用脂质体伊立替康治疗小细胞肺癌的制作方法
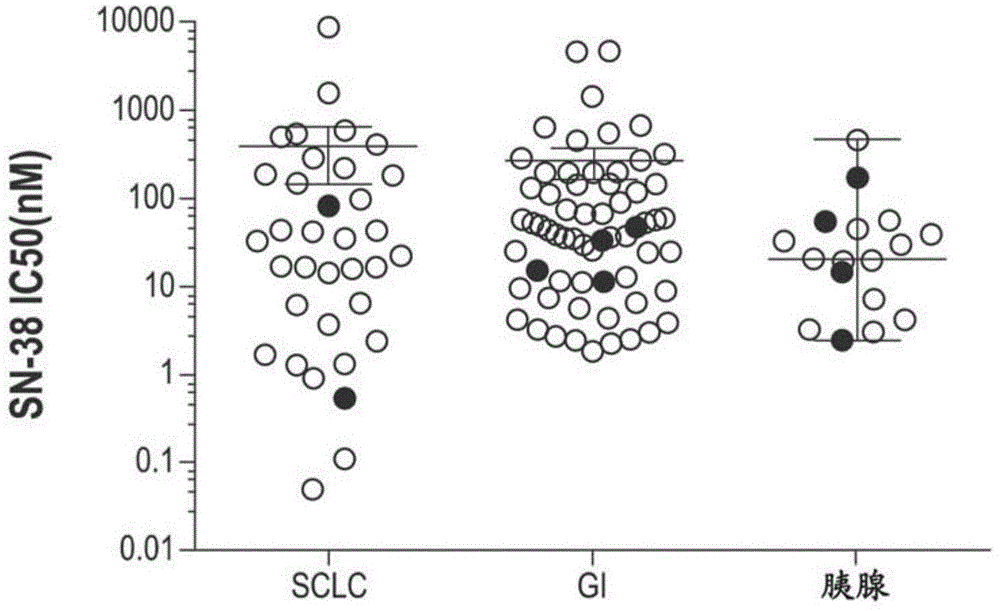
本申请要求美国临时申请号62/337,961(2016年5月18日提交)、美国临时申请号62/345,178(2016年6月3日提交)、美国临时申请号62/362,735(2016年7月15提交)、美国临时申请号62/370,449(2016年8月3日提交)、美国临时申请号62/394,870(2016年9月15日提交)、美国临时申请号62/414,050(2016年10月28日提交)、美国临时申请号62/415,821(2016年11月1日提交)、美国临时申请号62/422,807(2016年11月16日提交)、美国临时申请号62/433,925(2016年12月14日提交)、美国临时申请号62/455,823(2017年2月7日提交)以及美国临时申请号62/474,661(2017年3月22日提交)的优先权权益,每件所述美国临时申请以引用的方式整体并入本文。本发明涉及对诊断患有小细胞肺癌(sclc)的患者的治疗,包括在用基于铂的疗法治疗后具有sclc疾病进展的患者。发明背景小细胞肺癌(sclc)是在肺内最常发生的高度恶性癌症,虽然它也会在其他身体部位发生。sclc通常以大型、快速发展的病变形式存在,该病变起于位于中心的气管支气管气道并且入侵纵隔。典型地,患者表现为咳嗽或呼吸困难、哮鸣和/或胸痛。在高达三分之一的患者中出现体重减轻、疲劳和厌食。在诊断时,三分之二的sclc患者具有一种或多种临床可检测的远端转移。sclc的最初(一线)治疗可包括施用基于铂的疗法,诸如顺铂或卡铂组合依托泊苷或伊立替康的4-6个治疗周期。基于(owonikoko,tk等人,jthoraconcol.2012年5月;7(5):866-72),已报道sclc疾病进展(一线疗法之后)后的当前后续(二线)疗法提供约7.7个月(敏感患者)和5.4个月(疑难患者)的总存活期。一种二线疗法为在某些方案中报道的施用拓扑替康(例如,hycamtin,盐酸拓扑替康注射液)来提供7.8个月(敏感患者为9.9个月,疑难患者为5.7个月)的总存活期(owonikoko,tk等人,jthoraconcol.2012年5月;7(5):866-72)。例如,在三(3)周治疗周期中在第1-5天施用一次1.5mg/m2拓扑替康进行二线sclc治疗,提供约7%-24%的总响应率、约3.1-3.7个月的无进展存活期(pfs)以及5.0-8.9个月的总存活期(os)(伴随有3级或以上嗜中性白血球减少症比率28%-88%和3级或以上腹泻小于约5%)(pmid16481389、17135646、17513814、9164222、10080612、25385727)。另一种报道的sclc二线疗法为每三(3)周施用一次300mg/m2的非脂质体伊立替康,提供0-33%的混合型总响应率、1.7-2.8个月的pfs以及4.6-6.9个月的os(伴随有3级或以上嗜中性白血球减少症比率21%-23%和3级或以上腹泻小于约0-13%)(pmid19100647、1321891)。伊立替康为治疗sclc的活性剂(例如,列于nccn和esmo指南中),但它在美国或欧洲未被批准。此外,在iii期注册引导的研究中,在一线sclc上,在与铂组合的情况下它失败了(pmid:16648503)。至今还没有靶向治疗成功地显著改善患者疗效。因此,迫切需要对用于这种疾病的新型治疗的研究。发明概述本公开文本提供了通过施用治疗有效量的脂质体伊立替康来治疗在基于铂的疗法后疾病进展之后患有小细胞肺癌的患者的方法。具体地说,可每两周一次地向在基于铂的疗法后疾病进展之后诊断患有sclc的患者施用脂质体伊立替康诸如mm-398(onivyde)。在一些实施方案中,可向在基于铂的一线化学疗法(卡铂或顺铂)、免疫疗法和/或包括基于铂的化学疗法的化放疗以用于治疗局限期或广泛期sclc之时或之后诊断有sclc疾病进展的患者施用脂质体伊立替康。可用抗肿瘤疗法每两周一次地治疗在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者,该抗肿瘤疗法由包囊在伊立替康脂质体中的单一剂量的90mg/m2伊立替康(游离碱)组成。在另一个实施方案中,已知针对ugt1a1*28等位基因为纯合的并且在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者可用每两周施用一次的抗肿瘤疗法来治疗,该抗肿瘤疗法由包囊在脂质体中的单一减少剂量(例如,50-70mg/m2,包括50mg/m2或70mg/m2)的伊立替康(游离碱)组成。在另一个实施方案中,在诊断患有小细胞肺癌(sclc)后、并且在遵循用于sclc的基于铂的疗法疾病进展后接受脂质体伊立替康的同时或之后先前已经历3+级不良事件的人类患者可用每两周施用一次的抗肿瘤疗法来治疗,该抗肿瘤疗法由包囊在脂质体中的单一减少剂量(例如,50-70mg/m2,包括50mg/m2或70mg/m2)的伊立替康(游离碱)组成。脂质体伊立替康可为伊立替康的药学上可接受的脂质体制剂,其包含直径为约100nm的递送形式的伊立替康,诸如脂质体伊立替康(实施例1)。各种合适的脂质体伊立替康制剂可如本文所公开进行制备(实施例8)。优选地,脂质体伊立替康为产品mm-398(实施例9)。在本公开文本中,mm-398与mm-398脂质体伊立替康可互换地使用。附图简述图1为针对sclc、胃肠癌和胰腺癌细胞系(实施例2)绘制的示出了来自sanger数据库的sn-38药物敏感数据的图形。图2a和2b为在各种sn-38浓度下,经过88小时在incucyte仪器上获取的dms114和nci-h1048sclc细胞系的动态生长曲线。图3为示出了在sclc的dms114异种移植模型中mm-398的抗肿瘤活性的图形。在第23天开始以10或20mg/kg的三水盐酸伊立替康静脉内施用mm-398且每周给予持续4周,并且与盐水对照(黑色圆圈)比较。图4为在napoli-1的mm-398+5fu/lv组中超过阈值的未包囊sn-38(usn38)时间的四分位数的总存活率的kaplan-meier曲线图。q1-q4表示超过阈值的usn38时间的四分位数。q1表示最短时间并且q4表示最长时间。图5为示出了针对napoli-1中的mm-398+5fu/lv组,最佳响应与usn38>0.03ng/ml的持续时间之间的相关性的图形。图6a为示出了在用mm-398治疗的患者中,未包囊sn-38cmax与嗜中性白血球减少症≥3级之间的相关性的图形。图6b为示出了在用mm-398治疗的患者中,总伊立替康cmax与腹泻≥3级之间的相关性的图形。图7a为示出了羧酸酯酶(ces)活性的图形;如通过在sclc小鼠异种移植模型中施用后24h的肿瘤cpt-11所评价,增加的肿瘤sn-38水平与增加的肿瘤沉积相关。图7b为示出了羧酸酯酶(ces)活性的图形;sclcpdx肿瘤具有比得上其中伊立替康有活性的其他适应症的ces活性。图7c为示出了细胞敏感性的图形;nal-iri肿瘤沉积与h1048sclc细胞中的sn-38敏感性范围一致。图7d为示出了细胞敏感性的图形;topo1抑制剂的细胞毒性随着暴露而增加。图7e为示出了拓扑替康施用严重地受毒性限制,从而与安能得(onivyde)介导的延长sn-38暴露相比限制topo1的持续抑制的图表。图8a示出在sclc的dms-53异种移植模型中mm-398的抗肿瘤活性。图8b示出在sclc的hcl-h1048异种移植模型中mm-398的抗肿瘤活性。图8c示出在sclc的h841大鼠原位异种移植模型中的大鼠存活百分比,这些大鼠在接种后的数天里用对照、安能得(30或50mg/kg盐)、伊立替康(25mg/kg)或拓扑替康(4mg/kg)治疗。图9a和9b为示出了用mm-398和非脂质体伊立替康治疗的sclc异种移植模型中的肿瘤代谢物水平的图形。在注射后24小时,与30mg/kg(盐)的非脂质体伊立替康相比,针对用16mg/kg(盐)的mm-398治疗的小鼠,肿瘤中的(图9a)cpt-11和(图9b)活性代谢物sn-38显著更高。图10a和10b为示出了在从未接受治疗的sclc异种移植模型中,nal-iri优于所有比较治疗组的图形:图10a为示出了从未接受治疗的sclc模型ncl-h1048(nal-iri16mg/kg临床等效剂量,每bsa=1x~90mg/m2mm-398;拓扑替康0.83mg/kg/周,第1-2天,q2w临床等效剂量,每bsa=1x~1.5mg/m2拓扑替康,q3w,第1-5天)的图形;图10b示出完全响应(nal-iri)的数量。nci-h1048为化学敏感模型(由sclc的胸腔积液转移确立的)。所有nal-iri治疗的动物在2-3个剂量后具有完全响应(cr)-但在早期时间点观察到剂量响应。iri治疗的动物在初始响应于治疗后继续进展;而nal-iri治疗的动物至今保持cr。图11a和11b描述了通过用卡铂+依托泊苷治疗所创建的2lsclc异种移植模型。图11c为示出了从未接受治疗的sclc模型ncl-h1048(拓扑替康0.83mg/kg/周,第1-2天,q2w临床等效剂量,每bsa=1x~1.5mg/m2拓扑替康,q3w,第1-5天;1线依托泊苷(25mg/kg)和卡铂(30mg/kg)临床等效剂量,每bsa=1x~100mg/m2依托泊苷d1-3+auc6卡铂d1,q4w)的图形;图11b为1线和2线治疗的示意图。当在临床相关剂量下进行拓扑替康治疗时,1线方案产生类似的抗肿瘤活性(基于bsa/bw计算)。在3个周期的1线治疗后,随机化小鼠用于进一步的2线治疗。图12为示出了nal-iri仍有效于铂治疗的sclc肿瘤并且优于拓扑替康和伊立替康:2lsclc模型:ncl-h1048的图形。在铂治疗的sclc肿瘤中:nal-iri保持活性的并且倾向于完全响应;iri治疗是活性的,但在第3个周期后一些肿瘤倾向于再生长;在1-2个周期后拓扑替康(2x临床相关剂量)似乎是活性的,但在第3个剂量后快速进展;依托泊苷+卡铂到第5个周期是不耐受的。图13a和13b为示出了在另一种sclc异种移植模型(dms-114)中,nal-iri也优于拓扑替康和伊立替康的图形;图13a为示出了dms-114sclc小鼠异种移植物(皮下)的图形;图13b为示出nal-iri(第74天)肿瘤体积变化的图表。nal-iri优于临床相关剂量的伊立替康和拓扑替康。在早期,sclc肿瘤响应于伊立替康,但在2-3个周期后变得响应较少。图14a-14c为示出了用top1抑制剂治疗的sclc肿瘤仍对nal-iri有响应的图形。图14a.dms-114:从未接受治疗;图14b.dms-114:拓扑替康治疗的;图14c.dms-114:伊立替康治疗的。用拓扑替康治疗的dms114肿瘤对nal-iri(16mg/kg)有响应,但对伊立替康(33mg/kg)没有响应。图15a-15c为示出了暴露的持续时间可能对top1抑制剂活性非常重要的图形。图15a为dms-114sclc小鼠异种移植物(皮下);图15b为假设的肿瘤暴露;图15c为ncl-h1048小鼠异种移植物。在相同的剂量强度下,与分次的拓扑替康(第1天和第2天)相比,单次(在第1天给予的)拓扑替康具有较小的抗肿瘤活性。这可能指示,超过治疗阈值的top1抑制剂的延长暴露比高cmax更有益,因为伊立替康是前药(cpt-11),活性代谢物sn-38也可具有比拓扑替康更长的持续时间。图16a-16d示出ncl-h1048sclc小鼠异种移植物(皮下)图16a.肿瘤体积;图16b.存活率;图16c.体重变化;图16d.在第98天时的响应。图17a-7c示出ndmc-53sclc小鼠异种移植物(皮下)图17a.肿瘤体积;图17b.存活率;图17c,用对照、nai-iri(16mg/kg盐)或拓扑替康(0.83mg/kg/周,第1-2天)接种后第98天时的响应。图18a和18b为示出了在bxpc-3小鼠异种移植肿瘤中,nal-iri增加暴露并且持续递送伊立替康和sn-38(活性代谢物)的图形:图18a.血浆;图18b.肿瘤。图19为示出了在sclc的临床前模型中,nal-iri有效地向肿瘤递送伊立替康的图形。图20a和20b为示出了用top1抑制剂治疗的sclc肿瘤仍对nal-iri有响应的图形:图20a.dms-114:拓扑替康治疗的;图20b.dms-114:从未接受治疗。用拓扑替康治疗的dms114肿瘤对nal-iri(16mg/kg)有响应,但对伊立替康(33mg/kg)没有响应。图21a和21b为示出了在2lsclc模型:ncl-h1048中,nal-iri仍有效于铂治疗的sclc肿瘤并且优于拓扑替康和伊立替康的图形。图21a示出肿瘤体积的变化;图21b为存活率图形。图22a-22d为示出了在ht29crc异种移植模型-mm-39840mg/kg中mm-398具有改善的循环和肿瘤循环的临床前证据的图形:图22acpt-11血浆(持续血浆水平),图22b.sn-38血浆(中等持续血浆水平),图22ccpt-11肿瘤(持续肿瘤内水平),图22dsn-38肿瘤(对sn38的肿瘤内活化增强)。图23a-23f为示出了nal-iri具有比伊立替康和拓扑替康更大的抗肿瘤活性的图形。nod/scid小鼠具有皮下(图23a)dms-53、(图23b)dms-114或(图23c)nci-h1048。用静脉内nal-iri(16mg/kg;三角形)、静脉内伊立替康(33mg/kg;菱形)、腹腔内拓扑替康(0.83mg/kg/周,第1-2天;正方形)或赋形剂对照(圆形)治疗sclc异种移植肿瘤。针对dms-114和nci-h1048,所有组均n=10;针对dms-53,对于对照、拓扑替康和nal-iri分别为n=4、5和5。用静脉内nal-iri(16mg/kg;三角形)、静脉内伊立替康(33mg/kg;菱形)、腹腔内拓扑替康(0.83mg/kg/周,第1-2天;正方形)或赋形剂对照(圆形)治疗具有皮下人源性异种移植物(图23d)lun-182、(图23e)lun-081和(图24f)lun-164的balb/c裸鼠。针对所有pdx模型,所有组n=5。垂直的虚线指示每周给药的开始并且误差线指示平均值的标准误差。发明详述mm-398为提供sn-38的持续肿瘤暴露并且因此提供优于非脂质体伊立替康的某些优点的脂质体包囊伊立替康。在患有胰腺癌的患者中批准的mm-398方案为与5-fu/lv组合。然而,5-fu并非用于治疗sclc的活性剂。至今,尚未公开用mm-398治疗患有sclc的患者。申请人已发现mm-398单一疗法在患有sclc的患者中的某些方法和用途,包括本文公开的方法和用途。mm-398用于患有sclc的患者的这些方法和用途的发现部分地基于本文描述的临床前数据和临床药理学分析。该方法和用途被设计为平衡在更高剂量下预测的增加的疗效与增加的毒性。本文的临床前数据指示mm-398在sclc模型中的活性。临床药理学分析支持在增加的剂量下毒性增加,并且特别支持90mg/m2剂量的安全特性。最后,在等效于人类中的90mg/m2的小鼠剂量水平下的临床前疗效数据示出优于拓扑替康。可用抗肿瘤疗法治疗在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者,该抗肿瘤疗法由包囊在脂质体中的单一剂量的治疗有效量的伊立替康组成。脂质体伊立替康可为药学上可接受的伊立替康的脂质体制剂,其包含直径为约100nm的递送形式的伊立替康,诸如脂质体伊立替康(实施例1),包括peg化的脂质体。各种合适的脂质体伊立替康制剂可如本文所公开进行制备(实施例8)。优选地,脂质体伊立替康为产品mm-398(onivyde)(实施例9)。如本文所使用,90mg/m2伊立替康是指包囊在脂质体中的游离碱(基于伊立替康游离碱的量的剂量)并且等效于100mg/m2的盐酸伊立替康无水盐。将基于三水盐酸伊立替康的剂量转化成基于伊立替康游离碱的剂量是通过用伊立替康游离碱分子量(586.68g/mol)与三水盐酸伊立替康分子量(677.19g/mol)的比率乘以基于三水盐酸伊立替康的剂量来实现。这个比率为0.87,其可用作转化系数。例如,基于三水盐酸伊立替康的80mg/m2剂量等效于基于伊立替康游离碱的69.60mg/m2剂量(80x0.87)。在诊所里,这被四舍五入至70mg/m2以使任何潜在的给药误差最小化。在一些研究中,基于三水盐酸伊立替康(盐)的等效剂量计算nal-iri的剂量;在本说明书中,除非另有规定,否则剂量基于作为游离碱的伊立替康。因此,根据表1,基于作为游离碱的伊立替康的50mg/m2等效于基于作为三水盐酸盐的伊立替康的60mg/m2,基于作为游离碱的伊立替康的70mg/m2等效于基于作为三水盐酸盐的伊立替康的80mg/m2,基于作为游离碱的伊立替康的90mg/m2等效于基于作为三水盐酸盐的伊立替康的100mg/m2,以及基于作为游离碱的伊立替康的100mg/m2等效于基于作为三水盐酸盐的伊立替康的120mg/m2。表1盐游离碱180150120100100908070605050454035在施用作为单一试剂或组合疗法的部分的90mg/m2mm-398之后的总伊立替康和总sn-38的药物代谢动力学参数呈现在表2中。表2:患有实体瘤的患者中的总伊立替康和总sn-38药物代谢动力学参数。在50至150mg/m2的剂量范围内,总伊立替康的cmax和auc随着剂量而增加。另外,总sn-38的cmax随着剂量而成比例增加;然而,总sn-38的auc随着剂量而不到成比例增加。更高的血浆sn-38cmax与经历嗜中性白血球减少症的可能性增加相关。sn-38的cmax随着脂质体伊立替康剂量而成比例增加,但sn-38的auc随着剂量而不到成比例增加,从而实现了剂量调整的新方法。例如,与副作用相关的参数值(cmax)比与治疗有效性相关的参数值(auc)减小的程度相对更大。因此,当看出副作用时,可实施减少脂质体伊立替康的给药,使cmax减少与auc减少之间的差异最大化。该发现意味着在治疗方案中,给定的sn-38auc可取得令人惊奇地低的sn-38cmax。同样,给定的sn-38cmax可取得令人惊奇地高的sn-38auc。对伊立替康脂质体的直接测量表明,95%的伊立替康仍为包囊的脂质体,并且总形式与包囊形式之间的比率不随着给药后的0至169.5小时的时间而变化。在一些实施方案中,脂质体伊立替康可通过表2中的参数来表征。在一些实施方案中,脂质体伊立替康可为mm-398或生物等效于mm-398的产品。在一些实施方案中,脂质体伊立替康可通过表3中的参数来表征,包括cmax和/或auc值,该cmax和/或auc值为表2中的80%-125%的对应值。针对每两周一次施用90mg/m2伊立替康游离碱的各种替代脂质体伊立替康制剂,总伊立替康的药物代谢动力学参数提供在表3中。表3替代脂质体伊立替康制剂中的总伊立替康药物代谢动力学参数cmax:最大血浆浓度auc0-∞:外推至无限时间的血浆浓度曲线下的面积t1/2:末端排除半衰期在体外生长和活力测定中研究了针对各种sclc细胞系的伊立替康活性代谢物sn-38的活性(实施例2)。对这个数据的分析指示,sclc细胞系与胰腺癌和胃肠癌细胞系对sn-38具有类似的敏感性(图1)。另外,在四种测试的sclc细胞系中,sn-38诱导细胞活力减少>90%,ic50为可变的并且跨越了几个数量级。如在实施例2中所述,图2a和2b示出2种sclc细胞系中sn-38的细胞生长抑制动力学。在sclc的异种移植模型中研究了作为单一试剂的mm-398的活性(实施例3)。如图3所示,在dms-114模型中测试的所有剂量水平下看到抗肿瘤活性。在胰腺癌患者中评估mm-398暴露与疗效之间的估计相关性(实施例4)。针对mm-398+5fu/lv,os与时间(usn38>0.03ng/ml)的四分位数之间的关系提供在图4中。如在实施例6和7中所述,由药学上可接受的可注射形式的脂质体伊立替康组成的抗肿瘤疗法可每两周一次地向患有sclc疾病的患者施用,该sclc疾病在接受先前抗肿瘤疗法(例如,单独或与其他化学治疗剂一起的先前基于铂的疗法)之后有进展。可针对某些患者选择或修改脂质体伊立替康的剂量(例如,包囊在伊立替康脂质体中的50-90mg/m2伊立替康(游离碱))和脂质体伊立替康的给药频率(例如,每2周一次)。如在实施例6中所述,可将剂量选择成提供患者可耐受剂量,包括提供可接受的最低水平的3级或以上嗜中性白血球减少症(图6a)和/或腹泻(图6b)的剂量。在抗肿瘤疗法的过程中,患者可接受非抗肿瘤剂的其他试剂,诸如止吐剂。抗肿瘤疗法可在不存在拓扑替康的情况下施用。在一些实施方案中,本发明为一种治疗在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者的方法,该方法包括向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由提供包囊在伊立替康脂质体中的90mg/m2(游离碱)伊立替康的单一剂量的脂质体伊立替康组成。在一些实施方案中,本发明为一种治疗在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者的方法,该方法包括向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由提供包囊在伊立替康脂质体中的70mg/m2(游离碱)伊立替康的单一剂量的脂质体伊立替康组成。在一些实施方案中,本发明为一种治疗在遵循用于小细胞肺癌(sclc)的基于铂的疗法疾病进展后诊断患有sclc的人类患者的方法,该方法包括向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由提供包囊在伊立替康脂质体中的50mg/m2(游离碱)伊立替康的单一剂量的脂质体伊立替康组成。治疗方法可包括确定患者是否符合实施例7中规定的一种或多种入选标准,并且然后施用由脂质体伊立替康组成的抗肿瘤疗法。例如,抗肿瘤疗法可由向已针对sclc用基于铂的疗法(例如,单独地或与依托泊苷组合的顺铂和/或卡铂)治疗的患者施用治疗有效剂量(例如,包囊在脂质体中的50-90mg/m2伊立替康(游离碱))和给药频率(例如,每2周一次)的脂质体伊立替康组成。另外,治疗方法可包括确定患者是否符合实施例7中规定的一种或多种排除标准,并且不施用由脂质体伊立替康组成的抗肿瘤疗法。本文公开的治疗sclc的方法可包括向不符合实施例7中的一种或多种排除标准的患者施用抗肿瘤疗法。例如,抗肿瘤疗法可由向尚未针对sclc用伊立替康或拓扑替康治疗的患者施用治疗有效剂量(例如,包囊在脂质体中的50-90mg/m2伊立替康(游离碱))和给药频率(例如,每2周一次)的脂质体伊立替康组成。诊断患有sclc的患者的某些亚组可任选地用减少的剂量的脂质体伊立替康治疗,包括具有较高胆红素水平的患者或具有ugt1a1*287/7纯合等位基因的患者。减少的剂量是指向接受减少的剂量的患者每两周一次施用的包囊在脂质体中的小于90mg/m2的伊立替康(游离碱)的剂量。在一些例子中,减少的剂量可为向诊断患有sclc并且接受减少的剂量的患者每两周一次施用的50-90mg/m2的剂量,包括50mg/m2的减少的剂量、60mg/m2的减少的剂量、70mg/m2的减少的剂量、或80mg/m2伊立替康(游离碱)的减少的剂量。对于以70mg/m2开始的那些患者,第一次剂量减少应当减少至50mg/m2,然后减少至43mg/m2。适当剂量的准确确定将依赖于在所述子群中观察到的药物代谢动力学、疗效和安全性。在一些例子中,可向在免疫疗法之时或之后和/或在基于铂的一线化学疗法(卡铂或顺铂)或包括基于铂的化学疗法的化放疗以用于治疗局限期或广泛期sclc之后诊断有sclc疾病进展的患者施用脂质体伊立替康。在一些例子中,患者可接受用于sclc的一些形式的免疫疗法,之后施用脂质体伊立替康。免疫疗法的例子可包括阿特珠单抗、avelimumab、纳武单抗、帕姆单抗、易普利单抗、替西木单抗(tremelimumab)和/或度伐单抗。在一个例子中,患者接受用于sclc的纳武单抗(例如,根据nct02481830中的治疗方案),之后接受如本文所公开的脂质体伊立替康。在一个例子中,患者接受用于sclc的易普利单抗(例如,根据nct01331525、nct02046733、nct01450761、nct02538666或nct01928394中的治疗方案),之后接受如本文所公开的脂质体伊立替康。免疫疗法可包括结合ctla4、pdl1、pd1、41bb和/或ox40的分子,包括下表4中公开可用的化合物或结合相同表位或具有相同或类似生物功能的其他化合物。表4抗体抗体序列(参考文献)α-pdl110f.9g2,bioxcellα-41bblob12.3,bioxcellα-ctla49h10,bioxcellα-ox40ox-86,bioxcell使用脂质体伊立替康和免疫疗法的组合可用于治疗有需要的宿主中的癌症,以在所述癌症的治疗中治疗协同的量和施用方案来进行。免疫疗法可为结合和/或作用于α-pdl1、α-41bb、α-ctla4、α-ox40和/或pd1的抗体或抗体组合。在一些实施方案中,治疗有需要的宿主中的癌症包括在不施用类固醇的情况下施用mm-398。治疗方案可包括每两周或三周或者三周中有两周施用一次43、50、70、80或90mg/m2脂质体伊立替康(游离碱)的mm-398与免疫疗法组合(例如,与针对α-pdl1、pd1、α-41bb、α-ctla4和/或α-ox40的抗体组合)。例如,治疗方案可包括向诊断患有sclc的人类宿主施用(例如,28天)治疗周期,其中治疗周期包括每两周一次施用:总共43、50、70、80或90mg/m2脂质体伊立替康(游离碱),接着施用3mg/kg纳武单抗;并且重复所述治疗周期直到观察到进展或不可接受的毒性。在另一个例子中,治疗方案可包括向诊断患有sclc的人类宿主施用(例如,28天)治疗周期,其中治疗周期包括每两周或三周或三周中有两周施用一次:总共43、50、70、80或90mg/m2脂质体伊立替康(游离碱),接着每两周或三周一次施用2mg/kg帕姆单抗(其中第一次剂量的脂质体伊立替康和帕姆单抗在同一天给予);并且重复所述治疗周期直到观察到进展或不可接受的毒性。治疗方案可包括每两周一次施用90mg/m2脂质体伊立替康(游离碱)的mm-398。一种治疗在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者的方法,可由向人类患者每两周一次施用抗肿瘤疗法组成,该抗肿瘤疗法由提供包囊在伊立替康脂质体中的50、70或90mg/m2(游离碱)伊立替康的单一剂量的脂质体伊立替康组成。当已知患者针对ugt1a1*28等位基因为纯合的时,可减少每次伊立替康脂质体的剂量(例如,50或70mg/m2)。当患者针对ugt1a1*28等位基因并非纯合的时,每次伊立替康脂质体的剂量可为90mg/m2。该方法可进一步包括向患者使用皮质类固醇和止吐剂,之后施用伊立替康脂质体。一种治疗针对ugt1a1*28等位基因并非纯合的并且在先前的用于小细胞肺癌(sclc)的疗法后疾病进展之后诊断患有sclc的人类患者的方法,可包括向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由提供包囊在伊立替康脂质体中的90mg/m2伊立替康(游离碱)的单一剂量的脂质体伊立替康组成。该方法可进一步包括向患者使用皮质类固醇和止吐剂,之后施用伊立替康脂质体。在接受脂质体伊立替康的抗肿瘤疗法之前,患者可为在基于铂的方案上有进展并且还已经(任选地)接受维持或2线环境的单线免疫疗法的患者。患者可为在接受脂质体伊立替康抗肿瘤疗法之前未用针对sclc的拓扑替康治疗的患者。患者可先前接受免疫疗法诱导,接着和/或伴随有一个或多个维持剂量的化学疗法,之后施用脂质体伊立替康。治疗方案可包括每三周一次施用100-130mg/m2脂质体伊立替康(游离碱)的mm-398与免疫疗法组合(例如,与针对α-pdl1、pd1、α-41bb、α-ctla4和/或α-ox40的抗体组合)。例如,治疗方案可包括向诊断患有sclc的人类宿主施用治疗周期,其中治疗周期包括每三周一次施用:总共100、110、120或130mg/m2脂质体伊立替康(游离碱),接着施用3mg/kg纳武单抗;并且重复所述治疗周期直到观察到进展或不可接受的毒性。治疗方案可包括向诊断患有sclc的人类宿主施用治疗周期,其中治疗周期包括与每两周或三周一次施用3mg/kg纳武单抗组合而每三周一次施用:总共100、110、120或130mg/m2脂质体伊立替康(游离碱)(其中第一次剂量的脂质体伊立替康和纳武单抗在同一天给予);并且重复所述治疗周期直到观察到进展或不可接受的毒性。在另一个例子中,治疗方案可包括向诊断患有sclc的人类宿主施用治疗周期,其中治疗周期包括每三周一次施用:总共100、110、120或130mg/m2脂质体伊立替康(游离碱),接着施用2mg/kg帕姆单抗;并且重复所述治疗周期直到观察到进展或不可接受的毒性。治疗方案可包括向诊断患有sclc的人类宿主施用治疗周期,其中治疗周期包括与每两周或三周一次施用2mg/kg帕姆单抗组合而每三周一次施用:总共100、110、120或130mg/m2脂质体伊立替康(游离碱)(其中第一次剂量的脂质体伊立替康和帕姆单抗在同一天给予);并且重复所述治疗周期直到观察到进展或不可接受的毒性。治疗方案可包括向诊断患有sclc的人类宿主施用治疗周期,其中治疗周期包括与每两周或三周一次施用2mg/kg帕姆单抗组合而每三周中两周施用一次:总共100、110、120或130mg/m2脂质体伊立替康(游离碱)(其中第一次剂量的脂质体伊立替康和帕姆单抗在同一天给予);并且重复所述治疗周期直到观察到进展或不可接受的毒性。治疗方案可包括每三周一次施用110mg/m2脂质体伊立替康(游离碱)的mm-398与治疗有效量的免疫疗法组合(例如,与针对α-pdl1、pd1、α-41bb、α-ctla4和/或α-ox40的抗体组合)。治疗方案可包括每三周一次施用100mg/m2脂质体伊立替康(游离碱)的mm-398与治疗有效量的免疫疗法组合(例如,与针对α-pdl1、pd1、α-41bb、α-ctla4和/或α-ox40的抗体组合)。治疗方案可包括每三周一次施用120mg/m2脂质体伊立替康(游离碱)的mm-398与治疗有效量的免疫疗法组合(例如,与针对α-pdl1、pd1、α-41bb、α-ctla4和/或α-ox40的抗体组合)。治疗方案可包括每三周一次施用130mg/m2脂质体伊立替康(游离碱)的mm-398与治疗有效量的免疫疗法组合(例如,与针对α-pdl1、pd1、α-41bb、α-ctla4和/或α-ox40的抗体组合)。在一些实施方案中,脂质体伊立替康在用于sclc的基于铂的疗法与prexasertib、阿霉素、lurbinectedin和rova-t中的一种或多种组合后疾病进展之后施用。在一些实施方案中,可向先前接受pd-1导向的治疗剂(例如,纳武单抗、帕姆单抗)、pd-l1导向的治疗剂(例如,阿特珠单抗或度伐单抗)或notchadc化合物(例如,rova-t)作为sclc的一线(1l)疗法的患者施用脂质体伊立替康。在一些实施方案中,脂质体伊立替康可与chk1导向的治疗剂(例如,prexasertib)、topo-2导向的治疗剂(例如,阿霉素)、dna抑制剂(例如,lurbinectedin)或notchadc化合物(例如,rova-t)组合施用。在其他实施方案中,脂质体伊立替康可在不存在chk1导向的治疗剂(例如,prexasertib)、topo-2导向的治疗剂(例如,阿霉素)、dna抑制剂(例如,lurbinectedin)或notchadc化合物(例如,rova-t)的情况下施用。在一些实施方案中,可向先前接受用于sclc的顺铂或卡铂的患者施用脂质体伊立替康,并且脂质体伊立替康在不存在(即,没有)顺铂或卡铂的情况下施用(用于二线或后续疗法)。在一些实施方案中,治疗sclc的方法可包括向诊断患有sclc的人类宿主施用治疗周期,其中治疗周期包括每三周一次施用:总共90mg/m2脂质体伊立替康(游离碱)或120mg/m2脂质体伊立替康(游离碱),组合与第一次施用脂质体伊立替康同一天开始的每两周一次施用3mg/kg纳武单抗,并且重复所述治疗周期直到观察到进展或不可接受的毒性。在另一个例子中,治疗方案可包括向诊断患有sclc的人类宿主施用治疗周期,其中治疗周期包括每三周一次施用:总共90mg/m2脂质体伊立替康(游离碱)或120mg/m2脂质体伊立替康(游离碱),组合与第一次施用脂质体伊立替康同一天开始的每三周一次施用2mg/kg帕姆单抗;并且重复所述治疗周期直到观察到进展或不可接受的毒性。可在不施用另一种抗肿瘤剂(例如,不施用拓扑替康)的情况下,每两周一次向患者施用包含90mg/m2脂质体伊立替康的抗肿瘤疗法以用于治疗sclc。优选地,用于先前治疗(例如二线)的sclc的抗肿瘤疗法提供大于15周(例如,至少约20-25周,包括约21-24周、约22-24周、约23周或约24周)的无进展存活期的中位进展时间,大于30周(例如,至少约30-50周,包括约40-50周、约44-48周、约45-47周、约46周或约47周)的中位总存活期,其中危险比为小于1,并且优选地小于0.7、0.6或0.5(例如,包括约0.6-0.7的危险比)。优选地,该抗肿瘤疗法提供发生在>5%的群体中的以下主要不良事件(3+级):小于50%的嗜中性白血球减少症(例如,约10%-50%,包括约20%)、小于50%的血小板减少症(例如,小于10%,包括1%-10%、1%-5%,小于5%以及约2%、约3%和约4%)、以及小于30%的贫血症(例如,小于10%,包括1%-10%、1%-8%,小于8%以及约5%-7%、约6%和约5%)。一种治疗在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者的方法,可由向人类患者每两周一次施用抗肿瘤疗法组成,该抗肿瘤疗法由提供包囊在伊立替康脂质体中的90mg/m2(游离碱)伊立替康的单一剂量的脂质体伊立替康(或减少剂量的50-70g/m2(游离碱)作为脂质体伊立替康的伊立替康,向在先前施用脂质体伊立替康的过程中或之后已经历不良事件的患者和/或已知针对ugt1a1*28等位基因为纯合的患者)组成,其中至少300个患者(例如,约400-450个患者)的临床试验中的抗肿瘤疗法引起发生在>5%的群体中的以下主要不良事件(3+级):小于50%的嗜中性白血球减少症(例如,约10%-50%,包括约20%)、小于50%的血小板减少症(例如,小于10%,包括1%-10%、1%-5%,小于5%以及约2%、约3%和约4%)、以及小于30%的贫血症(例如,小于10%,包括1%-10%、1%-8%,小于8%以及约5%-7%、约6%和约5%)。一种治疗在用于小细胞肺癌(sclc)的基于铂的疗法后疾病进展之后诊断患有sclc的人类患者的方法,可由向人类患者每两周一次施用抗肿瘤疗法组成,该抗肿瘤疗法由提供包囊在伊立替康脂质体中的90mg/m2(游离碱)伊立替康的单一剂量的脂质体伊立替康(或减少剂量的50-70g/m2(游离碱)作为脂质体伊立替康的伊立替康,向在先前施用脂质体伊立替康的过程中或之后已经历不良事件的患者和/或已知针对ugt1a1*28等位基因为纯合的患者)组成,其中至少300个患者(例如,约400-450个患者)的临床试验中的抗肿瘤疗法引起下列情况中的一种或多种:大于15周(例如,至少约20-25周,包括约21-24周、约22-24周、约23周或约24周)的无进展存活期的中位进展时间,大于30周(例如,至少约30-50周,包括约40-50周、约44-48周、约45-47周、约46周或约47周)的中位总存活期,其中危险比为小于1,并且优选地小于0.7、0.6或0.5(例如,包括约0.6-0.7的危险比)。当已知患者针对ugt1a1*28等位基因为纯合的时,可减少每次伊立替康脂质体的剂量(例如,50或70mg/m2)。当患者针对ugt1a1*28等位基因并非纯合的时,每次伊立替康脂质体的剂量可为90mg/m2。该方法可进一步包括向患者使用皮质类固醇和止吐剂,之后施用伊立替康脂质体。在一些实施方案中,可向在用一种或多种喜树碱化合物或拓扑异构酶i(topo-1)抑制剂治疗后诊断有小细胞肺癌(sclc)疾病进展的患者施用脂质体伊立替康。喜树碱化合物或拓扑异构酶i(topo-1)抑制剂的例子包括但不限于喜树碱、9-氨基喜树碱、7-乙基喜树碱、10-羟基喜树碱、7-乙基10-羟基喜树碱、9-硝基喜树碱、10,11-亚甲二氧喜树碱、9-氨基-10,11-亚甲二氧喜树碱、9-氯-10,11-亚甲二氧喜树碱、伊立替康(cpt-11)、拓扑替康、勒托替康、斯类替康(silatecan)、etirinotecanpegol、鲁比替康、依喜替康、fl118、贝洛替康、吉马替康、indotecan、indimitecan、(7-(4-甲基哌嗪代亚甲基)-10,11-亚乙二氧-20(s)-喜树碱、7-(4-甲基哌嗪代亚甲基)-10,11-亚甲二氧-20(s)-喜树碱、以及7-(2-n-异丙基氨基)乙基)-(20s)-喜树碱。在一些实施方案中,可向用伊立替康(cpt-11)、拓扑替康或这两者治疗后诊断有sclc疾病进展的患者施用脂质体伊立替康。在一些实施方案中,可向用伊立替康(cpt-11)治疗后诊断有sclc疾病进展的患者施用脂质体伊立替康。在一些实施方案中,可向用拓扑替康治疗后诊断有sclc疾病进展的患者施用脂质体伊立替康。在一些实施方案中,可向用非脂质体伊立替康治疗后诊断有sclc疾病进展的患者施用脂质体伊立替康。在一些实施方案中,基于铂的疗法与依托泊苷或非脂质体伊立替康组合施用。在一些实施方案中,基于铂的疗法与依托泊苷组合施用。在一些实施方案中,基于铂的疗法与非脂质体伊立替康组合施用。一个实施方案为一种治疗在用于小细胞肺癌(sclc)的基于喜树碱的疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法,该方法包括向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由90mg/m2(游离碱)剂量的mm-398脂质体伊立替康组成。在一些实施方案中,基于喜树碱的疗法包括先前不连续施用拓扑替康或非脂质体伊立替康来治疗诊断患有sclc的人类患者。在一些实施方案中,基于喜树碱的疗法包括先前不连续施用非脂质体伊立替康,该非脂质体伊立替康以300mg/m2剂量每三周一次向人类患者施用。在一些实施方案中,基于喜树碱的疗法包括先前不连续施用非脂质体伊立替康,该非脂质体伊立替康以1.5mg/m2剂量在三周治疗周期中在第1天、第2天、第3天、第4天和第5天向人类患者施用。在一些实施方案中,诊断患有sclc的人类患者为铂敏感的。在一些实施方案中,诊断患有sclc的人类患者为具有铂抗性的。本公开文本的第一方面为一种治疗在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法。第一方面的一个实施方案为一种治疗在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法,该方法包括向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由90mg/m2(游离碱)剂量的mm-398脂质体伊立替康组成。在第一方面的一个实施方案中,基于铂的疗法包括先前不连续施用顺铂或卡铂来治疗诊断患有sclc的人类患者。在另一个实施方案中,在施用mm-398脂质体伊立替康之前,人类患者在不使用造血生长因子的情况下具有大于1,500个细胞/微升的血液anc。另一个实施方案为一种治疗在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法。又另一个实施方案为一种治疗在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法,该方法包括向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由90mg/m2(游离碱)剂量的mm-398脂质体伊立替康组成,其中在施用mm-398脂质体伊立替康之前,人类患者具有大于100,000个细胞/微升的血液血小板计数。在第一方面的一些实施方案中,在施用mm-398脂质体伊立替康之前,人类患者具有大于9g/dl的血液血红蛋白。在一些实施方案中,在施用mm-398脂质体伊立替康之前,人类患者具有小于或等于1.5xuln的血清肌酐以及大于或等于40ml/min的肌酐清除率。在第一方面的一些实施方案中,在施用mm-398脂质体伊立替康之前,人类患者不接受拓扑异构酶i抑制剂。在第一方面的又其他实施方案中,在施用mm-398脂质体伊立替康之前,人类患者只接受了基于铂的单一疗法。第一方面的实施方案可包括一种方法,其中该抗肿瘤疗法包括以下步骤:(a)通过将含有4.3mg伊立替康游离碱/ml分散体的mm-398脂质体伊立替康分散体与5%葡萄糖注射液(d5w)或0.9%氯化钠注射液组合以获得500ml最终体积和90mg/m2(游离碱)的mm-398脂质体伊立替康(±5%)的可注射组合物,制备药学上可接受的可注射组合物;以及(b)向患者以90分钟输注施用来自步骤(a)的含有mm-398伊立替康脂质体的可注射组合物。在第一方面的任何实施方案中,该方法可进一步包括在每次施用抗肿瘤疗法之前,向人类患者施用地塞米松和5-ht3阻断剂,并且任选地进一步向人类患者施用止吐剂。本公开文本的第二方面为一种治疗针对utg1a1*28等位基因并非纯合的并且在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法。第二方面的一个实施方案为一种治疗针对utg1a1*28等位基因并非纯合的并且在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法,该方法包括在六周的周期中向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由90mg/m2(游离碱)剂量的mm-398脂质体伊立替康组成。在第二方面的一些实施方案中,基于铂的疗法包括先前不连续施用顺铂或卡铂来治疗诊断患有sclc的人类患者。第二方面的一个实施方案为一种治疗针对utg1a1*28等位基因并非纯合的并且在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法,其中该方法包括在六周的周期中向人类患者每两周一次施用抗肿瘤疗法,该抗肿瘤疗法由90mg/m2(游离碱)剂量的mm-398脂质体伊立替康组成,其中在施用mm-398脂质体伊立替康之前,人类患者具有下列情况中的一种或多种:(a)在不使用造血生长因子的情况下大于1,500个细胞/微升的血液anc;(b)大于100,000个细胞/微升的血液血小板计数;(c)大于9g/dl的血液血红蛋白;以及(d)小于或等于1.5xuln的血清肌酐和大于或等于40ml/min的肌酐清除率。在第二方面的一些实施方案中,在施用mm-398脂质体伊立替康之前,人类患者未接受拓扑异构酶i抑制剂;并且在施用mm-398脂质体伊立替康之前,人类患者只接受了基于铂的单一疗法。在一些实施方案中,该方法包括施用抗肿瘤疗法持续至少三个六周的周期。在第二方面的一些实施方案中,抗肿瘤疗法包括以下步骤:(a)通过将含有4.3mg伊立替康游离碱/ml分散体的mm-398脂质体伊立替康分散体与5%葡萄糖注射液(d5w)或0.9%氯化钠注射液组合以获得500ml最终体积和90mg/m2(游离碱)的mm-398脂质体伊立替康(±5%)的可注射组合物,制备药学上可接受的可注射组合物;以及(b)向患者以90分钟输注施用来自步骤(a)的含有mm-398伊立替康脂质体的可注射组合物。这个实施方案可进一步包括在每次施用抗肿瘤疗法之前,向人类患者施用地塞米松和5-ht3阻断剂,并且任选地进一步向人类患者施用止吐剂。本公开文本的第三方面提供治疗在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法,该基于铂的一线疗法选自由顺铂或卡铂组成的组。第三方面的一个实施方案为一种治疗在用于小细胞肺癌(sclc)的基于铂的一线疗法之时或之后疾病进展后诊断患有sclc的人类患者的方法,该基于铂的一线疗法选自由顺铂或卡铂组成的组,该方法包括向人类患者每两周一次施用抗肿瘤疗法持续总共至少三个六周的周期,该抗肿瘤疗法由90mg/m2(游离碱)剂量的mm-398脂质体伊立替康组成;其中人类患者针对utg1a1*28等位基因并非纯合的,并且在施用每次mm-398脂质体伊立替康的抗肿瘤疗法之前具有下列情况:(a)在不使用造血生长因子的情况下大于1,500个细胞/微升的血液anc;(b)大于100,000个细胞/微升的血液血小板计数;(c)大于9g/dl的血液血红蛋白;以及(d)小于或等于1.5xuln的血清肌酐和大于或等于40ml/min的肌酐清除率。在第三方面的一些实施方案中,在施用mm-398脂质体伊立替康之前,人类患者未接受拓扑异构酶i抑制剂,并且在施用mm-398脂质体伊立替康之前,人类患者只接受了基于铂的单一疗法;并且该方法进一步包括在每次施用抗肿瘤疗法之前,向人类患者施用地塞米松和5-ht3阻断剂,并且任选地进一步向人类患者施用止吐剂。在第三方面的一个实施方案中,抗肿瘤疗法包括以下步骤:(a)通过将含有4.3mg伊立替康游离碱/ml分散体的mm-398脂质体伊立替康分散体与5%葡萄糖注射液(d5w)或0.9%氯化钠注射液组合以获得500ml最终体积和90mg/m2(游离碱)的mm-398脂质体伊立替康(±5%)的可注射组合物,制备药学上可接受的可注射组合物;以及(b)向患者以90分钟输注施用来自步骤(a)的含有mm-398伊立替康脂质体的可注射组合物。实施例实施例1:脂质体伊立替康脂质体伊立替康组合物优选地包含或由磷脂酰胆碱、胆固醇以及聚乙二醇衍生化的磷脂酰乙醇胺组成。脂质体伊立替康可包含单层的脂质双层囊泡,该囊泡包含磷脂酰胆碱和胆固醇,从而包囊伊立替康蔗糖八硫酸酯。脂质体伊立替康组合物中伊立替康脂质体的直径为110nm(±20%)。脂质体伊立替康可包含包囊在脂质体中的伊立替康蔗糖八硫酸酯,该脂质体具有单层的脂质双层囊泡,直径为大约110nm,该囊泡包囊含有胶凝化或沉淀状态下作为蔗糖八硫酸酯盐的伊立替康的水性空间;其中囊泡由以下构成:1,2-二硬脂酰-sn-丙三基-3-磷酸胆碱(dspc)(例如,约6.8mg/ml)、胆固醇(例如,约2.2mg/ml)以及甲氧基封端的聚乙二醇(mw2000)-二硬脂酰磷脂酰乙醇胺(mpeg-2000-dspe)(例如,约0.1mg/ml)。每ml还含有作为缓冲剂的2-[4-(2-羟乙基)哌嗪-1-基]乙磺酸(hepes)(例如,约4.1mg/ml)和作为等渗试剂的氯化钠(例如,约8.4mg/ml)。脂质体伊立替康的脂质膜可由处于合适摩尔比(例如,约3:2:0.015的摩尔比和/或针对200个磷脂分子为大约一个聚乙二醇(peg)的量)的磷脂酰胆碱、胆固醇以及聚乙二醇衍生化的磷脂酰乙醇胺构成。(在本文中又称为mm-398或nal-iri)为优选的脂质体伊立替康,其包含小的单层脂质双层囊泡(suv),直径为大约110nm,该囊泡包囊含有胶凝化或沉淀状态下作为蔗糖硫酸酯盐的伊立替康的水性空间。onivyde脂质体伊立替康包含包囊在脂质体中的伊立替康蔗糖八硫酸酯,该脂质体具有单层的脂质双层囊泡,直径为大约110nm,该囊泡包囊含有胶凝化或沉淀状态下作为蔗糖八硫酸酯盐的伊立替康的水性空间;其中囊泡由以下构成:1,2-二硬脂酰-sn-丙三基-3-磷酸胆碱(dspc)(6.8mg/ml)、胆固醇(2.2mg/ml)以及甲氧基封端的聚乙二醇(mw2000)-二硬脂酰磷脂酰乙醇胺(mpeg-2000-dspe)(0.1mg/ml)。每ml还含有作为缓冲剂的2-[4-(2-羟乙基)哌嗪-1-基]乙磺酸(hepes)(4.1mg/ml)和作为等渗试剂的氯化钠(8.4mg/ml)。onivyde为无菌的白色至浅黄色不透明等渗脂质体分散体。脂质体伊立替康可作为10ml一次性玻璃小瓶中的无菌的白色至浅黄色不透明脂质体分散体而供应,其含有43mg/10ml伊立替康游离碱。可在经过90分钟的静脉内输注之前,释稀小瓶中的脂质体分散体。本公开文本提供每两周一次使用脂质体伊立替康(例如,实施例9中描述的onivyde)以用于治疗sclc,为每2周(优选地,在6周的周期中)经过90分钟静脉内施用的包囊在脂质体中的总剂量90mg/m2伊立替康(游离碱)(基于伊立替康游离碱的量的剂量;等效于100mg/m2的盐酸伊立替康无水盐)。已知针对ugt1a1*28等位基因为纯合的患者中onivyde的建议起始剂量为经过90分钟由静脉内输注施用的50mg/m2(游离碱)。随着在后续周期中耐受,可将onivyde的剂量增加至70mg/m2。对于血清胆红素高于正常值上限的患者,没有onivyde的建议剂量。实施例2拓扑异构酶i抑制对广泛范围的癌症细胞系具有有效作用。在“癌症药物敏感性基因组学(genomicsofdrugsensitivityincancer)”计划的维尔康姆基金会桑格研究所数据库中的参考数据对于针对sn-38的敏感性筛选的663种癌症细胞系是可用的(urlwww.cancerrxgene.org/translation/drug/1003)。对这个数据的分析指示,sclc细胞系与胰腺癌和胃肠癌细胞系对sn-38具有类似的敏感性(图1)。在这个数据集中,胃肠(ht-29、hct-116、lovo、mkn45)或胰腺(aspc-1、bxpc3、cfpac-1、miapaca-2)来源的癌症细胞系由实心圆突出显示,对于这些癌症细胞系观察到显著的mm-398的体内抗肿瘤疗效。sclc细胞系dms114和nci-h1048(参见下文)也示为实心圆。在体外生长和活力测定中研究了针对各种sclc细胞系的伊立替康活性代谢物sn-38的活性。在四种测试的sclc细胞系(dms53、dms114、nci-h1048、sw1271)中,sn-38诱导细胞活力减少>90%,ic50为可变的并且跨越了几个数量级。图2a和2b示出了使用zoom系统,经过88小时的时间进程,在2种sclc细胞系(dms-114和nci-h1048)中的sn-38细胞生长抑制动力学。在1-10nm之间观察到有效的细胞生长抑制,而在浓度≥10nm下在延长的孵育时间之后观察到细胞杀死。这个sn-38治疗阈值的范围与由在mm-398施用后72h的患者肿瘤活检物测量的sn-38的量(范围:3-163nm)一致。这些数据表明,由于mm-398药理学特征而使肿瘤中的sn-38持续时间延长将提供sclc中的有效活性。临床前实验已证实mm-398大大地增加了肿瘤中sn-38的可用性,并且示出在比非脂质体伊立替康低很多的剂量下的剂量依赖性抗肿瘤疗效。实施例3在sclc的异种移植模型中研究了作为单一试剂的mm-398的活性。在ncrnu/nu小鼠中皮下接种dms114细胞。当肿瘤体积达到大约300mm3时,用10或20mg/kg的mm-398盐酸伊立替康治疗小鼠,该mm-398盐酸伊立替康按每周一次的标准静脉内施用4周。基于pk建模和与临床pk数据的比较,选择对应于被认为是临床相关小鼠剂量的剂量水平。如图3所示,在dms114模型中测试的所有剂量水平下看到抗肿瘤活性。接受10或20mg/kg的具有肿瘤的动物示出肿瘤消退,该肿瘤消退持续最后剂量的mm-398之后的大约20-27天(在10和20mg/kg剂量下分别为2/5和4/5的完全消退)。实施例4:暴露与疗效之间的相关性。虽然在sclc中有待研究mm-398暴露与疗效之间的相关性,但胰腺癌患者中的数据分析指示增加暴露于sn-38的益处。在napoli-1的mm-398+5fu/lv治疗组中,较长的总存活期(os)和无进展存活期(pfs)与较长时间usn38>0.03ng/ml以及较高tiri、tsn38和usn38的cavg相关,其中当usn38>0.03ng/ml时观察到最高的相关性。tiri、tsn38或usn38的cmax不能预测os(p=0.81-0.92)。针对mm-398+5fu/lv,os与时间(usn38>0.03ng/ml)的四分位数之间的关系提供在图4中。usn38>0.03ng/ml的较长持续时间与在mm-398+5fu/lv组中达到客观响应的概率较高相关(图5)。这种相关性在每3周一次以100mg/m2给药的mm-398单一疗法组中观察不到(p=0.62)。在单一疗法组中缺乏相关性可部分地归因于剂量间隔差异(在单一疗法组中mm-398剂量为每3周的100mg/m2,在mm-398+5-fu/lv组中mm-398剂量为每2周的70mg/m2)。实施例5:在mm-398的情况下暴露与安全性之间的相关性基于用安能得治疗的353位患者中的数据,评估暴露与安全性之间的相关性。较高的未包囊的sn-38cmax与嗜中性白血球减少症治疗紧急不良事件的发生率和严重性两者的较高概率相关(图6a)。较高的总伊立替康cmax与观察到3+级腹泻的概率较高相关(图6b)。此外,在存在和不存在用5fu/lv共同施用的情况下得到观察到3+级嗜中性白血球减少症的不同概率。使用这些相关性来评估针对有待在sclc中测试的替代剂量方案的预测安全性。实施例6:针对90mg/m2剂量的安全性预测基于针对嗜中性白血球减少症(图6a)和腹泻(图6b)的这些暴露-安全性相关性,预测的3+级嗜中性白血球减少症和腹泻比率提供在表5中。与作为单一疗法的70mg/m2(游离碱)剂量相比,预测90mg/m2(游离碱)剂量将3+级嗜中性白血球减少症从8.4%增加至11.1%,并且将腹泻从14.3%增加至20.0%。基于针对患有胰腺癌疾病的大部分(73%)患者的数据得到这些比率,这些患有胰腺癌疾病的患者与患有sclc的患者相比可具有更高的腹泻风险。表5通过伊立替康脂质体注射液剂量预测的3级或更高级嗜中性白血球减少症和腹泻实施例7:在基于铂的一线疗法之时或之后有进展的患有小细胞肺癌的患者中nal-iri(或mm-398)的随机化、开放标签3期研究研究设计的综述。这是在基于铂的一线疗法之时或之后有进展的患有小细胞肺癌的患者中伊立替康脂质体注射液与静脉内拓扑替康的开放标签、随机化的3期研究。该研究将分两个部分进行。部分1:部分1a部分1a的目的是:1)描述每2周施用的伊立替康脂质体注射液单一疗法的安全性和耐受性和2)为了确定本研究的部分1b和部分2的伊立替康脂质体注射液单一疗法剂量(每两周施用的90mg/m2或70mg/m2)。部分1b为出于表征伊立替康脂质体注射液和静脉内拓扑替康的初步疗效和安全性目的的nal-iri(n=25)和静脉内拓扑替康(n=25)的平行研究。部分1b的目的是为了描述1)12周的无进展存活期、2)客观响应率(orr)、3)无进展存活期(pfs)、4)总存活期(os)以及5)安全特性。部分2:nal-iri(n=210)与拓扑替康(n=210)的随机化的疗效研究。部分2的第一目的是为了将用伊立替康脂质体注射液治疗后的总存活期与用静脉内拓扑替康治疗后的总存活期进行比较。部分2的第二目的是为了将治疗组之间的下列内容进行比较:1)无进展存活期(pfs)、2)客观响应率(orr)、3)如通过欧洲癌症研究和治疗组织生活质量问卷(eortcqlq-c30)和肺癌13(lc13)所测量的在咳嗽、呼吸困难和疲劳上具有症状改善的患者比例以及4)安全特性。探索性目的(部分1和部分2)包括:1)为了描述用伊立替康脂质体注射液治疗后的qtcf(仅部分1),2)为了探索与用伊立替康脂质体注射液治疗后的疗效和安全性相关的生物标志物,3)为了描述ugt1a1基因型、sn-38浓度(仅伊立替康脂质体注射液治疗的患者)与安全性之间的相关性,4)为了评估本患者群体中的伊立替康脂质体注射液的血浆药物代谢动力学与疗效和安全性之间的关系,5)为了比较cns进展的发展/cns进展的发展时间与新cns转移的发展的比率,6)为了比较治疗组之间的治疗失败时间(ttf),以及7)为了比较使用eortc-qlq-c30、eortc-qlq-lc13和eq-5d-5l的治疗组之间的患者报告结果(pro)。部分1和部分2两者将由三个阶段组成:筛选阶段、治疗/主动随访阶段以及长期随访阶段。治疗/主动随访阶段是第一剂量研究药物到决定永久中止研究药物治疗的期间。长期随访阶段是针对总存活期的每月随访。部分1a有待在部分1a安全性部分招收的初始数量的患者为可评估安全性的6位患者。这个初始患者同期群将每2周用70mg/m2的伊立替康脂质体注射液进行治疗。在治疗的前28天(或如果存在治疗延迟,在第二次剂量的研究治疗之后14天)过程中,将评估剂量限制毒性(dlt)以确定该剂量是否耐受。如果每2周接受70mg/m2的伊立替康脂质体注射液的2位或更多位患者具有dlt,则该剂量将被宣告不耐受。在所有其他情况下,将招收用起始于90mg/m2的伊立替康脂质体注射液治疗的另一同期群6位患者。如果在70mg/m2同期群中治疗的初始6位患者的总体经历被断定为足够安全以合理期望90mg/m2剂量将在部分1研究者和发起人的评价中为耐受的,则将仅招收90mg/m2同期群。dlt的评估将遵循与第一同期群相同的准则。如果在90mg/m2剂量下,2位或更多位患者具有dlt,则该剂量将被认为超过最佳安全性和耐受性标准,并且70mg/m2将被指定为部分1b的剂量,并且部分1b将开始施用70mg/m2的伊立替康脂质体注射液。如果使用90mg/m2剂量,在安全性评估时期内存在0或1种dlt,则将由部分1研究者和发起人基于两个同期群的整体安全性经历来做出何种剂量用于部分1b的决定。·将对接受研究药物的所有患者评估dlt和安全性。如果在治疗的前28天(或如果根据章节6.2存在治疗延迟,在第二次剂量的研究治疗之后14天)中发生下列不良事件,它们应当被认为dlt,并且被研究者认为与研究治疗相关:在7天内不消退的4级嗜中性白血球减少症或血小板减少症以及任何持续时间的4级贫血症·由于药物相关毒性,计划日期的14天内不能开始后续疗程·并发发热≥38.5℃(即发热性嗜中性白血球减少症)和/或并发感染的3-4级嗜中性白血球减少症·除了下列情况以外的任何4级非血液学毒性:ο<2周的疲劳/乏力ο持续≤3天持续时间的恶心和呕吐(如果在用最佳止吐治疗进行治疗后它们持续>72小时,则仅认为是剂量限制的)ο≤3天持续时间的腹泻(如果在用最佳止泻方案治疗后腹泻持续>72小时,则仅认为是剂量限制的)·除了下列情况以外的3级非血液学毒性:ο任何胃肠失调和脱水(具有相关病征和症状),除非尽管在最佳医学管理>72小时的情况下3级毒性仍持续ο疼痛,除非尽管在最佳医学管理的情况下3级毒性仍持续ο疲劳、发热、流感样症状、感染和寄生虫侵扰ο输注反应(和相关症状),除非在类固醇术前用药之后发生ο肝肾功能异常和电解质异常,如果尽管在最佳医学管理的情况下它们仍持续将根据研究者与发起人之间的讨论来做出不良事件是否被认为是dlt的决定,并且由安全性检查委员会(即部分1a研究者和发起人医学监查员)确认。在安全性检查委员会的判定下,被认为与研究治疗相关的其他不良事件也可被认为是dlt事件。研究者与发起人之间的安全性检查会议将在研究的部分1a过程中有规律地进行,至少每月的会议,或如果需要,则更频繁。部分1b在确定部分1a中的nal-iri剂量之后,将开始研究的部分1b。在部分1b中,将大约50位合格的患者以1:1比率随机化在实验组(组1a:90mg/m2的nal-iri,每2周)与对照组(组1b:静脉内拓扑替康1.5mg/m2持续5天,每21天)之间。在中心位置使用交互式网络响应系统(iwrs),将患者随机化到治疗组。为了减少关于在部分2的随机化中用于分层的预后因素的不平衡,部分1b中的随机化将使用考虑部分2分层因素的最少化程序。铂抗性患者被定义为患有在含铂一线疗法过程中或在含铂一线疗法完成的90天内有进展的疾病的患者。铂敏感的患者被定义为患有在含铂一线疗法完成的90天之后进展的疾病的患者。根据先前公布的研究(vonpawel,2014),为了保持对一线治疗组的铂敏感性分布,将对来自部分1b中的铂敏感或铂抗性患者的不多于30位患者进行随机化。来自部分1b的安全性和疗效结果将确定研究是(或否)进行至部分2。如果符合下列停止标准中的两者,将停止该研究:伊立替康脂质体注射液在12周时的pfs(基于研究者评价)率小于50%,以及静脉内拓扑替康在12周时的pfs(基于研究者评价)率超过伊立替康脂质体注射液至少5个百分点。如果不符合停止标准,将由发起人经与研究的学术指导委员会磋商,在考虑来自研究的部分1的所有可用疗效和安全性数据之后做出进行至部分2的最终决定。部分2:如果不符合来自部分1b的停止标准并且做出决定进行至研究的部分2,将大约420位合格的患者以1:1比率随机化在实验组(组2a:90mg/m2的伊立替康脂质体注射液)与对照组(组2b:静脉内拓扑替康)之间。在中心位置使用交互式网络响应系统(iwrs),将患者随机化到治疗组。基于下列因素,将对随机化进行分层:·诊断时的疾病阶段(局限期与广泛期)·地区(北美与亚洲与其他地区)·铂敏感性(敏感与抗性)·性能状态(ecog0与1)·先前免疫疗法(是与否)·仅地区和铂敏感与抗性将用于疗效分析。将通过使用recist准则(版本1.1),每6周(+/-1周)测量并且记录肿瘤响应。基线处的肿瘤评价为使用造影剂的ct(所需要的胸部/腹部和如果有临床表现时的骨盆)以及使用造影剂的脑mri(脑ct是可接受的)。每次随访肿瘤评价应该使用与基线处所进行的相同的评价,除非医学上有所禁忌。在基线处和在每次评价时将所有患者进行脑成像。除了客观疾病进展以外的其他原因中止研究治疗的患者应该继续随访,直到放射学文档记载进展性疾病。发起人将收集并且存储整个研究过程中所有患者的所有肿瘤测量图像;然而,当地的放射科医师和/或pi评价将确定疾病进展。可由发起人进行扫描的检查以用于独立分析,包括pfs和/或orr的分析。所有患者将至少每月地随访直到死亡或研究结束,以先发生者为准。将仅在部分1b和部分2中使用eortc-qlq-c30、eortc-qlq-lc13以及euroqol五维、五级健康状况问卷(eq-5d-5l)进行生活质量评价。将在随机化之前并且在开始治疗后以6周间隔给药之前和在治疗中止以及在30天随访时,施用这两种手段。将根据美国国家癌症研究所的不良事件通用术语标准版本4.03(ctcaev4.03),评估不良事件(ae)。对于ae的概述,将使用最新meddr字典版本对事件进行编码。当已发生至少333种os事件时,计划初步分析。在已发生至少100种os事件之后,在30%信息时间里进行计划无用性的中期分析。在试验继续的情况中,当已发生至少210种os事件(63%信息时间,在50%的预期死亡事件下)时,将进行中期分析以评价由于实验治疗方案的疗效而提早停止的可能性。将通过独立的数据监测委员会(dmc)进行部分2的常规安全性数据检查。dmc将由独立于发起人的肿瘤学专家和统计学专家组成。在部分2中,在第30位患者治疗持续至少一个周期之后或第30位患者中止研究药物之后,以先发生者为准,将进行dmc的第一次安全性检查。后续数据检查的时间安排和细节将在dmc章程中详细描述。定期检查的项目将包括(但不限于)安全性事件、pk测试结果以及来自中心测试的ugt1a1*28基因型,该中心测试特别注意确定任何研究程序是否需要针对对于ugt1a1*28为纯合的患者而修改。药物代谢动力学仅在周期1中在下列时间点收集用于pk的血浆样品:部分1a和部分1b,组1a(nal-iri组;仅周期1):-第1天:给药前-第1天:nal-iri输注结束-第2天:输注结束后大约24小时-第8天:周期1,第8天(+/-1天),在这一天的任何时候-第15天:给药前-第15天:nal-iri输注结束部分1b,组1b(拓扑替康组;仅周期1):-第1天:给药前-第1天:拓扑替康输注结束-第1天、第2天或第3天:在开始输注后的1.5小时与4小时之间的两个另外的样品。每个样品必须分开至少1小时进行收集。优选在第1天收集这些样品;然而,这两个另外的样品可在第2天或第3天收集。部分2,组2a(伊立替康脂质体注射液组;仅周期1):-第1天:给药前-第1天:伊立替康脂质体注射输注结束-第1天:在开始输注后的2.5小时与6小时之间-第2-6天(任选的):在开始输注后的1天与5天之间的任何时候-第8天:周期1,第8天(+/-1天),在这一天的任何时候。研究群体入选标准疾病特异性入选标准1)根据国际肺癌研究协会(iaslc)组织病理学分类的组织病理学或细胞学确认的小细胞肺癌。根据iaslc的混合或组合的亚型是不允许的;2)如由recist版本1.1准则所定义的可评估疾病(仅具有非靶标病灶的患者为合格的);3)在用于治疗局限期或广泛期sclc的基于铂的一线化学疗法(顺铂或卡铂)或包括基于铂的化学疗法的化放疗之时或之后的进展;以及4)从任何先前化学疗法、外科手术、放射疗法或其他抗肿瘤疗法的作用中恢复(除了脱发以外,恢复至1级或更好)。血液学、生物化学和器官功能入选标准:如通过以下所证明的适当骨髓储备:·在不使用造血生长因子的情况下,anc>1,500个细胞/μl;以及·血小板计数>100,000个细胞/μl;以及·血红蛋白>9g/dl;输血是允许的。如通过以下所证明的适当肝功能:·该机构的正常范围内的血清总胆红素·天冬氨酸转氨酶(ast)和丙氨酸转氨酶(alt)≤2.5xuln(如果存在肝转移,≤5xuln是可接受的)如通过血清肌酐≤1.5xuln和肌酐清除率≥40ml/min所证明的适当肾功能。实际体重应该用于使用cockcroft-gault方程计算肌酐清除率(除了具有体重指数(bmi)>30kg/m2的患者应该替代使用去脂体重):其中对于男性,性别=1,并且对于女性为0.85。不具有任何临床显著发现的ecg从任何先前化学疗法、外科手术、放射疗法或其他抗肿瘤疗法的作用中恢复需要参与试验的转化研究部分,除非被当地监管机构所禁止,并且提供存档的肿瘤组织(如果可用的话)年龄至少18岁能够理解并且签署知情同意书(或具有能够做这些事情的法定代表人)患者必须符合以上列出的所有入选标准并且不符合下列排除标准中的任一种:一般的排除标准1)由研究者认为的可能影响患者签署知情同意书、合作并且参与该研究、或影响结果解释的任何医学或社会状况;2)妊娠或哺乳;有生育潜力的女性在招收时必须基于尿液或血清妊娠测试而测试为妊娠阴性。在研究过程中并且在最后一次剂量的研究药物之后4个月,具有生殖潜能的男性患者和女性患者两者必须同意使用高效节育方法。疾病特异性排除标准1)用伊立替康、拓扑替康或任何其他拓扑异构酶i抑制剂(包括研究的拓扑异构酶i抑制剂)进行的先前治疗方案;2)具有大细胞神经内分泌癌的患者;3)进行了多于一种先前细胞毒性化学疗法的方案的患者4)多于一线免疫疗法(例如纳武单抗、帕姆单抗、易普利单抗、阿特珠单抗、替西木单抗和/或度伐单抗)。一线免疫疗法被定义为下列内容:作为(i)与化学疗法组合,接着在一线环境中免疫疗法维持,(ii)仅作为对一线化学疗法响应后的维持而给予的单一疗法或免疫疗法试剂的组合或(iii)作为进展后二线治疗而给予的免疫疗法;5)具有免疫疗法诱导结肠炎病史的患者;6)除了如上所述的1线含铂方案或免疫疗法以外的任何先前全身性治疗;7)具有下列cns转移的患者:i)在预防性和/或治疗性头颅放射(全脑立体定向放射)之后发展了新的或进展性脑转移的患者。ii)具有症状性cns转移的患者(如果在头颅放射后≥2周神经症状为无症状的,接受头颅放射疗法的具有脑转移的患者为合格的并且不用皮质类固醇来治疗cns转移。具有无症状的脑转移的患者为合格的,直接招收到该研究中)。iii)具有癌性脑膜炎的患者;8)在接受第一次剂量的伊立替康脂质体注射液之前的至少1周,不能中止使用强cyp3a4或ugt1a1抑制剂,或在接受第一次剂量的伊立替康脂质体注射液之前的至少2周,不能中止使用强cyp3a4诱导剂;9)存在另一种活动性恶性肿瘤;或10)在本研究中的第一次计划给药日之前4周内、或小于研究试剂的至少5个半衰期的时间间隔内(取较小值)施用的研究疗法。血液学、生物化学和器官功能排除标准1)在入选之前小于6个月的严重动脉栓塞事件(例如心肌梗塞、不稳定性心绞痛、中风);2)iii类或iv类nyha充血性心脏衰竭、室性心律失常或不受控制的血压;3)在研究者看来可能危害患者参与试验或影响研究结果的活动性感染(例如急性细菌感染、肺结核、乙型活动性肝炎或活动性hiv);4)对伊立替康脂质体注射液的任何组分、其他脂质体产品或拓扑替康已知的超敏性;或临床显著的胃肠病症,包括肝脏病症、出血、炎症、闭塞或>1级的腹泻。研究时长意图为患者将被治疗直到疾病进展或不可接受的毒性。在治疗中止之后,患者将回到研究场所进行30天随访。在这次随访之后,患者将继续每月一次通过电话或拜访研究场所来跟踪总存活状态直到死亡或研究结束,以先发生者为准。将患者分配至治疗组的方法部分1a:在已完成所有筛选评价并且已完成第一次患者报告结果评价之后,合格的患者将进入部分1a。部分1b:部分1b将在部分1a中的剂量选择之后开始。在已完成所有筛选评价并且已完成第一次患者报告结果评价之后,使用计算机化的交互式网络响应系统(iwrs),以1:1比率将合格的患者随机化到下列治疗组之一:部分1b中的随机化将使用考虑部分2分层因素的最少化程序(mcentegart,2003)。组1a(实验组):伊立替康脂质体注射液组1b(对照组):静脉内拓扑替康随机化必须发生在计划给药的7天内。部分2:部分2将在通过停止标准后并且基于发起人经与学术指导委员会磋商的决定而开始。在已完成所有筛选评价并且已完成第一次患者报告结果评价之后,使用计算机化的交互式网络响应系统(iwrs),以1:1比率将合格的患者随机化到下列治疗组之一:组2a(实验组):伊立替康脂质体注射液组2b(对照组):静脉内拓扑替康随机化必须发生在计划给药的7天内。基于下列预后因素,将对随机化进行分层:-地区(北美与亚洲与其他地区)-铂敏感性(敏感与抗性)-诊断时的疾病阶段(局限期与广泛期)-性能状态(ecog0与1)-先前免疫疗法(是与否)铂抗性患者被定义为患有在含铂一线疗法过程中或在含铂一线疗法完成的90天内有进展的疾病的患者。铂敏感的患者被定义为患有在含铂一线疗法完成的90天之后进展的疾病的患者。伊立替康脂质体注射液的施用部分1a:在6周的周期中,每2周在70mg/m2(基于伊立替康游离碱表示的强度;大约等效于80mg/m2的无水盐)的剂量下经过90分钟静脉内施用伊立替康脂质体注射液。70mg/m2剂量应该被认为是耐受的并且探索90mg/m2,应该在6周的周期中,每2周在90mg/m2(基于伊立替康游离碱表示的强度;大约等效于100mg/m2的无水盐)的剂量下经过90分钟静脉内施用伊立替康脂质体注射液。部分1b和2:将在90mg/m2(基于伊立替康游离碱表示的强度;大约等效于100mg/m2的无水盐)的剂量下施用伊立替康脂质体注射液:在6周的周期中每2周,经过90分钟静脉内施用(除非在部分1中被认为是不可接受的)。在施用之前,必须将适当剂量的伊立替康脂质体注射液稀释在5%葡萄糖注射液(d5w)或0.9%氯化钠注射液中,最终体积为500ml。应该注意,不要使用除了d5w或0.9%氯化钠以外的任何稀释剂。ugt1a1*28监测将对所有患者收集ugt1a1*28基因型,并且集中评价。结果将向研究场所和发起人提供。研究场所也将被要求包括sae报告形式的来自ugt1a1*28基因型分型的结果。用伊立替康脂质体注射液治疗的所有患者,无论ugt1a1*28基因型的结果如何,将用相同起始剂量的伊立替康脂质体注射液治疗并且将遵循相同的剂量减少规则。在如将由发起人医学监查员和通过dmc(在部分2中)所进行的研究期间患者常规安全性监测的过程中,将ugt1a1*28纯合的患者的安全性和pk与针对ugt1a1*28并非纯合的那些患者进行比较,以便确定任何不同的给药策略(诸如伊立替康脂质体注射液的降低的起始剂量和/或不同的剂量减少)是否是ugt1a1*28纯合的患者所需要的。在第30位患者完成一次治疗周期或中止治疗之后,以先发生者为准,将进行第一次安全性dmc会议。在用拓扑替康治疗的患者中,预期到ugt1a1*28与安全性之间没有相关性。研究治疗伊立替康脂质体注射液:部分1a:(安全性部分)在6周的周期中每2周,经过90分钟静脉内施用伊立替康脂质体注射液70mg/m2(表示为伊立替康游离碱的强度;大约等效于80mg/m2的无水盐)或在6周的周期中每2周,经过90分钟静脉内施用伊立替康脂质体注射液90mg/m2(表示为伊立替康游离碱的强度;大约等效于100mg/m2的无水盐)。部分1b和部分2:组1a和2a(实验组):伊立替康脂质体注射液90mg/m2(表示为伊立替康游离碱的强度;大约等效于100mg/m2的无水盐):在6周的周期中每2周,经过90分钟静脉内(除非在部分1中被认为是不可接受的)。组1b和2b(对照组):拓扑替康1.5mg/m2:每天经过30分钟静脉内,持续连续5天,在6周的周期中每3周。伊立替康脂质体注射液:部分1a、部分1b组1a和部分2组2a:支持性护理措施应该遵循的处方信息中所概述的准则。高达两次伊立替康脂质体注射液的剂量减少为毒性所容许的。基于研究者判断,允许将预防性g-csf(基于研究者偏好,短期和长期作用的生长因子两者均为可接受的)与第二或更稍后的剂量的伊立替康脂质体注射液一起使用。拓扑替康:部分1b组1b和部分2组2b(静脉内拓扑替康)拓扑替康的预期剂量为每3周静脉内一次1.5mg/m2,持续连续5天。剂量、施用和剂量减少应该遵循静脉内拓扑替康的处方信息中所概述的准则。随机化到用拓扑替康进行的治疗中的患者应该被认为在所有周期中用于预防性g-csf,该预防性g-csf在最后一次剂量之后24小时开始(基于研究者偏好,短期和长期作用的生长因子两者均为可接受的)。每位患者高达两次拓扑替康的剂量减少为毒性所允许的。容许剂量延迟以允许从治疗相关的毒性中恢复。建议将预防性抗生素用于处于高风险感染并发症的患者。研究产品:伊立替康脂质体注射液(又称为nal-iri、聚乙二醇化的三水盐酸脂质体伊立替康、mm-398、pep02、bax2398和)为无菌、白色至浅黄色不透明等渗脂质体分散体。每10ml单一剂量的小瓶含有4.3mg/ml浓度的43mg伊立替康游离碱。脂质体为单层脂质双层囊泡,直径为大约110nm,该囊泡包囊含有胶凝化或沉淀状态下作为蔗糖八硫酸酯盐的伊立替康的水性空间。它将作为无菌一次性小瓶而供应,该小瓶含有4.3mg/ml浓度的43mg伊立替康游离碱。伊立替康脂质体注射液必须在避光的情况下冷藏存储(2℃至8℃,36°f至46°f)。不冷冻。部分1a如果在一个同期群6位患者中具有dlt的患者数量不超过1,将决定剂量为可接受用于进行至部分1b。基于这个规则,在作为dlt概率真率的函数的剂量下进行至部分1b的概率示于表6中。表6不可接受的毒性的真率发展到随机化的概率0.050.970.100.890.150.770.200.650.250.530.300.420.350.320.400.23部分1b部分1b的目的是为了在随机化环境中提供安全性和疗效数据的试点样品。如果针对益处/风险,观察到伊立替康脂质体注射液基本上不如拓扑替康,出于实用目的选择部分1b的样品大小以实现研究缩短。基于在12周观察到的pfs率的疗效规则在本方案中实施为正式停止规则,同时也将考虑另外的数据并且也可导致不进行至部分2的决定。下文描述了部分1b中的研究设计所给定的正式停止规则的操作特征。使用二项分布粗略估计并且将对照组内的12周时的无进展患者的真正比例假定为0.55,作为伊立替康脂质体注射液组真率的函数的研究将停止的概率示于表7中。表7当部分1b中的所有患者已完成肿瘤评价时,将通过对数秩检验进行pfs的最终治疗比较。如果审查率假定为10%,预期在最终分析时将发生45起事件。如果pfs危险比为0.64(例如,伊立替康脂质体注射液将中值pfs从3.5延长至5.5个月),则这种分析将具有大约75%的效能来用单侧水平0.20检验检测治疗差异。部分2初步终点为总存活期(os)。将总共420位患者以1:1比率随机化到两个治疗组。直到在两个治疗组中观察到至少333种os事件的随访为检测hr的真正危险比≤0.714提供至少85%的效能(mos:7.5个月与10.5个月),该检测使用了分层的对数秩检验(通过地区(北美与亚洲与其他地区)和铂敏感性(敏感与抗性)来分层),其中总单侧显著性水平为0.025(针对中期分析调整)。假定经过25个月,每月招收上升至21位患者并且两个治疗组中的失访率为5%,预期初步分析的时间在39个月。当在意向治疗(itt)群体中已观察到计划的最终os事件数量的大约30%(即,333种os事件中的100种)时,将进行无用性的中期分析。在研究进行的情况下,当已发生大约210种os事件(在整个研究群体中计划的os事件的63%和预期的事件的50%)时,将进行第二中期分析以评估无用性和疗效。综述:分类变量将通过频率分布(患者的数量和百分数)来概括,并且连续变量将通过描述性统计(平均值、标准偏差、中值、最小值、最大值)来概括。使用与部分2中相同的结果测量值,将描述性地报告部分1中nal-iri的疗效和安全性。另外,将详细描述研究的部分1中发生的不良事件。在部分1中招收并且用研究药物治疗的患者将包括部分1安全性群体。将描述性地呈现这些患者的安全性和疗效。部分2中随机化的患者将包括意向治疗(itt)群体。这将是比较地评估以评估实验组疗效的群体。在疗效的itt分析中,将根据随机化的治疗分配考虑每位患者。接受任何部分的任何研究药物的患者将定义部分2安全性群体。针对分层分析,分层因素将为地区(北美、亚洲、其他地区)和铂敏感性(敏感、抗性)的随机化分层因素。分层因素的分类将根据随机化而定。初步疗效分析(部分2):os被定义为自随机化日期至死亡日期的月数。在初步分析时没有观察到死亡的患者将具有根据最后一次记录的活着的日期审查的os。将使用对两个治疗组之间的os差异进行比较的分层对数秩检验进行初步分析,其中单侧显著性水平在0.025。分层因素将包括随机化分层因素并且分类将根据随机化而定。将使用kaplan-meier方法来估计中值os(具有95%置信区间)并且以图形形式展示os时间。将使用分层的cox比例危险模型来估计危险比并且其对应的置信区间为95%。将在统计分析计划(sap)中描述os的灵敏度分析。关键的二次分析(部分2):关键的二次终点为pfs、orr、在呼吸困难、咳嗽和疲劳上具有症状改善的患者比例。关键的二次终点将测试不多于一次。如果os的初步终点在中期为统计学显著的,将在中期检验二次终点的测试。否则,如果发现os在最终os分析时为统计学显著的,将在该分析时检验二次终点。将以分段层次结构方式进行关键二次终点的假设测试(glimm,e等人,statisticsinmedicine201029:219-228)。pfs比较的标称水平将取决于是否在中期或在计划的最终分析时进行检验,并且将并入类似于用于os的α损耗函数。如果os和pfs两者均为显著的,则将在单侧0.025水平(如针对pfs所述,基于损耗函数调整的标称α)下检验orr和eortc-qlq症状,其中针对4种计划比较的单侧α水平测试,每个p值使用benjamini-hochberg校正(benjamini和hochberg,j.royalstatisticalsoc.b200557,289-300)来调整。使用sasprocmulttest以fdr选项或等效算法,将报告调整的p值。任何非统计学显著的参数将被认为是描述性和探索性的。无进展存活期:无进展存活期为从随机化至使用recist版本1.1的第一次记载客观疾病进展(pd)或由于任何原因引起的死亡(以先发生者为准)的时间。根据研究者评价确定pfs。如果既没有观察到死亡也没有观察到进展,将在最后一次观察到的肿瘤评价日的日期对数据进行审查。将对随机化时不具有有效肿瘤响应评估的患者在随机化日期进行审查。将对在记载的pd之前开始新的抗癌治疗的患者在开始新治疗之前的最后一次观察到的肿瘤评价日期进行审查。将对在不可接受的长间隔(即,2次或更多次漏服或不确定的计划评价)之后具有记载的pd或死亡的患者在进展或死亡之前的最后一次观察到的非pd肿瘤评价日的时间进行审查。将使用分层对数秩检验来评估治疗之间的pfs差异。将使用kaplan-meier方法来估计中值pfs(具有95%置信区间)并且以图形形式展示pfs时间。将使用分层的cox比例危险模型来估计pfs危险比并且其对应的置信区间为95%。将使用分层的对数秩检验(通过地区和铂敏感性来分层)来评估治疗之间的pfs差异。将使用kaplan-meier方法来估计中值pfs(具有95%置信区间)并且以图形形式展示pfs时间。将使用分层的cox比例危险模型来估计pfs危险比并且其对应的置信区间为95%。将在sap中描述pfs的灵敏度分析。客观响应:客观响应率(orr)为根据recist版本1.1准则的实现部分响应或完全响应的患者比例。将计算orr的估计值及其95%ci。将使用通过地区和铂敏感性来分层的cochran-mantel-haenszel方法,比较治疗组之间的orr差异。具有肺癌症状改善的患者比例:这种二次分析将考虑咳嗽、呼吸困难和疲劳的患者报告的eortc-qlq-lc13症状量表,因为这些被最清楚地认为是疾病相关的并且可评估关于具有改善的患者比例的治疗影响。剩余的eortc-qlq症状域将在探索性分析中评价。症状改善被定义为实现并且6周维持症状分量表得分低于基线至少10个量表百分点(在转换至0-100量表之后)。将通过治疗组把响应分类制成表,并且统计分析将比较针对给定症状的响应者的比例。对于每种症状,将通过治疗组把具有改善的患者比例制成表,其中基于正态近似的置信区间为95%。具有症状改善的患者比例的差异将以对应的95%置信区间来呈现。将使用通过地区和铂敏感性来分层的cochran-mantel-haenszel方法,比较治疗方案之间的具有症状改善的患者比例。安全性分析:将使用被定义为接受任何研究药物的所有患者的安全性群体来进行安全性分析(不良事件和实验室分析)。治疗分配将根据所接受的实际治疗。将使用最新meddr字典对不良事件进行编码。将根据ncictcae版本4.03来对严重性分级。治疗突发不良事件(teae)被定义为从第一次研究药物暴露至最后一次研究药物暴露日期后的30天所报告的任何不良事件。将针对以下概括患者的频率和百分数:任何级别的teae、3级或更高级teae、研究药物相关的teae、严重teae、导致剂量修改的teae、以及导致研究药物中止的teae。将通过系统器官类别和优选术语来概括不良事件。将由患者列出所有不良事件数据。将根据参数类型概括实验室数据。在适用的情况下,将基于ncictcae版本4.03标准来分配实验室安全性参数的毒性分级。qtcf分析:将在本研究的部分1中接受伊立替康脂质体注射液的患者中评估使用伊立替康脂质体注射液治疗的qtcf延长的潜力。对于初步qtcf延长分析,将使用混合效应建模由暴露-qtcf关系获得qtcf的预测变化。将通过按时间点评估和分类分析来进行灵敏度分析。eortc-qlq结果将根据eortc准则(fayers,2001)进行eortc-qlq-c30问卷的分析。将基于eortc评分手册对eortcqlq-c30和qlq-lc13的分量表进行评分。将得分标准化,使得eortcqlq-c30或qlq-lc13的更高得分将表示起作用的水平更高(“更好”)和/或症状的水平更高(“更差”)。用于具有症状改善的患者的比例的分析方法如在关键的二次分析(章节11.5.2.3)中所述。针对每个qlq-c30和qlq-lc13分量表,将报告按治疗组划分的具有症状改善的患者比例的频率表。另外的eortc-qlq分析的细节将提供在统计分析计划中。将报告随时间推移的原始标准化分量表得分和距离基线的变化。将描述性地比较治疗组之间的平均变化得分,并且可经由纵向建模(即,协变量分析和重复测量建模)来进行探索。eq-5d-5l:将报告随时间推移的原始得分和距离基线的变化。将描述性地比较治疗组之间的平均变化得分,并且经由纵向建模(即,协变量分析和重复测量建模)来进行探索。cns进展时间:被定义为从随机化至cns进展出现的时间,如由rano-bm工作组所定义(lin等人,lancetoncology2015)。将通过kaplan-meier方法来描述cns进展时间,并且将使用分层的对数秩检验来比较治疗。药物代谢动力学(pk)和药效动力学(pd)分析:将使用非线性混合效应建模,由样品浓度定量总伊立替康、sn-38和拓扑替康的血浆药物代谢动力学(pk)。初始pk分析将使用经验贝叶斯估计,然而,将进行另外的协变量分析以评估特异于sclc的替代基线因素。所得的pk估计值将用于评估pk与pd(疗效和安全性终点)之间的相关性。拓扑替康pk将用于提供另外的数据以理解通过将本研究中的pk分布和pk与疗效/安全性的相关性与历史值进行比较得到的来自部分1b的结果。剂量修改所有剂量修改应该基于先前最差的毒性。表8:伊立替康脂质体注射液的建议剂量修改a所提到的所有剂量均基于伊立替康游离碱b美国国家癌症研究所的不良事件通用术语标准,版本4.03注射用拓扑替康应该仅在具有大于或等于1,500/mm3(1.5x109/l)的基线嗜中性白血球计数和大于或等于100,000/mm3(100x109/l)的血小板计数的患者中开始拓扑替康。不应该在后续周期中施用拓扑替康,除非嗜中性白血球计数为≥1x109/l,血小板计数为≥100x109/l,并且血红蛋白水平为≥9g/dl(如果必要,在输血后)。应该延迟治疗以允许足够时间来恢复,并且在恢复后,应该根据下表9中的准则施用治疗。拓扑替康的剂量减少应该发生在下列毒性的情况下:-4级嗜中性白血球减少症(anc<500/mm3或<0.5x109/l);-4级血小板减少症(血小板计数<25,000/mm3或<0.5x109/l);-3级或4级非血液学毒性,除了恶心和呕吐。在恶心和呕吐的情况下,如果尽管在最佳医学管理的情况下仍发生3级或4级毒性,应该发生剂量减少剂量减少决定应该基于先前最差的毒性。容许从剂量水平0至剂量水平2的改动。建议将预防性抗生素用于处于高风险感染并发症的患者。如表9中所示,每位患者高达两次拓扑替康的剂量减少为毒性所容许的。如果需要第三次剂量减少来管理毒性,应该中止拓扑替康治疗。表9:后续周期的建议拓扑替康剂量修改方案如果肌酐清除率在20与39ml/min之间,应该将患者中的拓扑替康的剂量减少至0.75mg/m2/天,持续连续五天。如果新诊断为间质性肺病,应该中止拓扑替康。实施例8:脂质体伊立替康制备可用多步法来制备脂质体伊立替康。首先,将脂质溶解于加热的乙醇中。脂质可包含以3:2:0.015摩尔比组合的dspc、胆固醇和mpeg-2000-dspe。优选地,脂质体可包囊伊立替康蔗糖八硫酸酯盐(sos),包囊在由以3:2:0.015摩尔比组合的dspc、胆固醇和mpeg-2000-dspe组成的囊泡中。将所得的乙醇-脂质溶液分散在含有取代的胺和聚阴离子的水性介质中,在有效于形成适当大小(例如80-120nm)的含有取代的胺(处于铵形式)和聚阴离子的基本上单层的脂质体的条件下,该脂质体包囊在由分散的脂质形成的囊泡内。可例如通过在高于脂质转变温度例如60℃-70℃下使乙醇脂质溶液与含有取代的胺和聚阴离子的水性溶液混合,并且通过一个或多个径迹蚀刻(例如聚碳酸酯)膜过滤器在压力下挤出所得的水合脂质混悬液(多层脂质体)来进行分散,该膜过滤器具有限定的孔径,例如50nm、80nm、100nm或200nm。取代的胺可为三乙胺(tea),并且聚阴离子可为浓度约0.4-0.5n的以化学计量比组合的蔗糖八硫酸酯盐(sos)(例如,tea8sos)。然后去除(例如,通过凝胶过滤、渗析或超滤)所有或基本上所有的未包埋的tea或sos,之后在有效于允许伊立替康进入脂质体与离开脂质体的tea交换的条件下,使脂质体与伊立替康接触。该条件可包括选自由以下组成的组的一种或多种条件:将渗透剂(例如,5%葡萄糖)加入脂质体外部介质中以平衡包埋tea-sos溶液的渗透压和/或防止载荷过程中脂质体渗透破裂,调节和/或选择ph(例如,达到6.5)以在载荷过程中减少药物和/或脂质降解,以及增加温度至高于脂质体脂质的转变温度(例如,达到60℃-70℃)以加速tea和伊立替康的跨膜交换。通过在整个脂质体上与tea交换载荷伊立替康优选地继续直到所有或基本上所有的tea从脂质体中去除,从而耗尽tea在整个脂质体上的浓度梯度。优选地,伊立替康脂质体载荷过程继续直到伊立替康与蔗糖八硫酸酯盐的克当量比为至少0.9、至少0.95、0.98、0.99或1.0(或在约0.9-1.0、0.95-1.0、0.98-1.0或0.99-1.0的范围内)。优选地,伊立替康脂质体载荷过程继续直到至少90%、至少95%、至少98%、至少99%或更多的tea从脂质体内部中去除。伊立替康可在脂质体内形成伊立替康蔗糖八硫酸酯盐,诸如以约8:1的摩尔比的伊立替康和蔗糖八硫酸酯盐。接下来,使用例如凝胶(大小排阻)色谱法、渗析、离子交换或超滤方法将任何剩余的脂质体外伊立替康和tea去除。用可注射的药理学上可接受的流体,例如缓冲等渗盐水来替换脂质体外部介质。最后,例如通过0.2微米过滤对脂质体组合物进行灭菌,将其分散到剂量小瓶中,贴标签,并且例如在2℃-8℃下冷藏后存储直到使用。在去除剩余脂质体外伊立替康和tea的同时,可用药理学上可接受的流体来替换脂质体外部介质。例如通过在约6.5-8.0的ph、或其间的任何合适ph值(包括,例如7.0-8.0和7.25)下制备组合物,可对组合物的脂质体外ph进行调节或者选择,以便提供希望的存储稳定性特性(例如,以便减少在4℃存储期间180天中脂质体内溶血磷脂(lyso-pc)的形成)。如本文所述更详细地提供的内容,可制备具有脂质体外ph值、伊立替康游离碱浓度(mg/ml)和各种浓度蔗糖八硫酸酯盐的伊立替康脂质体。以分别对应于3:2:0.015摩尔比的量(例如,1264mg/412.5mg/22.44mg)称重dspc、胆固醇(chol)和peg-dspe。将脂质溶解于氯仿/甲醇(4/1v/v)中,仔细混合并且分成4个等分式样(a-d)。使用旋转蒸发仪在60℃下蒸发每个样品至干燥。通过放置在室温下在真空(180μtorr)下持续12h,从脂质中去除残余氯仿。在60℃下将干燥的脂质溶解于乙醇中,并且加入适当浓度的预先温热的tea8sos以使最终醇含量为10%(v/v)。脂质浓度为75mm。在约65℃下,使用lipex热筒挤出器(加拿大北部脂质公司(northernlipids))将脂质分散体挤出穿过2个堆叠的0.1μm聚碳酸酯膜(nucleopore)10次,产生具有95-115nm的典型平均直径的脂质体(通过准弹性光散射来确定的)。必要时,用1nnaoh将挤出的脂质体的ph调节至ph6.5。通过离子交换色谱法和大小排阻色谱法的组合来纯化脂质体。首先,用1nnaoh来处理dowextmira910树脂,接着用去离子水洗涤3次,然后接着的是3nhci洗涤3次,然后用水洗涤多次。使脂质体穿过准备好的树脂,并且通过使用流通池电导率仪(瑞典乌普萨拉的法玛西亚公司(pharmacia))来测量洗脱的级分的电导率。如果电导率小于15μs/cm,级分被认为是可接受用于进一步纯化。然后将脂质体洗脱液应用至用去离子水平衡的sephadexg-75(法玛西亚公司)柱,并且测量收集的脂质体级分的电导率(典型地小于1μs/cm)。通过加入40%葡萄糖溶液至最终浓度为5%(w/w)和加入来自储备溶液(0.5m,ph6.5)的缓冲液(羟乙基哌嗪乙硫磺酸(hepes))至最终浓度为10mm,实现跨膜等渗。通过将三水盐酸伊立替康溶解在去离子水中达到15mg/ml的无水盐酸伊立替康来制备伊立替康的储备溶液,考虑从每个批次的分析证书中获得的含水量和杂质水平。通过在500g/mol脂质体磷脂下加入伊立替康并且在热水浴中加热至60℃±0.1℃持续30min来开始药物载荷。在从水浴中移除后,通过浸入冰水浴来快速冷却溶液。使用由羟乙基哌嗪乙硫磺酸缓冲的盐水(10mm羟乙基哌嗪乙硫磺酸,145mmnacl,ph6.5)平衡和洗脱的sephadexg75柱,通过大小排阻色谱法来去除脂质体外药物。通过hplc分析样品的伊立替康,并且通过bartlett方法(参见磷酸盐确定)分析样品的磷酸盐。为了存储,将样品分成4ml的等分试样,并且使用1nhcl或1nnaoh如在结果中所示调节ph,在无菌条件下无菌过滤,并且填充到无菌透明玻璃小瓶中,该小瓶在氩气下用内具螺纹盖密封并且在4℃下放置在恒温控制的冰箱中。在限定的时间点,从每种样品中移出等分试样并且测试外观、大小、药物/脂质比以及药物和脂质化学稳定性。使用库尔特纳米粒度仪(coulternano-sizer),在90°的角度下通过动态光散射在稀释的样品中确定脂质体大小,并且该脂质体大小以通过累积量方法获得的平均值±标准偏差(nm)呈现。实施例9:onivyde(mm-398)脂质体伊立替康本文描述的存储稳定的脂质体伊立替康的一个优选例子为作为onivyde(伊立替康脂质体注射液)市售的产品。onivyde为用三水盐酸伊立替康配制成用于静脉内使用的脂质体分散体的拓扑异构酶抑制剂。onivyde指示用于治疗在基于吉西他滨的疗法后疾病进展之后的胰腺转移性腺癌。onivyde为ph约7.25的存储稳定的脂质体。onivyde产品含有包囊在脂质体中的伊立替康蔗糖八硫酸酯盐,该脂质体由三水盐酸伊立替康起始材料获得。伊立替康的化学名称为(s)-4,11-二乙基-3,4,12,14-四氢-4-羟基-3,14-二氧1h-吡喃并[3’,4’:6,7]-吲哚嗪并[1,2-b]喹啉-9-基-[1,4’二哌啶]-1’-甲酸酯。可基于用于制备伊立替康脂质体的三水盐酸伊立替康起始材料的等效量或基于脂质体中的伊立替康的量来计算onivyde的剂量。每克三水盐酸伊立替康存在约866mg的伊立替康。例如,基于三水盐酸伊立替康起始材料的量的80mgonivyde剂量实际上在最终产品中含有约0.866x(80mg)的伊立替康游离碱(即,基于盐酸伊立替康起始材料的重量的80mg/m2onivyde剂量等效于最终产品中的约70mg/m2的伊立替康游离碱)。onivyde为无菌的白色至浅黄色不透明等渗脂质体分散体。每10ml单一剂量的小瓶含有4.3mg/ml浓度的43mg伊立替康游离碱。脂质体为单层脂质双层囊泡,直径为大约110nm,该囊泡包囊含有胶凝化或沉淀状态下作为蔗糖八硫酸酯盐的伊立替康的水性空间。囊泡由以下构成:6.81mg/ml1,2-二硬脂酰-sn-丙三基-3-磷酸胆碱(dspc)、2.22mg/ml胆固醇以及0.12mg/ml甲氧基封端的聚乙二醇(mw2000)-二硬脂酰磷脂酰乙醇胺(mpeg-2000-dspe)。每ml还含有4.05mg/ml作为缓冲剂的2-[4-(2-羟乙基)哌嗪-1-基]乙磺酸(hepes)和8.42mg/ml作为等渗试剂的氯化钠。每个onivyde小瓶含有呈单一剂量小瓶中的白色至浅黄色不透明脂质体分散体的43mg/10ml伊立替康游离碱。在一个例子中,onivyde单位剂型为包含如下量的包囊伊立替康的脂质体的药物组合物,该量提供总量为约90mg/m2的伊立替康游离碱或等效于100mg/m2三水盐酸伊立替康的量的伊立替康。单位剂型可为通过将浓度约4.3mg伊立替康游离碱/ml可注射流体的单位剂型(例如,小瓶)稀释成约500ml的总体积而获得的静脉内制剂。制备onivyde用于如下从小瓶中稀释等渗脂质体分散体进行施用:从小瓶中取出计算的体积的onivyde。onivyde被稀释成500ml5%葡萄糖注射液(usp)或0.9%氯化钠注射液(usp),并且通过轻轻翻转混合稀释的溶液;使稀释的溶液避光并且当存储在室温下时在制备的4小时内或当存储在冷藏条件[2℃至8℃(36°f至46°f)]时在制备的24小时内施用稀释的溶液。实施例10:与人源性异种移植(pdx)模型(crc、sclc和胰腺)相比,在sclc细胞系来源的异种移植(cdx)模型(nci-h1048、dms-114、h841)中评估nal-iri递送伊立替康和sn-38至肿瘤的能力。向具有异种移植肿瘤的小鼠静脉内施用伊立替康脂质体注射液。施用后的24小时,处死小鼠并且收获肿瘤。通过高效液相色谱法(hplc)测量肿瘤中的伊立替康和sn-38。将数据归一化成注射的剂量/肿瘤重量。图7a示出如通过在sclc小鼠异种移植模型(h841、h1048和dms-53)中施用后24小时的肿瘤cpt-11所评价,增加的肿瘤sn-38水平与增加的肿瘤沉积相关。图7b示出crc、sclc和胰腺pdx肿瘤中的羧酸酯酶(ces)活性,示出了sclcpdx肿瘤具有比得上其中伊立替康为活性的其他适应症的ces活性。在sclc细胞系(dms114、nci-h1048)中,用sn-38进行的治疗使细胞活力减少>90%。如图7c所示(针对nci-h1048细胞),在1-10nm之间观察到有效的细胞生长抑制,其中随着时程高达88小时的暴露时间增加,细胞杀死增加。开始发生细胞杀死的sn-38浓度范围与在施用伊立替康脂质体注射液后72小时从患有各种实体瘤的患者取得的肿瘤活检物中测量的sn-38的量一致(范围:3-163nm;ramanathan等人,eur.j.cancer,2014年11月;50:87),其覆盖时间依赖性sn-38生长抑制曲线(以虚线内的区域示出)。在dms-114细胞中看到类似的作用。使用zoom系统来确定细胞系中sn-38的细胞生长抑制动力学。图7d为示出了细胞敏感性的图形;topo1抑制剂的细胞毒性随着暴露而增加。图7e为示出了拓扑替康施用严重地受毒性限制,从而与安能得介导的延长sn-38暴露相比限制topo1的持续抑制的图表。实施例11.用于在患有小细胞肺癌的患者中评估拓扑替康脂质体注射液(nal-iri,mm-398)的临床前支持在dms-53和nci-h1048异种移植模型中评估作为单一疗法的nal-iri的抗肿瘤活性。将细胞皮下植入到nod-scid小鼠的右翼中;当肿瘤达到大约280mm3时开始治疗。在16mg/kg盐q1w的剂量下给药nal-iri,该剂量等效于提出的90mg/m2游离碱q2w的临床剂量。在0.83mg/kg/周,每7天的第1-2天给药拓扑替康,该剂量近似1.5mg/m2(每21天第1-5天)的临床剂量强度。使用先前确立的高效液相色谱法,在注射后24小时测量nal-iri和非脂质体伊立替康的肿瘤代谢物水平。在dms-53中单一疗法治疗的结果示于图8a中并且在nci-h1048中的结果示于图8b中。在图8a和8b中,垂直的虚线指示给药的天数并且基于自基线的肿瘤体积变化来确定响应率:cr:肿瘤体积(tv)变化<-95%;pr:-95%≤tv变化<-30%;sd:-30%≤tv变化<30%;pd:tv变化≥30%。基于肿瘤生长动力学和总存活期,nal-iri展示出比拓扑替康显著更大的抗肿瘤活性。此外,在用nal-iri治疗的nci-h1048模型中,7只小鼠中有7只在4个治疗周期后经历完全肿瘤消退,并且在最后一次剂量后持续至少50天,与用拓扑替康治疗的7只小鼠中0只小鼠相比。sclc模型中的羧酸酯酶活性和对sn-38的敏感性比得上其中nal-iri或盐酸伊立替康证明为临床有效的适应症(例如胰腺癌、结肠直肠癌)中的情况。发现在sclc肿瘤中nal-iri递送伊立替康至肿瘤的程度与其他肿瘤类型类似或更大。nal-iri(16mg/kg盐)的肿瘤伊立替康和sn-38水平分别比非脂质体伊立替康(30mg/kg盐)高12至57倍和5至20倍。nal-iri证实了sclc的两种异种移植模型中临床相关剂量水平下的抗肿瘤活性,并且在4个治疗周期后产生完全或部分响应,与具有受限的肿瘤生长控制的拓扑替康相比。在sclc的h841大鼠原位异种移植模型中的mm-398(安能得)的抗肿瘤活性示于图8c中,该图为示出在接种后的数天里用对照、安能得(30或50mg/kg盐)、伊立替康(25mg/kg)或拓扑替康(4mg/kg)治疗的大鼠的存活百分比的图形。在30和50mg/kg下用安能得治疗的大鼠示出比用对照、伊立替康或拓扑替康治疗的那些大鼠更长的存活时间。在多种sclc异种移植模型中,mm-398具有抗肿瘤活性。在临床相关的剂量(16mg/kg/周mm-398、0.8mg/kg/周拓扑替康)下,mm-398具有比拓扑替康更大的抗肿瘤活性和延长的存活期。这些研究证实了在sclc临床前模型中,在临床相关剂量下,nal-iri比拓扑替康更有活性,并且因此支持在先前基于铂的疗法上有进展的患有sclc的患者中提出的nal-iri与拓扑替康的随机化3期试验。实施例12在具有sclc肿瘤的异种移植模型dms-53和nci-h1048中,将nal-iri的肿瘤代谢物水平与非脂质体伊立替康进行比较(图9a和9b)。基于体表面积给药并且按体重比例,小鼠中的nal-iri和非脂质体盐酸伊立替康的临床相关剂量分别为约16mg/kg(盐)和30mg/kg(盐)。在16mg/kg盐(q1w)下给药的nal-iri等效于提出的90mg/m2游离碱q2w的临床剂量。以30mg/kgq1w给予的盐酸伊立替康近似300mg/m2q3w的临床剂量强度,其在二线sclc患者中产生与拓扑替康(当前护理标准)类似的疗效(zhaoml,biq,renhx,tianq,baoml.clinicalobservationofirinotecanortopotecanassecond-linechemotherapyontreating43patientswithsmall-celllungcancer.chinoncol.2011;21:156–158)。使用高效液相色谱法,在注射(经由尾静脉的静脉内)后24小时测量cpt-11(图9a)和活性代谢物sn-38(图9b)的肿瘤水平。在两种sclc模型中,nal-iri递送伊立替康至肿瘤的程度大于非脂质体盐酸伊立替康。nal-iri(16mg/kg盐)的肿瘤cpt-11和sn-38水平分别比非脂质体伊立替康(30mg/kg盐)高12至57倍和5至20倍。由nal-iri递送的肿瘤cpt-11和sn-38增加归因于由脂质体包囊引起的延长循环以及肿瘤中的脂质体伊立替康的局部激活(pmid25273092:preclinicalactivityofnanoliposomalirinotecanisgovernedbytumordepositionandintratumorprodrugconversion.kalraav1,kimj1,klinzsg1,pazn1,cainj1,drummonddc1,nielsenub1,fitzgeraldjb)。实施例13:体内伊立替康脂质体注射液介导的伊立替康和sn-38的肿瘤递送与其他肿瘤类型的cdx和人源性异种移植(pdx)模型相比,在sclc细胞系来源的异种移植(cdx)模型(nci-h1048、dms-114、h841)中评估mm-398递送伊立替康和sn-38至肿瘤的能力。向具有异种移植肿瘤的小鼠静脉内施用伊立替康脂质体注射液。施用后的24小时,处死小鼠并且收获肿瘤。通过高效液相色谱法(hplc)测量肿瘤中的伊立替康和sn-38。将数据归一化成注射的剂量/肿瘤重量。如图19所示,如通过伊立替康含量所评价,来源于sclc细胞系的肿瘤与其他肿瘤类型相比具有类似或更高水平的伊立替康脂质体注射液沉积。此外,sn-38水平的分析指示伊立替康递送的增加与sn-38水平的增加相关。这些发现与提出的脂质体沉积机理和肿瘤内伊立替康向sn-38的局部转化一致。实施例14:在二线sclc的临床前模型中伊立替康脂质体注射液、非脂质体伊立替康和拓扑替康的抗肿瘤活性nal-iri被设计用于相对于非脂质体伊立替康延长循环,并且探索用于增强的向肿瘤药物递送的渗漏肿瘤血管。在肿瘤沉积之后,巨噬细胞吞噬nal-iri,接着伊立替康释放并且在肿瘤中转化成其活性代谢物sn-38。假设通过延长的sn-38递送引起的拓扑异构酶1(top1)持续抑制实现与常规top1抑制剂相比优越的抗肿瘤活性。拓扑替康(一种top1抑制剂)为当前用于小细胞肺癌(sclc)的二线治疗的护理标准。如下所述,用sclc中的一线方案卡铂加上依托泊苷来治疗具有nci-h1048sclc肿瘤的小鼠。一旦肿瘤逃脱了由卡铂加上依托泊苷引起的生长控制,将小鼠随机化继续用卡铂加上依托泊苷治疗或切换至用伊立替康脂质体注射液、非脂质体伊立替康或拓扑替康进行的二线治疗。每周用30mg/kg卡铂加上25mg/kg依托泊苷的组合治疗具有ncih1048sclc异种移植肿瘤的nod/scid小鼠。当肿瘤达到大约1200mm3时,将小鼠随机化以接受用拓扑替康(在第1天和第2天以相等分量腹腔内施用1.66mg/kg/周)、非脂质体伊立替康(在第1天静脉内施用33mg/kg/周)、伊立替康脂质体注射液(在第1天静脉内施用16mg/kg/周)进行的每周治疗,继续用卡铂加上依托泊苷或赋形剂对照进行治疗。垂直的虚线指示每周给药的开始。基于盐酸伊立替康,示出伊立替康脂质体注射液剂量。在肿瘤于用卡铂加上依托泊苷进行的一线治疗上有进展之后,伊立替康脂质体注射液与拓扑替康和伊立替康相比展示出显著的抗肿瘤活性(分别针对拓扑替康和伊立替康,在第70天p=0.0002和第84天p=0.0002)。在卡铂加上依托泊苷治疗的sclc肿瘤中:nal-iri保持活性并且倾向于完全响应;非脂质体伊立替康治疗是活性的,但在第3个周期后一些肿瘤倾向于再生长;在1-2个周期后拓扑替康(2x临床相关剂量)似乎是活性的,但在第3个剂量后快速进展;卡铂加上依托泊苷到第5个周期是不耐受的。如图21a所示,在二线环境中伊立替康脂质注射液具有抗肿瘤活性,并且此外具有比非脂质体伊立替康和拓扑替康两者显著更大的抗肿瘤活性。图21b为每种治疗的小鼠的存活率图形。实施例15:与非脂质体盐酸伊立替康和拓扑替康相比,伊立替康脂质体注射液在体内具有改善的抗肿瘤活性。在两种cdx模型(dms-114和nci-h1048)中,在临床相关剂量下直接比较伊立替康脂质体注射液、非脂质体伊立替康和拓扑替康的活性,以及在一种cdx模型(dms-53)中伊立替康脂质体注射液的活性。根据nci准则通过使用标准表面积与重量比率转换来计算临床相关剂量。图23呈现了每周用伊立替康脂质体注射液、拓扑替康和非脂质体伊立替康(三种中的两种)治疗的具有sclc异种移植肿瘤的小鼠的肿瘤生长动力学。在dms-114和nci-h1048模型中,伊立替康脂质体注射液展示出比非脂质体伊立替康和拓扑替康两者显著更大的抗肿瘤活性。在dms-53模型中,伊立替康脂质体注射液展示出比拓扑替康显著更大的抗肿瘤活性。此外,在用伊立替康脂质体注射液治疗的nci-h1048模型中所治疗的10只小鼠中有10只经历其肿瘤的完全消退,与用拓扑替康治疗的10只小鼠中0只相比。图23示出了从具有皮下(图23a)dms-53、(图23b)dms-114或(图23c)nci-h1048的nod/scid小鼠获得的数据。用静脉内nal-iri(16mg/kg;三角形)、静脉内伊立替康(33mg/kg;菱形)、腹腔内拓扑替康(0.83mg/kg/周,第1-2天;正方形)或赋形剂对照(圆形)治疗sclc异种移植肿瘤。针对dms-114和nci-h1048,所有组均n=10;针对dms-53,对于对照、拓扑替康和nal-iri分别为n=4、5和5。垂直的虚线指示每周给药的开始并且误差线指示平均值的标准误差。基于盐酸伊立替康,示出伊立替康脂质体注射液剂量。治疗后,与拓扑替康(针对dms-114,第52天,p<0.0001并且针对nci-h1048,第59天,p<0.0001;非参数t-检验)和伊立替康(针对dms-114,第65天,p<0.0001并且针对nci-h1048,第84天,p<0.0001;非参数t-检验)相比,伊立替康脂质体注射液展示出显著的抗肿瘤活性。除了cdx模型以外,也使用皮下人源性异种移植物来检验pdx模型。用静脉内nal-iri(16mg/kg;三角形)、静脉内伊立替康(33mg/kg;菱形)、腹腔内拓扑替康(0.83mg/kg/周,第1-2天;正方形)或赋形剂对照(圆形)治疗具有皮下人源性异种移植物(图23d)lun-182、(图23e)lun-081和(图24f)lun-164的balb/c裸鼠。针对所有pdx模型,所有组n=5。垂直的虚线表明每周给药的开始并且误差线指示平均值的标准误差。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1