物(在WO2006/048768中描述),其待与前体药物5-氟胞嘧啶(5-FC))。如果使用了转基因,其可以与本发明的任何核酸分子相融合的形式表达,或在适合的调控元件控制下独立地表达。此外,其可插入本发明载体的任何位置,或在与本发明的组合物的载体组合使用的独立载体中。
一方面,本发明提供了感染性病毒颗粒以及包含这样的感染性病毒颗粒的组合物,所述病毒颗粒包含本发明的核酸分子或载体。
一般地,这样的病毒颗粒通过包括以下步骤的方法产生:
a)将本发明的病毒载体导入合适的细胞系。
b)在合适的条件下培养所述细胞系使得所述感染性病毒颗粒产生。
c)由所述细胞系的培养物回收所产生的感染性病毒颗粒,和
d)任选地纯化所述回收的感染性病毒颗粒。
当病毒载体是缺陷型时,感染性颗粒通常在互补细胞系中或通过使用辅助病毒来产生,该辅助病毒提供了反式非功能性病毒基因。例如,用于互补E1缺失腺病毒载体的合适的细胞系包括293细胞(Graham,1997,J.Gen.Virol.36,59-72)以及HER-96和PER-C6细胞(如,Fallaux等人,1998,Human Gene Ther.9,1909-1917;WO97/00326)。用于繁殖痘病毒载体的适合的细胞为鸟类细胞,并最优选鸡胚原代成纤维细胞(CEF),其由获得自受精卵的鸡胚制备。
感染性病毒颗粒可由培养物上清或裂解后的细胞回收。它们可根据标准技术进一步纯化(例如在WO96/27677、WO98/00524、WO98/22588、WO98/26048、WO00/40702、EP1016700和WO00/50573中描述的层析法、氯化铯梯度离心法)。
本发明还涵盖载体或病毒颗粒,其被修饰以允许优先靶向于特定靶细胞(见例如Wickam等人,1997,J.Virol.71,8221-8229;Amberg等人,1997,Virol.227,239-244;Michael等人,1995,Gene Therapy 2,660-668;WO94/10323;WO02/96939和EP1146125)。本发明的靶向载体和病毒颗粒的标志性特征为在其表面上存在配体,所述配体能够识别并结合细胞和表面暴露的组分,如细胞特异性标记物(如,HBV感染的细胞)、组织特异性标记物(如,肝脏特异性标记物)以及病毒(如,HBV)抗原。合适的配体的实例包括针对HBV抗原结构域的抗体或其片段。细胞靶向可通过将配体遗传学插入存在于病毒表面的多肽(如,腺病毒纤维、腺病毒亚单位、pIX或痘苗p14基因产物)而实施。
本发明还涉及宿主细胞以及包含这样的宿主细胞的组合物,所述宿主细胞包含本发明的核酸分子、载体或感染性病毒颗粒。就本发明来说,宿主细胞包括原核细胞,低等真核细胞如酵母,和其它真核细胞如昆虫细胞、植物和哺乳动物(如人或非人)细胞,以及能够互补本发明的复制缺陷型载体(如腺病毒载体)的至少一种缺陷功能的互补细胞如293和PERC.6细胞。
根据本发明特定的实施方案,宿主细胞还可被封装(encapsulated)。细胞封装技术以前已被描述(Tresco等人,1992,ASAIO J.38,17-23;Aebischer等人,1996,Human Gene Ther.7,851-860)。
本发明的另一方面为用于采用本发明的载体、感染性病毒颗粒和/或宿主细胞产生重组聚合酶、核心和/或包膜部分的方法。本发明的方法包括(a)将本发明的载体或感染性病毒颗粒导入合适的宿主细胞以产生被转染的或感染的宿主细胞,(b)在适合于所述宿主细胞生长的条件下体外培养所述被转染的或感染的宿主细胞,(c)由细胞培养物回收所述聚合酶、核心和/或包膜部分,和(d)任选地,纯化回收的多肽。
可预计,本领域技术人员了解大量的可得到的表达系统(用于在合适的宿主细胞中产生HBV部分)和用于将载体或感染性病毒颗粒导入宿主细胞的方法。这样的方法包括,但不限于,显微注射(Capechi等人,1980,Cell22:479)、CaPO4介导的转染(Chen和Okayama,1987,Mol.Cell Biol.7:2745)、DEAE葡聚糖介导的转染、电穿孔法(Chu等人,1987,Nucleic Acid Res.15:1311)、脂质体转染/脂质体融合(Feigner等人,1987,Proc.Natl.Acad.Sci.USA 84:7413)、粒子轰击(Yang等人,1990,Proc.Natl.Acad.Sci.USA87:9568)、基因枪、转导、病毒感染以及通过不同方法直接向宿主生物体施用。
为了便于将载体引入宿主细胞,本发明的载体可与转染试剂联合使用,如聚阳离子聚合物(如,壳聚糖、聚甲基丙烯酸酯、PEI等)和阳离子脂质(如,DC-Chol/DOPE、可从Promega得到的transfectam lipofectin)。此外,如上文描述,重组DNA技术可用于改善核酸分子在宿主细胞或生物体中的表达,如通过使用高拷贝数载体、取代或修饰一或多种转录调控序列(如,启动子、增强子等)、优化宿主细胞对核酸分子的密码子使用,和抑制可破坏转录的阴性序列。
本发明的宿主细胞可在常规发酵生物反应器、烧瓶和培养皿平板中培养。培养可在适合给定宿主细胞的温度、pH值和氧含量下进行。此处不尝试描述已知用于在原核和真核细胞中产生蛋白质的不同方法的细节。
然后HBV部分可通过众所周知的纯化方法纯化,其包括硫酸铵沉淀、酸萃取、凝胶电泳;过滤和色谱方法(如,反相、尺寸排阻、离子交换、亲和、磷酸纤维素、疏水相互作用、羟基磷灰石或高效液相色谱法)。所用的条件和技术取决于如静电荷、分子量、疏水性、亲水性的因素,并对本领域技术人员是显而易见的。此外,纯化的水平将取决于所需的用途。
另一方面,本发明提供了组合物,其包含本发明的聚合酶、核心和/或包膜部分,编码核酸分子,载体,感染性的病毒颗粒,或宿主细胞(本文中也指“活性剂”)中至少一种,或其任何组合(如,多肽或载体/病毒颗粒的组合,其编码如本文中描述的多种HBV部分或不同基因型的组合)。优选地,所述组合物为药物学组合物,其在药物学有效量的活性剂外还包含药物学可接受的载体。
合适地,本发明的组合物包含适合于人或动物用途的稀释剂。其优选为等渗的、低渗的或弱高渗的,并具有相对低的离子强度。代表性的实例包括,无菌水、生理盐水(如,氯化钠)、林格氏(Ringer's)液、葡萄糖、海藻糖或蔗糖溶液(见例如最新版本的Remington:The Science and Practice of Pharmacy,A.Gennaro,Lippincoft,Williams和Wilkins)。本发明的组合物被合适地缓冲以适合于在生理或弱碱性pH下用于人(如,从大约pH值7到大约pH值9)。合适的缓冲液包括但不限于磷酸盐缓冲液(如,PBS)、碳酸氢盐缓冲液和/或Tris缓冲液。
所述组合物还可含有其它药物学可接受的赋形剂以提供所需的药物学或药效学特性,其包括,例如,修饰或维持配方的pH值、渗透压、粘度、澄清度、颜色、无菌性、稳定性、制剂的溶解速率,修饰或维持向人或动物生物体中的释放或吸收,促进跨血液屏障的运输或在特定器官(如,肝脏)中的渗透。合适的赋形剂包括氨基酸。
本发明的组合物中包含的药物学可接受的载体还必须允许在冷冻(如,-70℃、-20℃)、冷藏(如,4℃)或常温的制造和长期储存条件下保持其稳定性。这方面,特别适于本发明的组合物的配方包括:
o1M蔗糖,150mM NaCl,1mM MgCl2,54mg/l吐温80,10mM pH值8.5的Tris(尤其当活性剂为腺病毒载体时),和
o10mg/ml甘露醇、1mg/ml HSA,20mM pH7.2的Tris,和150mM NaCl
o生理盐水
另外,本发明的组合物可包含一或多种适合于在人中全身或粘膜应用的佐剂。优选地,所述佐剂能够刺激对本发明组合物的免疫,尤其是T细胞介导的免疫,如,通过Toll样受体(TLR)如TLR-7、TLR-8和TLR-9。有用的佐剂代表性的实例包括但不限于,明矾,矿物油乳剂如弗氏完全和不完全佐剂(IFA),脂多糖或其衍生物(Ribi等人,1986,Immunology and Immunopharmacology of Bacterial Endotoxins,Plenum Publ.Corp.,NY,407-419页),皂苷如QS21(Sumino等人,1998,J.Virol.72:4931;WQ98/56415),咪唑喹啉类化合物如咪喹莫特(Suader,2000,J.Am.Acad.Dermatol.43:S6)、S-27609(Smorlesi,2005,Gene Ther.12:1324)和如在WO2007/147529中描述的相关化合物,胞嘧啶磷酸鸟嘌呤寡脱氧核苷酸如CpG(Chu等人,1997,J.Exp.Med.186:1623;Tritel等人,2003,J.Immunol.171:2358)和阳离子肽如IC-31(Kritsch等人,2005,J.Chromatogr Anal.Technol.Biomed.Life Sci.822:263)。
本发明的组合物适合于多种施用模式,包括全身、外用和局部施用。可通过任何手段完成注射,例如,通过皮下、皮内、肌肉内、静脉内、腹腔、肿瘤内、血管、动脉注射或直接注射入供养肝脏的动脉(如,肝动脉灌注)或静脉(如,注射入门静脉)。注射可用常规注射器和针头,或本领域可得到的任何其它合适的设备来完成。另外,本发明的组合物可通过粘膜途径,如口腔/消化道、鼻腔内、气管内、肺内、阴道内或直肠内途径施用。呼吸道内的施用可通过使用压力容器(如,用合适的推进剂如二氯二氟甲烷、丙烷、氮气等等),或非压力分配器中雾化或气化液滴、喷雾剂、或干粉组合物来完成。局部施用还可使用透皮方法(如,贴片等)完成。就本发明来说,优选的组合物是配置为用于肌内和皮下途径的。
本发明的组合物可为多种形式的,如,固体、液体或冷冻。固体(如,干粉或冻干物)组合物可通过涉及真空干燥及冷冻干燥的方法获得。对粘膜施用,所述组合物可配制为肠溶制剂胶囊、和用于口服施用的冲剂、用于直肠或阴道施用的栓剂,其最终与提高粘膜膜孔径大小的有用的吸收增强剂组合。这样的吸收增强剂一般为与粘膜膜磷脂酶结构域具有相似结构的物质,如脱氧胆酸钠、甘氨胆酸钠、二甲基-β-环糊精、十二烷基-1-溶血磷脂胆碱。
合适的剂量可按不同参数的函数修饰,尤其是施用模式;采用的组合物;宿主生物体的年龄、健康和体重;症状的性质和程度;并行治疗的种类;治疗的频率;和/或预防或治疗的需要。确定合适的治疗剂量所必须的进一步完善的计算由医生依照相关环境例行做出。作为一般性的指导,含病毒的组合物的合适的剂量从约105到约1013vp(病毒颗粒)、iu(感染单位)或pfu(斑形成单位)变化,其取决于所用的载体和定量技术。在样本中评估vp、iu和pfu的数量的可用技术在本领域是常规的。例如,通常通过测量A260吸光度确定腺病毒颗粒(vp)的数目,通过定量DBP免疫荧光确定iu滴度和通过感染允许细胞后的斑计数确定pfu。按照FDA指南,vp/iu比优选低于100。对于基于MVA的组合物,优选约5×105至约109pfu的剂量,特别优选约107、约5×107、约108或约5×108pfu的剂量。对于基于Ad的组合物,优选含有约106至约1012vp的剂量,特别优选约109、约5×109、约1010或约5×1010vp或约1011vp的剂量。基于载体质粒的组合物可能以10μg及20mg之间,最好在100μg和2mg之间的剂量施用。蛋白质组合物可能以10ng和20mg之间的一或多种剂量施用,特别优选从每公斤体重约0.1μg至约2mg的治疗性蛋白质的剂量。施用可能以单一剂量或以一定的时间间隔重复一或若干次的剂量进行。
本发明的组合物可能在用于治疗多种疾病和病理学状态(尤其是由HBV感染引起的或与之相关的疾病和病理学状态)的方法中采用。如本文中所用,术语“治疗(treatment)”或“治疗(treating)”涵盖预防和/或治疗。其对HBV感染的患者中治疗HBV慢性感染和/或包括肝硬化和肝癌的肝脏病变特别有用。优选地,根据本文描述的模式导入宿主生物体后,与治疗前相比,本发明的组合物提供了治疗性好处来治疗宿主。所述治疗性的好处可通过许多方式证实,例如在被感染对象的血液、血浆、血清或肝脏中检测到HBV病毒载量的下降,和/或通过检测到抗HBV的免疫应答(如,抗HBV抗体的产生和/或T细胞介导的免疫)或通过与HBV感染相关的症状的延迟(如,肝硬化或肝癌发展的延迟),或通过通常伴随着HBV感染的肝脏炎症/脂肪化/纤维化状况的减少,或通过改善的个体对常规治疗的应答。
因此,本发明还涵盖本发明的HBV部分、核酸分子、载体、感染性的病毒颗粒、宿主细胞或组合物中的至少一种在制备用于根据本文中描述的模式治疗或预防HBV感染、HBV相关的疾病和病理学状况的药物中用途。
本发明还提供了用于治疗或预防HBV感染,尤其是慢性HBV感染,HBV相关疾病和病理学状态的方法,其包括向有需要的人或动物生物体施用治疗有效量的至少一种本发明的HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞或组合物。
本发明的方法或用途包含治疗有效量的所述活性制剂的一或多次施用(1、2、3、4、5、6、7、8、9、10等),该施用通过合适的时间相互隔开,并且通过同样的施用途径或不同的施用途径(如,肌内或皮下途径),在同样的部位或不同的部位完成。相互隔开3至10天(如,3次每周一次的施用)的三次施用特别适合基于MVA的组合物和载体。该第一系列的施用可跟着再使用同样的活性剂进行一或多次施用,其可进行一或若干个月以便恢复由3次连续施用刺激的抗HBV免疫应答。对基于Ad的组合物和载体,优选的方法或用途包括一次施用,最终跟着在一个月和六个月后进行一或两次施用。
如果需要,本发明的方法或用途可与一或多种常规治疗模式(如,放射疗法、化学疗法和/或手术)组合完成。多重治疗途径的使用提供给患者更广阔基础的治疗。在一个实施方案中,本发明的方法可先于或晚于手术治疗。在另一个实施方案中,其可先于或晚于放射治疗(如,γ射线)。本领域技术人员可容易地配制合适的可使用的放射疗法方案和参数(见例如Perez和Brady,1992,Principles and Practice of Radiation Oncology,第二版,JB Lippincott Co;使用合适的改变和修饰对本领域技术人员将会是显而易见的)。
在另一个实施方案中,本发明的方法或用途与常规地用于治疗或预防HBV感染、HBV相关的疾病和病理学状态的使用一或多种HBV药物的化学疗法相关。这些药物的施用可能先于、同时、或晚于本发明所用的活性制剂的施用。HBV药物代表性的实例包括但不限于聚合酶抑制剂、RNase H抑制剂、核苷类似物、核苷酸类似物、TLR激动剂、N糖基化抑制剂、siRNA、反义寡核苷酸、抗HBV抗体、免疫调节剂、治疗性疫苗和通常用于治疗HBV相关的肝癌的抗肿瘤药物(如,阿霉素、含碘油或索拉菲尼(sorasenib)的阿霉素)。合适的治疗性疫苗的实例包括但不限于重组抗原、病毒样颗粒(VLP)、基于或编码HBV蛋白(核心、前S1、前S2、S和/或聚合酶)的载体或合成肽,所述HBV蛋白尤其适合于触发抗HBV的体液应答。根据若干小时、天和/或星期的标准方案、剂量和疗程,这样的HBV药物可以单一剂量提供,或可选地,以多剂量提供。本发明特别适合的方法或用途为用于与标准治疗(standard of care)组合,该治疗可先于、同时或晚于本发明的方法或用途。尽管这样的标准治疗可能在患者与患者间不同,其通常包括用细胞因子(如,干扰素α、聚乙二醇干扰素α2)和/或用核苷酸或核苷类似物如拉米夫定、恩替卡韦、替比夫定、阿德福韦酯或替诺福韦治疗。
在另一个实施方案中,本发明的方法或用途根据初始加强(prime boost)治疗模式完成,该治疗模式包括依次的一或多次初始组合物和一或多次加强组合物的施用。一般地,初始和加强组合物使用不同的载体,其至少包含或编码共同的抗原结构域。另外,初始和加强组合物可在相同的部位或另外的部位以相同的施用途径或不同的施用途径来施用。例如,基于多肽的组合物可通过粘膜途径施用而基于载体的组合物优选注射施用,如MVA载体用皮下注射、DNA质粒用肌内注射和腺病毒载体用皮下或肌内注射。
本发明还提供了在宿主生物体中诱导或刺激针对HBV的免疫应答的方法,包含向所述生物体施用至少一种本发明的HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞或组合物以诱导或刺激所述免疫应答。所述免疫应答可以为特异性的和/或非特异性的,体液的和/或细胞的,并且在此情况下,其可为CD4+或CD8+或CD4+和CD8+介导的。所述免疫应答优选针对HBV抗原的T细胞应答。
本发明的方法在动物或人生物体中施用后能诱导或刺激抗HBV的免疫应答的能力可以用多种本领域标准化的测定在体外或体内评估。对于可用的评估免疫应答的发生和激活的一般的描述,见例如Coligan等人(1992和1994,Current Protocols in Immunology;编辑J Wiley和Sons Irac,National Institute of Health)。细胞免疫的测量可通过测量由活化的效应细胞分泌的细胞因子谱(包括源自CD4+和CD8+T细胞的那些)(如,通过酶联免疫斑点(ELIspot)法定量IL-10或IFNg产生细胞)、通过确定活性状态的免疫效应细胞(如,通过经典的[3H]胸腺嘧啶摄取法的T细胞增殖试验)、通过测定致敏的对象中的抗原特异性T淋巴细胞(如,在细胞毒性试验中的肽特异性裂解)来完成。刺激体液应答的能力可通过抗体结合和/或竞争性结合确定(见例如Harlow,1989,Antibodies,Cold Spring Harbor Press)。本发明的方法还可在用合适的感染性或肿瘤诱导剂(如,表达了HBV基因产物的痘苗病毒或李斯特菌(Listeria Monocytogenes))攻击的动物模型中确定对感染性或肿瘤诱导剂的中和,以及反映了抗HBV免疫应答的诱导或增强的最终对相关症状的部分抵抗而进一步证实。本发明的组合物的测试和验证也在所附实施例部分说明。
另一方面,本发明还提供了用于根据本文中描述的模式治疗或预防HBV感染(包括慢性HBV感染)、HBV相关疾病和病理学状态,更具体地用于在感染HBV的对象中诱导或产生免疫应答的试剂盒,其中所述试剂盒包含多种活性剂,其选自本文中描述的HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞或组合物。理想地,所述多种活性剂以分离多肽或分离载体的形式提供,并且每种活性剂的施用可同时(在相同的时间)或分别(一种在一定的时间间隔后接着另一种)进行,其通过同样的途径或不同的途径施用,并且在同样的(或紧邻的)部位或不同的部位,并且使用同样的剂量或不同的剂量。
本发明特别感兴趣的是试剂盒,所述试剂盒包含第一载体和第二载体,所述第一载体包含编码如本文中定义的聚合酶部分的核酸分子,所述第二载体包含编码如本文中定义的核心部分和/或包膜部分的核酸分子。
根据优选的实施方案,所述第一载体为MVA载体,其包含置于痘苗病毒启动子如7.5K启动子控制下并编码聚合酶部分的核酸分子,所述聚合酶部分包含SEQ ID NO:7、8或9所示的氨基酸序列,或包含在494位的Asp残基被His残基取代以及在672位的Glu残基被His残基取代的SEQ ID NO:7所示氨基酸序列;并且所述第二载体为MVA载体,其包含置于痘苗病毒启动子如pH5r启动子控制下以及编码包含SEQ ID NO:18或19所示氨基酸序列的核心部分和包膜部分的核酸分子。
根据另一优选的实施方案,所述第一载体为腺病毒载体,其包含置于合适的启动子如CMV启动子控制下并编码聚合酶部分的核酸分子,所述聚合酶部分包含如SEQ ID NO:7或SEQ ID NO:8所示的氨基酸序列,或包含包含在494位的Asp残基被His残基取代以及在672位的Glu残基被His残基取代的SEQ ID NO:7所示氨基酸序列;并且所述第二载体为腺病毒载体,其包含置于合适的启动子如CMV启动子控制下并编码核心部分和包膜部分的核酸分子,所述核心部分和包膜部分包含SEQ ID NO:18、19或20中所示的的氨基酸序列,或SEQ ID NO:20从残基1开始并在残基251结束的部分(core-env1-env2)或SEQ ID NO:20从残基1开始并在残基221结束的部分(core-env1)。
本发明的试剂盒还可包含表达如上文定义的免疫调节剂的第三载体。为了说明性的目的,试剂盒中包含的每种活性成分优选的剂量与上文描述的本发明的组合物为同样的数量级(order),特别优选每种痘病毒或MVA载体5×105至109pfu,以及每种腺病毒载体约106约1012vp。
本发明还提供了选择性结合本发明所用的HBV部分或其肽片段的抗体。如本文中所用的,当抗体结合靶肽而不显著地结合无关蛋白时,其为选择性地结合靶肽。某些情况下,应理解尽管存在某种程度的交叉活性,结合肽的抗体仍然是选择性的。但是,本发明的抗体优选不以高亲和力或高选择性地结合HBV天然蛋白质。
如本文中所用,抗体依本领域公认的一致定义。本发明的抗体包括多克隆抗体和单克隆抗体,以及这样的抗体的片段,其包括但不限于Fab或F(ab').sub.2,和Fv片段。本发明的抗体可使用本领域常规技术产生,如遵循向动物施用有效量的任何本文中描述的HBV部分和/或其肽片段。抗体优选由包含唯一的序列的HBV部分的区域或离散片段制备,如涉及本文中描述的引入至天然HBV蛋白质中的修饰的那些。
本发明的抗体在本发明的范围内拥有多种潜在的用途。例如,这样的抗体可用作(a)作为检测本发明的第一或第二多肽的测定中的试剂,(b)作为检测生物学样本中HBV病毒存在的测定中的试剂,和/或(c)作为从蛋白质混合物和其它污染物中回收重组产生的HBV部分的工具(如,允许通过亲和层析或免疫共沉淀从培养的宿主细胞中纯化)。
本发明还涉及用于在取自怀疑被所述HBV病毒感染的个体的生物学样本(如,血浆、血清、组织)中检测和/或定量HBV病毒或抗HBV抗体的方法,其使用至少一种本发明的HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞、组合物或抗体。
在一个实施方案中,该方法尤其适合于在生物学样本中检测和/或定量HBV病毒,并且至少包括在使得形成病毒和抗体间复合物的条件下将所述生物学样本与至少一种本发明的抗体相接触,并通过任何适合的方法检测和/或定量所述复合物形成的步骤。
在另一个实施方案中,该方法尤其适合于在生物学样本中检测和/或定量抗HBV抗体,并且至少包括在使得抗HBV抗体与本发明的HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞、组合物之间形成复合物的条件下将所述生物学样本与本发明的HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞、组合物中的至少一种相接触,并通过任何适合的方法检测和/或定量所述复合物形成的步骤。
本领域技术人员将容易地确定本发明的方法所用的抗体、HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞、组合物的量。检测和/或定量病毒的方法为常规的和本领域技术人员熟知的。举例来说,可能提及印迹、ELISA、所谓的夹心技术、竞争技术、和PCR技术,尤其是所谓的“实时”技术。可通过偶联(如,物理连接)于可检测的物质便于本发明的抗体、HBV部分、核酸分子、载体、感染性病毒颗粒、宿主细胞或组合物用作试剂的用途。可检测的物质的实例包括各种酶(如,辣根过氧化物酶、碱性磷酸酶、β半乳糖苷酶或乙酰胆碱酯酶)、辅基(如,链霉亲和素/生物素或抗生物素蛋白/生物素)、荧光材料(如,伞形花内酯、荧光素或荧光素衍生物)、发光材料、生物发光材料(如,萤光素酶、萤光素或水母发光蛋白)、和放射性材料(如,125I、131I、35S或3H)。
最后,本发明涉及本发明的HBV部分、核酸分子、载体、感染性的病毒颗粒、宿主细胞、组合物或抗体中的至少一种用于体外诊断生物学样本中HBV感染的用途。
本发明以说明性的方式描述,并且应该理解所用的术语是描述性质而非限制性。明显地,根据上述技术,本发明的许多修饰和变化可能的。因此应理解,在所附权利要求的范围之内,本发明可能以不同于本文中明确描述的方法实行。
上述引用的所有专利公开、出版物和数据库条目明确地以其整体以相同的程度通过引用并入本文,就如同每一单个专利、出版物或条目被明确和单独地说明被引用并入本文一样。
附图说明
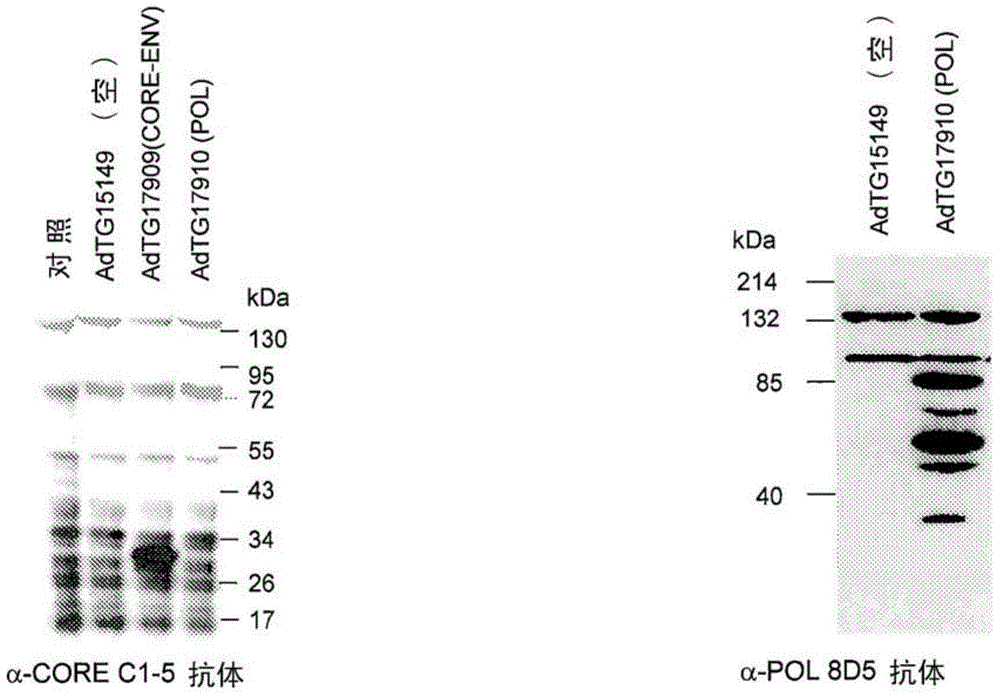
图1表明了来自腺病毒和MVA感染的细胞表达的HBV多肽。A549细胞或鸡胚成纤维细胞以对腺病毒的MOI 10或50,或对MVA的MOI 0.2或1感染,并且在感染48小时后裂解细胞。然后对获得自不同的Ad(图1A)或MVA(图1B)构建体感染的细胞的细胞裂解产物实行蛋白印迹以检测特异的HBV蛋白质。使用抗核心抗体(C1-5或13A9,1/200稀释)检测含核心多肽,并且用抗聚合酶抗体检测含聚合酶多肽(8D5,1/200稀释),其作为一抗,并且二抗偶联HRP。由Ad TG17909和Ad TG17910表达的蛋白质的预期大小分别为31.6kDa和88.5kDa。由MVA TG17971、MVA TG17972、MVA TG17993和MVA TG17994表达的蛋白质的预期大小分别为20.2kDa、15.8kDa、20kDa和23.5kDa。由MVA TG17842和MVA TG17843表达的蛋白质的预期大小分别为88.5kDa和98.2kDa。
图2表明了Elispots IFNγ测定中由腺病毒编码的HBV多肽的免疫原性。5只独立的小鼠(HLA-A2转基因小鼠)用Ad TG17909单独(黑色柱)、Ad TG17910单独(白色柱)或组合免疫(Ad TG17909+Ad TG17910)(灰色柱)一次。图2A表明使用HLA-A2限制性肽SLY(SEQ ID NO:55)或无关肽(没有显示)的靶向聚合酶蛋白质的特异性T细胞应答。图2B表明使用HLA-A2限制性肽FLP(SEQ ID NO:56)或ILC(SEQ ID NO:57)的靶向核心蛋白的特异性T细胞应答。图2C表明使用HLA-A2限制性肽VLQ(SEQ ID NO:58)、FLG(SEQ ID NO:59)或GLS(SEQ ID NO:60)的靶向包膜蛋白的特异性T细胞应答。每个柱代表了独立的接种疫苗的小鼠并且阴影的柱代表了每组的中间值。结果显示为106个脾脏细胞中观察到的斑的数目平均值,其获得自三个孔。如果斑数目高于50斑每106细胞(该截值由粗黑线条表示),即认为该应答为阳性的。
图3表明了细胞内细胞因子染色试验中由腺病毒载体编码的HBV多肽的免疫原性。5只独立的小鼠(HLA-A2转基因小鼠)用Ad TG17909(图3B)、Ad TG17910(图3A)或Ad TG17909和Ad TG17910的组合免疫一次(图3C)。脾细胞在含高尔基体抑制剂(Golgi-Plug),并存在每种HLA-A2限制性肽(用于聚合酶的SLY,用于核心的FLP、ILC和用于包膜的VLQ、FLG)或无关肽下培养5小时。可产生特异地针对每一HLA-A2限制性表位的细胞因子(IFNg和/或TNFα)的CD8+细胞的百分比通过ICS测定评估。每一柱代表了个体接种疫苗的小鼠,产IFNg细胞由黑色柱代表、产TNFα细胞由白色柱代表和产IFNg+TNFα细胞由阴影柱代表,并且对每一小鼠所有的这些细胞群体进行总计。
图4表明了由腺病毒载体编码的HBV多肽诱导CD8和CD4细胞应答的能力,其由细胞内细胞因子染色试验检测。5只独立的小鼠(HLA-A2转基因小鼠)用Ad TG17909和Ad TG17910的混合物免疫一次。脾细胞在存在每一HLA-A2限制性肽(用于聚合酶的SLY,用于核心的FLP、ILC和用于包膜的VLQ、FLG)或覆盖整个抗原结构域的重叠肽池(15氨基酸长,重叠11个氨基酸,核心2个肽池和包膜2个肽池)或无关肽下和高尔基体抑制剂(Golgi-Plug)培养5小时。诱导出的产生IFNγ和/或TNFα(图4A)的特异性CD8T细胞和诱导出的产生IFNγ和/或TNFα(图4B)或产生IFNγ和/或SL2(图4C)的特异性CD4T细胞通过ICS试验检测。每一柱代表了独立的接种疫苗的小鼠,产IFNg细胞由灰色柱代表、产TNFα或IL2细胞由白色柱代表和产IFNg+TNFα或IFNg+IL2细胞由阴影柱代表,并且对每一小鼠所有的这些细胞群体进行总计。还显示了每组的中间值。
图5表明了编码HBV多肽的腺病毒载体在体内诱导针对装载了HBV HLA-A2限制性表位的靶细胞的功能性细胞溶解的能力。3只独立的小鼠(HLA-A2转基因小鼠)用Ad TG17909和Ad TG17910组合免疫一次(M1至M3),并且一只小鼠用空腺病毒载体免疫一次作为阴性对照(M0)。来自同系小鼠的CFSE染色的脾脏细胞,不论是否(阴性对照)装载了HBV HLA-A2表位,被静脉注射至接种免疫后的小鼠。24小时后,每只小鼠的染色后细胞的体内溶解通过流式细胞术评估并按照材料与方法中所述进行计算。计算并显示每一肽在3只用AdHBV接种免疫后的小鼠中观察到的特异性溶解的平均值(平均值M1-M3)。
图6表明了通过Elispots IFNγ测定而确定的由MVA载体编码的HBV多肽的免疫原性。独立的小鼠(HLA-A2转基因小鼠)用MVA TG17842或MVA TG17843(图6A),或MVA TG17971(图6B),或MVA TG17972或阴性对照MVA TGN33.1(数据未显示)以一周的间隔免疫3次。图6A表明了用MVA TG17842(深灰色柱)或MVA TG17843(浅灰色柱)免疫后的靶向聚合酶蛋白的特异性T细胞应答,其使用了HLA-A2限制性肽SLY(SEQ ID NO:55)、覆盖聚合酶蛋白C末端部分的肽池8(长15个氨基酸,重叠11个氨基酸的25个肽/池)、无关肽或培养基(阴性对照)。图6B表明了用MVA TG17971免疫后的靶向核心蛋白的特异性T细胞应答,其使用了HLA-A2限制性肽FLP(SEQ ID NO:56)、ILC(SEQ ID NO:57)、“核心1和核心2”肽池(15个氨基酸长,重叠11个氨基酸的21至22个肽/池)、无关肽或培养基(阴性对照)。每个柱代表了独立的接种疫苗的小鼠并且阴影的柱代表了每组的平均值。结果显示为106脾脏细胞中观察到的斑的数目平均值,其获得自三个孔。如果斑数目高于50斑每106细胞(该截值由黑色虚线表示),即认为该应答为阳性的。
图7表明了共同注射进小鼠的由MVA载体编码的HBV多肽的免疫原性,其通过Elispots IFNγ测定而确定。独立的小鼠(HLA-A2转基因小鼠)用MVA TG17843与MVA TG17972(图7A)、或MVA TG17993(图7B)、或MVA TG17994(图7C)混合地,或单独用MVA TGN33.1作为阴性对照(数据未显示),以一周的间隔免疫3次。测定使用HLA-A2限制性肽SLY(SEQ ID NO:55)的靶向聚合酶蛋白的特异性T细胞应答,和使用HLA-A2限制性肽GLS(SEQ ID NO:60)或覆盖了Env2结构域的肽池(长15个氨基酸,重叠11个氨基酸的池)的靶向包膜蛋白的特异性T细胞应答。无关肽和培养基被用作阴性对照。每个柱代表了独立的接种疫苗的小鼠并且阴影的柱代表了每组的平均值。结果显示为106脾脏细胞中观察到的斑的数目平均值,其获得自三个孔。如果斑数目高于92斑每106细胞(该截值由黑色虚线表示),即认为该应答为阳性的。
实施例
1.材料与方法
下文描述的构建根据Maniatis等人(1989,Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor NY)中详述的普通基因工程和分子克隆技术或根据产生商推荐(当使用商业化的试剂盒时)来完成。PCR扩增技术对本领域技术人员是已知的(见例如PCR protocols-A guide to methods and applications,1990,Innis,Gelfand.Sninsky和White,Academic Press出版)。携带氨苄青霉素抗性基因的重组质粒在补充了100μg/ml抗生素的琼脂或液体培养基中的大肠杆菌C600(Stratagene)中复制。重组痘苗病毒的构建根据上文引用的文献和Mackett等人(1982,Proc.Natl.Acad.Sci.USA 79,7415-7419)以及Mackett等人(1984,J.Virol.49,857-864)中本领域的常规技术进行。使用了大肠杆菌选择性基因gpt(黄嘌呤鸟嘌呤磷酸转移酶)以方便重组痘苗病毒的选择。
1.1.载体构建和产生
1.1.1.选择的抗原和HBV序列株系
下文中举例的载体已被工程化以表达聚合酶、核心多肽和包膜蛋白的免疫原性结构域。其都源自HBV株系Y07587,其序列已在国际性的数据库(GenBank Y07587)和不同的出版物中描述。其为血清型ayw的D基因型病毒。
核心多肽为野生型(aa 1-183)或缺失了氨基酸77至84的核心多肽(即,包含氨基酸1至76和85至183的核心,命名为core*)或C末端截短的多肽(1-148)或另外缺失了氨基酸77至84的C末端截短的核心(1-148)(即,包含氨基酸1至76和85至148的核心,命名为core*t)。
聚合酶多肽为野生型或缺少了前47个氨基酸的N末端截短的多肽(48-832)或另外在位点540(D突变为H)和718(E突变为H)突变的N末端截短的聚合酶(48-832)(给出的位置450和718是对于野生型聚合酶而言的)或与狂犬病毒糖蛋白的信号和跨膜结构域肽融合的截短的(48-832)和突变的聚合酶(D540H和E718H)(Pol*TMR)。
选择的包膜结构域为:S蛋白的氨基酸14至51的结构域(Env1)和HBs蛋白的氨基酸165至194的结构域(Env2)和HBs蛋白的氨基酸202至226的结构域(Env4)。
1.1.2.表达截短的和突变的HBV聚合酶(Pol*)的MVA TG17842的构建和产生。
编码修饰的HBV聚合酶多肽的核苷酸序列由Geneart公司使用合成的寡核苷酸和PCR产物合成。修饰的HBV聚合酶对应为了消除天然HBV聚合酶展现的RNase H和RTase活性而在540位(D突变为H)和718位(E突变为H)突变的HBV Y07587(SEQ ID NO:1)的聚合酶(得到SEQ ID NO:8所示的氨基酸序列和SEQ ID NO:22所示的核苷酸序列)。重组装的聚合酶序列随后克隆进质粒载体,得到了pGA15-pol(SEQ ID NO:27)。缺失存在于天然HBV聚合酶N末端的前47个氨基酸的截短版本通过PCR由pGA15-pol质粒扩增,其使用下述引物OTG19037(GAGCGATATCCACCATGAATGTTAGTATTCCTTGGAC)(SEQ ID NO:28)和OTG19038(GATCGCTAGCTCACGGTGGTCTCCATGCGAC)(SEQ ID NO:29)。得到的片段插入到MVA转移质粒的p7.5K启动子下游的NheI和EcoRV限制性酶切位点中(Cochran等人,1985,J.Virol.54:30),得到了pTG17842。突变的和截短的聚合酶在下文命名为pol*。
MVA转移质粒设计为允许将插入的核苷酸序列通过同源重组转移进MVA基因组的缺失III中。该质粒源自质粒pTG1E(在Braun等人,2000,Gene Ther.7:1447中描述),其中克隆了围绕MVA缺失III(Slitter和Moss,1992,Proc.Natl.Acad.Sci.USA 89:10847))的侧翼序列(BRG3和BRD3)。转移质粒还包含维多利亚多管发光水母(Aequorea victoria)增强的绿色荧光蛋白(eGFP基因,分离自pEGP-C1,Clontech)和大肠杆菌黄嘌呤鸟嘌呤磷酸转移酶基因(gpt基因)之间在早晚期痘苗病毒合成启动子p11K7.5控制下(由洛桑大学(University of Lausanne)的R.Wittek友好地提供)的融合。黄嘌呤鸟嘌呤磷酸转移酶的合成使GPT+重组MVA在含有霉酚酸、黄嘌呤和次黄嘌呤的选择性培养基上可形成斑(Falkner等人,1988,J.Virol.62,1849-54)并且eGFP使得可观察重组MVA斑。选择性的标记物eGFP-GPT放置于两个相同方向的同源序列之间。当克隆的选择完成后,选择标记物可通过若干允许eGFP-GPT重组MVA生长的无选择性的传代而容易地消除。
MVA TG17842病毒的产生通过在鸡胚原代成纤维细胞(CEF)中用MVA感染并用pTG17842(根据标准的磷酸钙DNA沉淀的)转染的同源重组来完成。病毒的选择通过含有霉酚酸、黄嘌呤和次黄嘌呤的选择性培养基存在下的三轮斑纯化完成。如上文提及,选择标记物随后通过在非选择性培养基上传代消除。通过PCR证实不存在亲代MVA污染。
HBV聚合酶表达的分析通过蛋白质印迹法完成。A549细胞(ATCC CCL-185)用的MVA TG17842(Pol*)以1MOI在存在或缺失添加如培养基的蛋白酶体抑制剂MG-132(10μM)下感染。24小时后,收获细胞。蛋白质印迹分析使用商业化的单克隆抗聚合酶抗体Hep B Pol(8D5,Santa Cruz,#sc-81591)。
1.1.3.表达融合于狂犬病毒包膜糖蛋白的膜锚定结构域截短的和突变的HBV聚合酶(Pol*TMR)的MVA TG17843的构建和产生。
HBV Pol*序列然后通过在其N末端融合肽信号(SS)和在其C末端融合膜锚定序列(TMR)修饰,膜锚定序列源自狂犬病毒的糖蛋白(ERA分离株;在GenBank N°M38452中描述的)。SS和TMR序列通过PCR从质粒pTG8042(在WO99/03885中描述)扩增,分别使用引物对OTG19045(SEQ ID NO:30)(GAGTGATATCCACCATGGTTCCTCAGGCTCTCCTG)/OTG19047(SEQ ID NO:31)(GTCCAAGGAATACTAACATTAATAGGGAATTTCCCAAAACACAATG)和OTG19049(SEQ ID NO:32)(GTCGCATGGAGACCACCGTATGTATTACTGAGTGCAGGG/OTG19050(SEQ ID NO:33)(GAGTGCTAGCTCACAGTCTGGTCTCACCC)。Pol*序列通过PCR从质粒pGA15-聚合酶扩增,其使用引物对OTG19046(SEQ ID NO:34)(GTTTTGGGAAATTCCCTATTAATGTTAGTATTCCTTGGACTC)/OTG19048(SEQ ID NO:35)(CTGCACTCAGTAATACATACGGTGGTCTCCATGCGACGTGC)。然后,SS-Pol*-TMR序列通过三重PCR重新装配,其使用下述引物OTG19045(SEQ ID NO:30)和OTG19050(SEQ ID NO:33)。得到的片段插入到痘苗病毒转移质粒的p7.5K启动子下游的NheI和EcoRV限制性酶切位点中(Cochran等人,1985,J.Virol.54:30),得到了pTG17843。
MVA TG17843病毒的产生通过如上文描述的同源重组在CEF进行。
Pol*-TMR表达的分析通过蛋白质印迹法进行。A549细胞用MVA TG17843以1MOI在存在或缺失添加如培养基的蛋白酶体抑制剂MG-132(10μM)下感染。24小时后,收获细胞。蛋白质印迹分析使用商业化的单克隆抗聚合酶抗体Hep B Pol(8D5,Santa Cruz,#sc-81591)进行。
1.1.4.表达缺失的核心多肽(Core*)的MVA TG17971的构建和产生。
Core*对应于缺失氨基酸77至84的HBV Y07587的核心序列(SEQ ID NO:2)。
Core*编码序列通过pGA4-Core质粒的双重PCR进行重构。该质粒由Geneart公司制造。其包含修饰的HBV核心基因全长编码序列,其由合成的寡核苷酸和/或PCR产物装配。编码序列的最后的两个密码子CAA TGT被修饰为CAG TGC以避免与Pol(SEQ ID NO:36)的序列同源性。
1至76位的核心序列通过PCR扩增,其使用下述引物OTG19290(SEQ ID NO:37)(GACTGTTAACCACCATGGACATTGATCCTTATAAAGAATTTG)和OTG19292(SEQ ID NO:38)(GTTGACATAACTGACTACCAAATTACCACCCACCCAGGTAG)。85至183位的核心序列通过PCR扩增,其用下述引物OTG19291(SEQ ID NO:39)(GTGGGTGGTAATTTGGTAGTCAGTTATGTCAACACTAATATG)和OTG19080(SEQ ID NO:61)(GACTCTCGAGTTAGCACTGAGATTCCCGAGATTG)。使用OTG19290(SEQ ID NO:37)和OTG19080(SEQ ID NO:61)以及两个后产生的扩增子进行双重PCR。得到的片段插入到痘苗病毒转移质粒的pH5R启动子下游的XhoI和HpaI限制性酶切位点中(Rosel等人,1986,J.Virol.60:436),得到了pTG17971。
MVA TG17971病毒的产生通过如上文描述的同源重组在CEF进行。
Core*表达的分析通过蛋白质印迹法进行。鸡胚成纤维细胞用MVA TG17971以0.2MOI感染。24小时后,收获细胞。蛋白质印迹分析使用商业化的单克隆抗核心抗体Hep B cAg(13A9)(Santa Cruz,#sc-23946)进行。
1.1.5.表达缺失的和截短的核心多肽(Core*t)的MVA TG17972的构建和产生。
Core*t对应于截短了148位之后的氨基酸并缺失了氨基酸77至84的HBV Y07587的核心序列(SEQ ID NO:2)。
Core*t编码序列通过对含有编码全长HBV核心基因的序列的pGA4-Core质粒的双重PCR进行重构,该基因由合成的寡核苷酸和PCR产物装配,除了编码序列的最后两个密码子CAA TGT被修饰为CAG TGC以避免与Pol(SEQ ID NO:36)的序列同源性。
1至16位的核心序列用PCR扩增,其使用下述引物OTG19290(SEQ ID NO:37)(GACTGTTAACCACCATGGACATTGATCCTTATAAAGAATTTG)和OTG19292(SEQ ID NO:38)(GTTGACATAACTGACTACCAAATTACCACCCACCCAGGTAG)。85至148位的核心序列通过PCR从pGA4-core扩增,其用下述引物OTG19291(SEQ ID NO:39)(GTGGGTGGTAATTTGGTAGTCAGTTATGTCAACACTAATATG)和OTG19299(SEQ ID NO:40)(GACTCTCGAGTTAAACAGTAGTCTCCGGAAGTG)。使用OTG19290(SEQ ID NO:37)和OTG19299(SEQ ID NO:40)进行双重PCR。得到的片段插入到痘苗病毒转移质粒的pH5R启动子下游的XhoI和HpaI限制性酶切位点中(Rosel等人,1986,J.Virol.60:436),得到了pTG17972。
MVA TG17972病毒的产生通过如上文描述的同源重组在CEF进行。
Core*t表达的分析通过蛋白质印迹法进行。鸡胚成纤维细胞用MVA TG17972以0.2MOI感染。24小时后,收获细胞。蛋白质印迹分析使用商业化的单克隆抗核心抗体Hep B cAg(13A9)(Santa Cruz,#sc-23946)进行。
1.1.6.表达融合于Env1免疫原性结构域的缺失的和截短的核心多肽(Core*t-env1)的MVA TG17993的构建和产生。
Core*t融合于HBs蛋白从氨基酸14至51的Env1结构域。
Core*t-Env1序列通过双重PCR进行重构。Core*t序列通过PCR从pTG17972扩增,其用下述引物OTG19371(SEQ ID NO:41)(GACGGGATCCACCATGGACATTGATCCTTATAAAGAATTTGG)和OTG19319(SEQ ID NO:42)(GCCTGCTTGCAGGACAACAGTAGTCTCCGGAAGTGTTG)。Env1序列通过PCR从pMK-C/E(SEQ ID NO:43)扩增,其使用下述引物OTG19318(SEQ ID NO:44)(CCGGAGACTACTGTTGTCCTGCAAGCAGGCTTCTTC)和OTG19320(SEQ ID NO:45)(GAGTCATTCTCGACTTGCGGCCGCTTACTGACCCAGGCAAACCGTGG)。双重PCR使用OTG19317(SEQ ID NO:41)和OTG19320(SEQ ID NO:45)进行。得到的片段插入到痘苗病毒转移质粒的pH5R启动子下游的BamHI和NotI限制性酶切位点中(Rosel等人,1986,J.Virol.60:436),得到了pTG17993。
为了说明性的目的,pMK-C/E质粒由Geneart制造并且含有由三种HBV包膜结构域插入进核心序列(SEQ ID NO:43)组成的嵌合序列。天然核心和包膜核苷酸序列为简并的以避免与HBV聚合酶序列的序列同源性以及起因于多聚T或多聚GC区段的序列不稳定性。另外,核心序列缺失了氨基酸77至84并在氨基酸148截短。选择的包膜结构域为:S蛋白的氨基酸14至51的结构域(Env1)和S蛋白的氨基酸165至194的结构域(Env2)和S蛋白的氨基酸202至226的结构域(Env4)。此三个结构域被分别插入核心序列的位置nt127、nt 22和nt 416。必须注意的是该序列向MVA载体的插入在表达细胞中导致细胞毒性,其强调了包膜-核心融合的设计是不容易的。
MVA TG17993病毒的产生通过如上文描述的同源重组在CEF中进行。
Core*t-env1表达的分析通过蛋白质印迹法进行。鸡胚成纤维细胞用MVA TG17993以0.2MOI感染。24小时后,收获细胞。蛋白质印迹分析使用商业化的单克隆抗核心抗体Hep B cAg(13A9)(Santa Cruz,#sc-23946)进行。
1.1.7.表达融合于Env1和Env2免疫原性结构域的缺失的和截短的核心多肽(Core*t-env1-env2)的MVA TG17994的构建和产生。
1.1.5.中描述的Core*t多肽然后与HBs蛋白质从氨基酸14至51(Env1)和从氨基酸165至194的(Env2)两个免疫原性结构域相融合。
Core*-Env1-Env2编码核苷酸序列通过三重PCR重新装配。Core*t序列通过PCR从pTG17972扩增,其使用下述引物OTG19371(SEQ ID NO:41)和OTG19319(SEQ ID NO:42)。Env1从pMK-C/E质粒中扩增,其使用下述引物OTG19318(SEQ ID NO:44)和OTG19322(SEQ ID NO:46)(GCGTGCGCTTGCCCACTGACCCAGGCAAACCGTGG)。Env2从pMK-C/E质粒中扩增,其使用下述引物OTG19321(SEQ ID NO:47)(CGGTTTGCCTGGGTCAGTGGGCAAGCGCACGCTTTAGC)和OTG19323(SEQ ID NO:48)(GAGTCATTCTCGACTTGCGGCCGCTTACACGCTCAGCCACACG GTTGG)。三重PCR使用OTG19317(SEQ ID NO:41)和OTG19323(SEQ ID NO:48)进行。得到的片段插入到痘苗病毒转移质粒的pH5R启动子下游的BamHI和NotI限制性酶切位点中(Rosel等人,1986,J.Virol.60:436),得到了pTG17994。
MVA TG17994病毒的产生通过如上文描述的同源重组在CEF中进行。
Core*t-env1-env2表达的分析通过蛋白质印迹法进行。鸡胚成纤维细胞用MVA TG17994以MOI 0.2感染。24小时后,收获细胞。蛋白质印迹分析使用商业化的单克隆抗核心抗体Hep B cAg(13A9,Santa Cruz,#sc-23946)进行。
1.1.8.表达CORE-Env1-Env2-Env4的腺病毒载体MVA TG17909的构建和产生。
编码CORE-Env1-Env2-Env4融合的合成基因(831核苷酸)通过双重PCR重构。CORE通过PCR从pGA4-Core中扩增,其使用下述引物OTG19152(SEQ ID NO:49)(GGGGGGCTAGCAAGCTTCCACCATGGACATTGATCCTTATAAA GAATTTG)和OTG19154(SEQ ID NO:50)(GAAAGAATCCAGCTTGCAGGACGCACTGAGATTCCCGAGATT GAG)。Env1-Env2-Env4通过PCR从pGA4-Env中扩增,其使用下述引物OTG19153(SEQ ID NO:51)(CTCAATCTCGGGAATCTCAGTGCGTCCTGCAAGCTGGATTCTT TC)和OTG19159(SEQ ID NO:52)(GAGTCATTCTCGACTTGCGGCCGCTTAGAT ATAAACCCACAAGC)。使用OTG19152(SEQ ID NO:49)和OTG19159(SEQ ID NO:52)进行双重PCR。得到的片段插入进腺病毒穿梭质粒的NheI和NotI限制性酶切位点中,其包含由腺病毒序列(分别为腺病毒核苷酸1-454和核苷酸3513-5781)围绕的CMV驱动表达框以允许载体基因组同源重组(Chartier等人,1996,J.Virol.70:4805)。得到E3(核苷酸28593-30464)和E1(核苷酸455-3512)缺失的的腺病毒载体pTG17909,其E1区域被5'至3'含有CMV即刻早期增强子/启动子、嵌合的人β珠蛋白/IgG内含子(如在Promega可获得的pCI载体中发现的)、编码CORE-Envl-Env2-Env4的序列和SV40晚期多聚腺苷化信号的表达框架代替。重组腺病毒通过将PacI线性化的病毒基因组转染进E1互补的细胞系中产生。病毒增殖、纯化和滴定依前述进行(Erbs等人,2000,Cancer Res.60:3813)。
融合蛋白的表达通过蛋白质印迹评估。106A549细胞(ATCC CCL-185)用Ad TG17909或作为阴性对照的空腺病毒以MOI 10或50感染48小时。收集细胞沉淀并用抗核心的小鼠单克隆抗体检测(C1-5,sc-23945,Santa Cruz)。
1.1.9.表达Pol*的腺病毒载体MVA TG17971的构建和产生。
编码Pol*(截短了除甲硫氨酸起始子外的前47个氨基酸(48-832)并在位置540(D突变为H)和718(E突变为H)突变(对于野生型聚合酶来说)的聚合酶蛋白质)的基因被插入腺病毒载体中。Pol基因(2364个核苷酸)通过PCR从pGA15-Pol(1.1.2中描述)扩增,其使用引物OTG19155(SEQ ID NO:53)(GGGGGGCTAGCAAGCTTCCACCATGAATGTTAGTATTCCTTGG ACTCATAAG)和OTG19156(SEQ ID NO:54)(GAGTCATTCTCGACTTGCGGCCGCTCACGGTGGTCTCCATGCG ACGTGC)。得到的片段插入进腺病毒穿梭质粒的NheI和NotI限制性酶切位点中,其包含由腺病毒序列(分别为腺病毒核苷酸1-454和核苷酸3513-5781)围绕的CMV驱动表达框以允许载体基因组同源重组(Chartier等人,1996,J.Virol.70:4805)。得到E3(核苷酸28593-30464)和E1(核苷酸455-3512)缺失的的腺病毒载体pTG17910,其E1区域被5'至3'含有CMV即刻早期增强子/启动子、嵌合的人β珠蛋白/IgG内含子(如在Promega可获得的pCI载体中发现的)、编码截短并突变的Pol的序列和SV40晚期多聚腺苷化信号的表达框架代替。重组腺病毒通过将PacI线性化的病毒基因组转染进E1互补的细胞系中产生。病毒增殖、纯化和滴定依前述进行(Erbs等人,2000,Cancer Res.60:3813)。
融合蛋白在腺病毒感染的细胞中的表达通过蛋白质印迹评估。A549细胞(106细胞)(ATCC CCL-185)用腺病毒Ad TG17910,以及作为阴性对照的空腺病毒以MOI 10或50感染48小时。收集细胞沉淀并用抗Pol的小鼠单克隆抗体(8D5,sc-81591,Santa Cruz)检测。
1.2.抗原免疫原性评估
抗原免疫原性通过HLA转基因小鼠接种免疫后的Elispot IFNγ和胞内细胞因子染色(ICS)测定体内评估。
1.2.1.小鼠模型
本研究所用的HLA-A2.1转基因小鼠由Pascolo等人(1997,J.Exp.Med.185:2043)描述。该小鼠具有H-2Db和小鼠β2-微球蛋白基因敲除并且表达转基因的单链I类组织相容性分子(HHD分子),其中人β2m的C末端共价连接于嵌合重链的N末端(HLA-A*0201αl-α2,H-2Dbα3跨膜和胞质结构域)。7-10周大的小鼠(雄性和雌性)被免疫。小鼠的平均体重为25-30克左右。
1.2.2.免疫方案
小鼠分为4组;组1用Ad TG17909(编码与env1、env2和env4免疫原性结构域融合的HBV核心)免疫,组2用Ad TG17910(编码截短并突变的Pol*)免疫,组3用两种载体免疫和组4用空腺病毒(Ad TG15149)免疫作为阴性对照。所有的动物通过在尾底部皮下注射免疫,组1和2动物接受一次皮下注射108IU每一腺病毒(TG17909或TG17910),组3一次皮下注射含108IU每一腺病毒的混合物(总共2.108IU:108IU Ad TG17909+108IU Ad TG17910)并且阴性对照接受一次皮下注射2.108IU的Ad TG15149。细胞免疫应答通过免疫后两周的IFGg Elispot和胞内细胞因子染色(ICS)测定评估。
1.2.3.肽
用于体外刺激细胞的肽为9-10个氨基酸的短肽,其已被描述或预测为HLA-A2限制性表位,或15氨基酸的长肽,其包含在覆盖了所有感兴趣的抗原的肽文库中。
对应于已描述或预测的HLA-A2聚合酶蛋白、核心蛋白或包膜结构域的限制性表位的短肽由Eurogentec(Belgium)合成,并且以10mM的浓度溶解于100%DMSO(sigma,D2650)。
覆盖了整个聚合酶、核心和包膜蛋白的肽文库由ProImmune(Oxford,United Kingdom)合成。聚合酶、核心和包膜文库由11个氨基酸重叠的15肽组成。每一粗肽以50mg/ml的浓度溶解于100%DMSO(sigma,D2650)中。对每一文库,汇总肽到每肽2mg/ml的浓度:
HBV Pol蛋白被来自聚合酶文库的24-25肽的8个池覆盖(池1:覆盖残基45-151的24肽;池2:覆盖残基140-251的24肽;池3:覆盖残基240-347的24肽;池4:覆盖残基337-447的24肽;池5:覆盖残基437-543的24肽;池6:覆盖残基533-639的24肽;池7:覆盖残基629-735的24肽;池8:覆盖残基725-832的25肽);
HBV核心蛋白被来自核心文库的21-22肽的2个池覆盖(池1:覆盖残基1-100的22肽;池2:覆盖残基89-183的21肽);
HBV包膜蛋白被来自包膜文库的6-10肽的3个池覆盖(池1:覆盖HBs残基9-59的10肽;池2:覆盖HBs残基157-194的9肽;池4:覆盖HBs残基193-226的6肽)。
1.2.4.IFNg Elispot测定
收集来自免疫后小鼠的脾脏细胞,并且裂解血红细胞(Sigma,R7757)。每孔2.105个细胞一式三份在包被有溶于MEM培养基(Gibco,22571)的抗鼠IFNγ单克隆抗体(BD Biosciences;10μg/ml,551216)的多孔滤膜板(Multiscreen plates)(Millipore,MSHA S4510)中培养40小时,该培养基补充含有10%FCS(Sigma,F7524or JRH,12003-100M)、80U/mL青霉素/80μg/mL链霉素(PAN,P06-Q7-100)、2mM L-谷氨酰胺(Gibco,25030)、1×非必需氨基酸(Gibco,11140)、10mM 4-羟乙基哌嗪乙磺酸(HEPES)(Gibco,15630)、1mM丙酮酸钠(Gibco,31350)和50μMβ-巯基乙醇(Gibco,31350),并且存在10单位/ml的重组小鼠IL2(Peprotech,212-12),单独作为阴性对照,或含有:
-10μM的一种HLA-A2限制性肽或无关肽,所述HLA-A2限制性肽存在于由Ad载体编码的HBV抗原中(聚合酶中的SLY,核心的FLP、ILC,包膜的VLQ、FLG和GLS);
-肽池,终浓度为每肽5μg/ml;
-5μg/ml的伴刀豆球蛋白A(Sigma,C5275),其用作阳性对照。
通过如前文描述(Himoudi等人,2002,J.Virol.76:12735)的Elispot(细胞因子特异的酶联免疫斑点)测定来定量产生IFNg的T细胞。阴性对照孔中斑的数目(对应于产生IFNg的T细胞)被从含有HBV肽的实验孔的斑数目中减去。结果显示为获得自三份孔的平均值。观察到应答的实验阳性阈值(或截值)通过计算对应于用单独培养基所观察到的斑的平均值+2个标准差的阈值而确定,报告为106个细胞。连接于CTL Elispot读数器的技术性的截值还定义为50斑点/106个细胞(超过该值读数器的CV系统性地低于20%)。在技术性的截值和为每一实验计算的实验阈值之间的最高值被考虑来定义每一实验的截值。通过使用Mann-Whitney检验进行Elispot反应的统计学分析。P值等于或低于0.05被视为有显著性的。
1.2.5.胞内细胞因子染色(ICS)测定
ICS在来自每一组的每一动物的脾脏细胞上进行。用裂解缓冲液(Sigma,R7757)裂解血红细胞后,在平底96孔板中每孔2×106个细胞在完全αMEM培养基(Gibco BRL,22571)中在存在10单位/ml的小鼠重组IL-2(Peprotech,212-12)下温育,单独作为阴性对照或含有每肽终浓度5μg/ml的肽池或含有10μM无关肽。立即加入终浓度1μl/ml的GolgiPlug(BD Biosciences,555029)5小时。然后,在V底96孔板中收获细胞并用1%FCS-PBS洗涤。使用针对CD3(仓鼠MAb抗-CD3e-PE,1/200稀释)、CD 8(大鼠MAb抗CD8a-APC,1/600稀释)和CD4(大鼠MAb抗-CD4-PerCP,1/600稀释)(都来自BD Biosciences,分别为553063、553035和553052)的单克隆抗体,在50μl 1%FCS-PBS中室温15分钟进行染色。洗涤后,用Cytofix/Cytoperm固定并透明化细胞,并且用Perm/Wash溶液(BD Biosciences,554714)洗涤。然后,加入抗鼠IFNg-PE抗体(BD Biosciences,554412557724)和抗鼠TNFa-Alexa488抗体(BD Biosciences,557719)或抗鼠IFNg-PE抗体(BD Biosciences,554412557724)和抗鼠IL2-Alexa488抗体(BD Biosciences,557719),室温15分钟,并且在用Perm/Wash洗涤后,细胞在1%FCS-PBS中重悬并通过使用FacsCalibur(Becton Dickinson)的流式细胞法分析。CD3e+、CD8a+细胞或CD3e+、CD4+细胞被门控以确定IFNg+CD8+或IFNg+CD4+T,或TNFα+CD8+或TNFa+CD4+T,或IL2+CD8+或IL2CD4+T,或IFNg+TNFα+CD8+或IFNg+TNFα+CD4+或IFNg+IL2+CD8+或IFNg+IL2+CD4+T细胞群的百分比。在仅含培养基中获得的百分比被认为是背景。
1.2.6.体内CTL测定
体内CTL测定如已描述(Foumillier等人,2007)的进行。脾细胞悬液从同系小鼠脾脏获得并且在血红细胞裂解后调整至20×106cells/ml。三分之一的细胞与HBV特异性肽之一温育,第二个三份之一的细胞与另一HBV肽温育,都以10μM终浓度于37℃,1小时,而最后的部分保留为不脉冲处理的。向未脉冲处理的细胞加入16μM(高CFSE),向ILC或VLQ肽脉冲处理的细胞加入4μM(中CFSE)并向SLY或FLP肽脉冲处理的细胞加入1μM(低CFSE)的5(6)-羧基荧光素乙酰乙酸琥珀酰亚胺酯(CFSE)(分子探针,C1157),10分钟。用PBS洗涤后,3个群体(未脉冲处理的、ILC和SLY肽脉冲处理的细胞或未脉冲处理的、FLP和ILC肽脉冲处理的细胞)被混合,并且总的30.106个细胞通过眼窝静脉注射入麻醉后的小鼠(使用氯胺酮-赛拉嗪-PBS混合(氯胺酮,Virbac,Centravet KET204,终浓度25mg/ml;赛拉嗪盐酸盐Rompun Bayer,Centravet,终浓度5mg/ml))。因此,低CFSE和中CFSE群体代表应被细胞毒性T细胞裂解的特异性靶而高CFSE群体为内部对照,其允许标准化测定。来自受体小鼠的脾细胞在24小时后由流式细胞术分析以检测CFSE标记的细胞。门控淋巴细胞(SSC/FSC)后,基于事件数目/CFSE荧光(FL1)进行第二门控,其显示3个峰,第一个对应低CFSE细胞、第二个对应中CFSE细胞和第三个对应高CFSE细胞。对每一动物,计算CFSE+肽脉冲处理的靶与CFSE+未脉冲处理的靶之间的比值(R=低CFSE细胞数目/高CFSE细胞数目)。两只幼鼠用于确定R参考值。对每一动物的特异性裂解的百分比通过下述公式确定:
%裂解=(1-R小鼠/R参考)×100
如果特异性裂解的百分比高于10%(截值),该应答被认为是阳性的。
2.结果
2.1通过病毒载体表达抗原
2.1.1.由腺病毒构建体Ad TG17909和Ad TG17910表达抗原
core-envl-env2-env4融合蛋白的表达通过蛋白质印迹评估。A549细胞(106细胞)用腺病毒Ad TG17909或作为阴性对照的空腺病毒以MOI 10或50感染48小时。收集细胞沉淀并用抗核心的小鼠单克隆抗体检测(C1-5,sc-23945,Santa Cruz)。如图1A中表明,收集于用Ad TG17909感染的细胞的样品显示具有预期分子量(31.6kDa)的主要条带。
Ad TG17910感染A549细胞后的Pol*多肽的表达通过蛋白质印迹评估。然后收集细胞沉淀并用抗聚合酶的小鼠单克隆抗体(8D5,sc-81591,Santa Cruz)检测。如图1A中表明,从用Ad TG17910感染的细胞收集的样品显示了具有预期分子量(88.5kDa)的主要条带与某些子产物(部分的聚合酶蛋白质)。
2.1.2.由MVA构建体表达抗原
Pol*、Pol*TMR、Core*t、Core*tEnv1、Core*t-Env1-Env2表达的分析通过蛋白质印迹进行。A549细胞或CEF分别用MVATG17842、MVATG17843、MVATG17971、MVATG17972、MVATG17993和MVATG17994以MOI 1或0.2在存在或不存在添加至用于MVATG17842和MVATG17843的生长培养基的蛋白酶体抑制剂MG-132的情况下感染。24小时后收集细胞。
对于MVATG17842,使用商品化的单克隆抗聚合酶抗体Hep B Pol(8D5,Santa Cruz,#sc-81591)进行蛋白质印迹分析。如图1B所示,仅在MG-132存在下检测到具有表观分子量88.5kDa的蛋白的表达。该条带具有Pol*蛋白预期的分子量。
对于MVATG17843,使用商品化的单克隆抗聚合酶抗体Hep B Pol(8D5,Santa Cruz,#sc-81591)进行蛋白质印迹分析。如图1B所示,在MG-132存在或不存在下检测到具有表观分子量98.2kDa的蛋白的表达。该条带具有Pol*-TMR蛋白预期的分子量。应注意在MG-132存在下检测到更多的产物以及额外的超过200kDa的高分子量产物。
对于MVATG17971,使用商品化的单克隆抗核心抗体Hep B cAG(13A9,Santa Cruz,#sc-23946)进行蛋白质印迹分析。如图1B所示,检测到具有表观分子量21kDa的表达,对应于Core*预期的分子量。
对于MVATG17972,使用商品化的单克隆抗核心抗体Hep B cAG(13A9,Santa Cruz,#sc-23946)进行蛋白质印迹分析。如图1B所示,检测到具有表观分子量15.8kDa的表达,对应于Core*t预期的分子量。
对于MVATG17993和MVATG17994,使用商品化的单克隆抗核心抗体Hep B cAG(13A9,Santa Cruz,#sc-23946)进行蛋白质印迹分析。如图1B所示,分别检测到具有表观分子量19.9kDa和23.4kDa的蛋白表达。该条带具有Core*t-Env1和Core*t-Env1-Env预期的分子量。
2.2从腺病毒载体AdTG17909和Ad17910表达的抗原的免疫原性
在单独用Ad TG17909或Ad TG17910,或2种腺病毒组合免疫的HLA-A2转基因小鼠中,评估由腺病毒载体表达的HBV多肽的免疫原性。一次皮下注射后所诱导的特异性T细胞应答通过Elispot IFNg,ICS和体内细胞裂解测定评估,其使用了存在于聚合酶、核心或包膜结构域或/和覆盖了感兴趣的HBV抗原的重叠肽池中的已知(描述为患者中特异性T细胞应答的靶标)HLA-A2表位。
2.2.1.通过Elispot测定评估产生HBV特异性IFGg的细胞
Elispot IFNg测定显示Ad TG17910能够诱导产生特异于HLA-A2限制性表位(SLYADSPSV)(SEQ ID NO:55,定位于HBV聚合酶内位置816-824)(图2A)的IFNg的细胞。用Ad TG17909免疫还导致了高频率诱导产生特异于2种核心HLA-A2限制性表位(在位置18-27的FLPSDFFPSV(SEQ ID NO:56)和位置99-108的ILCWGELMTL(SEQ ID NO:57))的以及三种包膜HLA-A2限制性表位(在位置14-22的VLQAGFFLL(SEQ ID NO:58)和位置45-49的FLGGTTVCL(SEQ ID NO:59),两者都存在于Env1中,和位置185-194的GLSPTVWLSV(SEQ ID NO:60),存在于Env2中)的IFNg的细胞(图2B和C)。用Ad TG17909和Ad TG17910的混合物免疫还可诱导类似水平的产生特异性IFNg的细胞,其靶向3种抗原的相同表位,即存在于聚合酶中的SLY表位、核心蛋白质中的FLP和ILC表位,以及包膜结构域中的3个表位(VLQ、FLG和GLS)(图2A、B和C)。用单一Ad或两者混合物免疫后检测到T细胞应答的频率为类似的,显示了由所描述的载体表达的3种抗原之间没有主要的优势免疫表位。
2.2.2.通过胞内染色评估产生HBV特异性IFNg/TNFα的细胞
能够单独产生IFNg或产生IFNg+TNFα靶向存在于聚合酶(SLY)中、核心中(FLP和ILC)和包膜结构域中(VLQ、FLG和GLS)的HLA-A2限制性表位的CD8T细胞的数目通过ICS测定评估。所有这些表位是双重和简单分泌细胞的靶。结果示于图3。用Ad TG17909单独或与Ad TG17910组合免疫的动物装载了大致相当的核心和包膜特异的CD8T细胞应答(相同百分比的特异性CDS T细胞,其如图3B和3C所示,在FLP、ILC、VLQ、FLG和GLS肽重新刺激后产生IFNg或IFNg+TNFα)。另一方面,考虑到聚合酶特异性的CDS T细胞应答(SLY表位),在用表达Pol*的Ad TG1710处理的小鼠(Figure 3A)中,以及用Ad TG17910和Ad TG17909混合物免疫的小鼠(尽管水平较低)(Figure 3C)中检测到非常高百分比的产生IFNg或IFNg+TNFα的CD8+细胞。
2.2.3.通过胞内染色评估用腺病毒载体混合物免疫后产生HBV特异性IFNg/TNFα的CD8和CD4T细胞
能够单独产生IFNg或产生IFNg+TNFα或IFNg+IL2靶向存在于聚合酶(SLY)中、核心中(FLP和ILC)和包膜结构域中(VLQ、FLG和GLS)或覆盖了核心蛋白质和包膜结构域的重叠肽池的HLA-A2限制性表位的CD8和CD4T细胞的百分比通过ICS测定评估。所有被测试的HLA-A2限制性表位为单一或双重分泌细胞(IFNg和IFNg+TNFa)的靶标,而一些重叠肽池也是单一或双重分泌细胞(IFNg和IFNg+TNF和IFNg+IL2)的靶标。结果示于图4。5只HLA-A2转基因小鼠用Ad TG17909和Ad TG17910混合免疫而3只HLA-A2转基因小鼠用Ad TG15149(阴性对照)免疫。用Ad TG15149免疫的小鼠不显示HBV特异性的T细胞应答(数据未显示)。用Ad TG17909组合Ad TG17910免疫的动物显示了强烈的特异的靶向HBV抗原的CD8T细胞免疫应答(图4A),其具有高百分比的单一(IFNg)和双重(IFNg+TNFα)产生特异于存在于聚合酶、核心和包膜结构域并且特异于“核心1”肽池和覆盖了Env1和Env2结构域的肽池的HLA-A2表位的细胞。如图4B和4C中说明,这些免疫接种的小鼠还显示了CD4T细胞应答,其特异于HBV抗原,尤其是单一(IFNg)和双重(IFNg+TNFa和IFNg+IL2)产生特异于“核心2”肽池和覆盖Env2的肽池的细胞。
2.2.4.通过体内CTL测定测量体内诱导的细胞裂解
腺病毒载体Ad TG17909和Ad TG17910体内诱导针对存在HBV HLA-A2表位的细胞裂解的能力通过体内CTL试验评估。四种HLA-A2表位被测试,分别为SLY(聚合酶)、FLP和ILC(核心)以及VLQ(包膜结构域)。6只动物用Ad TG17909+Ad TG17910混合免疫而2只动物用Ad TG15149(阴性对照)免疫。每组的一半用于测试其体内裂解用SLY肽脉冲处理后的细胞和用ILC肽脉冲处理后的细胞的能力。另一半用于测试疫苗接种的动物体内裂解用FLP肽脉冲处理后的细胞和用VLQ肽脉冲处理后的细胞的能力。结果示于图5。
如预期的,用Ad TG15149免疫的小鼠检测不到HBV特异性体内细胞裂解(数据未显示)。针对2个核心表位FLP和ILC的体内细胞裂解在用Ad TG17909+Ad TG17910免疫的小鼠中是微弱的。然而,相反地,用Ad TG17909和Ad TG17910混合物免疫的动物显示了强烈的针对聚合酶表位SLY(图5A)和Env1表位VLQ(图5B)的体内细胞裂解,其在两种情况都达到了高于50%的特异性裂解。
有趣的是,当共-注射时,表达聚合酶、核心和包膜部分的Ad载体的组合允许诱导靶向3种HBV抗原的特异性T细胞应答。被诱导的T细胞能够产生一或两种细胞因子并体内裂解装载了一些HBV肽的细胞。所有这些数据一起证实了所描述的组合物的免疫原性活性和当通过Ad载体化时,其诱导CD8和CD4T细胞应答的能力。
2.3.由MVA载体MVA TG17842、MVA TG17843、MVA TG17971、MVA TG17972、MVA TG17993和MVA TG17994表达的抗原的免疫原性
在用实施例1.1.2-1.1.7中描述的MVA载体(MVA TG17842,MVA TG17843,MVA TG17971或MVA TG17972单独的)或用2种MVA混合物(MVA TG17843+MVA TG17972,MVA TG17843+MVA TG17993或MVA TG17843+MVA TG17994)免疫后的HLA-A2转基因小鼠中评估基于MVA的组合物的免疫原性活性小鼠用间隔一周的三次皮下注射免疫,并且特异性T细胞应答通过Elispot IFNg和ICS评估,其使用上述的存在于聚合酶、核心或包膜结构域或/和覆盖了感兴趣的HBV抗原的重叠肽池中的已知(描述为患者中特异性T细胞应答的靶标)HLA-A2表位。
2.3.1.用表达聚合酶的MVA免疫后通过Elispot测定评估产生HBV特异性IFGγ的细胞
用表达截短的和突变的聚合酶抗原的MVA TG17842或表达膜定位版本的相同截短的和突变的聚合酶的MVA TG17843或MVA N33.1(阴性对照)免疫三只小鼠。通过使用SLY HLA-A2限制性表位和覆盖了聚合酶的肽池的IFNg Elispot测定评估聚合酶特异的T细胞应答。对用MVA N33.1和用MVA TG17842免疫的小鼠,无HBV特异的T细胞应答被检测到(数据未显示)。然而,如图6A所示,产生IFNg的细胞在用MVA TG17843免疫后被诱导,其对HLA-A2限制性表位SLY和覆盖了聚合酶的C末端部分的肽池8特异(在测试实验条件下,不能检测到针对另外的肽池1-7的特异性应答)。这些数据突出了至少在基于MVA的组合物中将聚合酶表达为膜锚定抗原的好处。
2.3.2.用表达核心的MVA免疫后通过Elispot测定评估产生HBV特异性IFGγ的细胞
用表达缺失了残基77-84的核心部分的MVA TG17971或表达其截短版本(从残基149截短C末端)的MVA TG17972或MVA N33.1(阴性对照)免疫八只小鼠。通过使用了HLA-A2限制性表位(FLP和ILC肽)和上述核心1和核心2肽池的IFNg Elispot测定确定核心特异的T细胞应答。如图6B表明,用MVA TG17971免疫能够诱导零星的针对HLA-A2限制性的FLP和ILC肽,以及覆盖了核心抗原的两个肽池(Figure 6B)的T细胞应答。在测试实验条件下,在用MVA TG17972免疫的小鼠中无HBV特异的T细胞应答被检测到(数据未显示)。
2.3.3.用MVA组合免疫后通过Elispot测定评估产生HBV特异性IFGγ的细胞
用MVA TG17843和MVA TG17972或MVA TG17993和MVA TG17994混合物免疫3只小鼠,通过使用上述肽的Elispot IFNg测定评估HBV特异性T细胞应答。
-在绝大多数用MVA TG17843+MVA TG17972(如图7A所示2/3动物中阳性应答)、MVA TG17843+MVA TG17993(如图7B所示3/3动物中阳性应答)和MVA TG17843+MVA TG17994(如图7C所示2/3动物中阳性应答)组合接种免疫的动物中检测到了聚合酶特异的T细胞应答。产生IFNg的细胞的频率似乎与单独注射MVA TG17843(图6A)时相似,其表明载体组合没有不利于诱导免疫应答。
-使用HLA-A2或肽池在任何接种免疫后的动物中测试的实验条件下,不能检测到核心特异的应答(数据未显示)。
-在使用Env1 HLA-A2或肽池在任何用含MVA TG17993的组合物接种免疫的动物中测试的实验条件下,不能检测到包膜特异的应答(数据未显示)。
-如图7C中表明,在3只用含MVA TG17994的组合物免疫的动物中,2只检测到了Env2特异的T细胞应答,其被导向针对HLA-A2限制性的GLS表位和覆盖Env2的肽池(在这些实验条件下,没有观察到检出针对Env1结构域的T细胞应答;数据未显示)。
在相同的条件下进行的ICS测定证实了Elispot测定中观察到的结果(数据未显示)。
有趣的是,当共-注射时,表达聚合酶、核心和包膜部分的MVA载体的组合允许诱导靶向聚合酶和env2抗原的特异性T细胞应答。
所有这些数据一起证实了所描述的组合物的免疫原性活性和其诱导针对主要HBV抗原的T细胞应答的能力。
- 还没有人留言评论。精彩留言会获得点赞!