肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒及其制备方法与流程
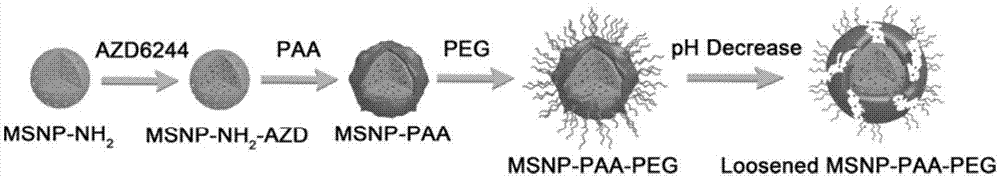
本发明属于靶向载药材料领域,涉及肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒及其制备方法。
背景技术:
基因改变引起的丝裂原活化蛋白激酶(mapk)通路的过度活化在各种癌症中都很常见。小分子靶向治疗被认为是最有前景的治疗癌症与遗传病变的方法之一。约60%的黑色素瘤中均含有brafv600e突变,特异性靶向brafv600e突变的靶向药物维罗非尼(vemurafenib,plx4032)在治疗转移性黑色素瘤治疗中显示出了前所未有的应答率。在一系列的临床试验中,维罗非尼治疗的患者客观总体反应率为48%-53%,平均受益时间为5.3-6.7个月。但是,在靶向治疗一段时间后肿瘤会出现耐药性,随后肿瘤会很快复发。研究表明braf耐药的机理之一是mapk通路的再激活,包括nras突变、braf基因扩增、braf可变剪接、mek突变、nf1下调等。此外,braf抑制剂(brafi)还可以诱导野生型的braf过度激活,诱导鳞状细胞癌(scc)的发生,严重影响病人的生活质量。
为了解决这些问题,研发出了新的靶向mapk通路下游mek激酶的抑制剂司美替尼,其在治疗耐药性和黑色素瘤中取得了较好的反应率,可显著延长患者的生存期。在brafi和mek抑制剂(meki)联合治疗的临床试验中,联合治疗组无进展生存期中位数为9.4个月,而brafi单药治疗组为5.8个月;scc的发生率从单药治疗的19%下降到联合治疗的7%。因此,brafi联合meki双药治疗取代了brafi单药治疗,成为一线临床方案。虽然联合治疗提高了病人总体反应率,延长了病人生存期和减轻了副作用,但疾病进展仍然普遍。因此,迫切需要寻求新的长期抗肿瘤策略。
在过去的几年里,免疫疗法,尤其是抗体介导的免疫检查点阻断,在转移性黑色素瘤的治疗中显示出持久的肿瘤抑制作用。抗体介导的免疫检查点阻断的代表药物包括阻断ctla-4的ipilimumab,阻断pd-1/pd-l1通路的pembrolizumab和opdivo。在临床研究中,通过pembrolizumab和opdivo抗体阻断pd-1/pd-l1,可显著延长患者无进展生存期和总体生存期,且毒性较低。但是,抗体介导的pd-1/pd-l1免疫检测点阻断总体效应率低,大约只有10%-40%的患者对抗pd1抗体治疗有效。
无论是brafi联合meki的双药联合靶向治疗,还是免疫检查点阻断,均不能持久地惠及广大患者。有趣的是,它们的优点和缺点恰好是互补的。因此,将免疫治疗与靶向治疗联合用于治疗肿瘤,将会是一项有前景的抗肿瘤策略。然而,由于靶向药物对免疫系统潜在的毒副作用,使得二者的联合受到了阻碍。一方面,brafi和meki可以通过促进肿瘤抗原呈递机制促进免疫识别,促进肿瘤浸润性t细胞的比例;另一方面,meki可能通过抑制激活的t细胞mapk通路来抑制其功能,抑制肿瘤免疫反应。在brafi与meki联合联合免疫检测点阻断的临床实验中,其联合效果并不理想。虽然meki在免疫检查点阻断联合方案中的作用仍有争议,但meki对t细胞的毒副作用需要去避免。
纳米靶向载体具有优异的生物相容性、生物安全性和精确的纳米孔径,因而被广泛应用于药物传递系统。基于肿瘤的高通透性和滞留效应(epr效应),以及肿瘤微环境与正常组织微环境的差异,例如肿瘤组织的ph值低于正常组织,还原性高于正常组织,酶的种类及含量的变化,以及肿瘤细胞通透性增强等特征,可以设计具有肿瘤组织和肿瘤细胞特异性靶向、刺激响应的药物载体。对于抗癌药物的载体而言,以脂质体、胶束为代表的纳米载体是目前应用最多的。但是,脂质体和胶束的载药量低、稳定性不佳等缺点限制了其进一步的发展。此外,脂质体介导的跨膜药物传递是通过膜融合、内吞作用和小分子扩散的促进作用,导致脂质体既可被肿瘤细胞内吞,也能被淋巴细胞内吞,会影响淋巴细胞的功能。因此,开发能够实现肿瘤组织和肿瘤细胞精准靶向、并能使淋巴细胞免受靶向药物损伤的新型纳米载药系统,对于实现肿瘤靶向治疗与免疫治疗的协同治疗具有重要的意义。
技术实现要素:
针对靶向药物引起的免疫毒性的问题,本发明提供了一种肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒及其制备方法,以避免靶向药物对淋巴细胞造成损伤,提高靶向治疗与免疫治疗对肿瘤的联合治疗效果。
本发明提供的肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒,由表面被聚丙烯酸和聚乙二醇修饰的介孔二氧化硅纳米颗粒和药物组成,所述表面被聚丙烯酸和聚乙二醇修饰的介孔二氧化硅纳米颗粒由表面被氨基修饰的介孔二氧化硅纳米颗粒,通过静电作用包覆表面被氨基修饰的介孔二氧化硅纳米颗粒的聚丙烯酸膜层,以及与聚丙烯酸膜层中的聚丙烯酸通过酰胺键连接的聚乙二醇组成,聚丙烯酸膜层作为封堵阀门,能响应ph值的变化使表面被氨基修饰的介孔二氧化硅纳米颗粒的孔道处于封堵或打开状态,所述药物位于表面被氨基修饰的介孔二氧化硅纳米颗粒的孔道结构中;该纳米载药颗粒具有避免药物造成淋巴细胞毒性的能力。
上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的技术方案中,纳米载药颗粒的粒径为100~200nm。
上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的技术方案中,纳米载药颗粒中,聚丙烯酸的含量优选为7.2wt.%~7.5wt.%,但不限于该范围,具体的含量可根据实际应用需求进行调整。
上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的技术方案中,纳米载药颗粒中,与聚丙烯酸膜层中的聚丙烯酸通过酰胺键连接的聚乙二醇的含量优选为1.8wt.%~2.8wt.%,但不限于该范围,具体的含量可根据实际应用需求进行调整,聚乙二醇的含量进一步优选为2.2wt.%~2.5wt.%。
上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的技术方案中,纳米载药颗粒中,介孔二氧化硅纳米颗粒表面的氨基修饰基团的含量应使表面被氨基修饰的介孔二氧化硅纳米颗粒的zeta电位优选为10~30mv。
上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的技术方案中,所述表面被氨基修饰的介孔二氧化硅纳米颗粒是在介孔二氧化硅纳米颗粒的表面接枝基团r得到的,基团r优选为
上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的技术方案中,所述的药物为具有淋巴细胞毒性的靶向药物或具有淋巴细胞毒性的化疗药物,包括但不限于azd6244、plx4032、bez235以及azd8055。硅纳米载药颗粒中药物的含量和种类根据实际应用需求影响确定,药物的含量优选为5wt.%~15wt.%,但药物含量不限于该范围。
上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的技术方案中,纳米载药颗粒具有被动靶向肿瘤组织的能力,具有特异靶向肿瘤细胞、避免被淋巴细胞内吞的能力,具有在体外和体内避小分子药物造成淋巴细胞毒性的能力,具有在体内与肿瘤免疫药物协同抗肿瘤和刺激免疫系统的能力。
本发明还提供了上述肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的制备方法,步骤如下:
(1)制备介孔二氧化硅纳米颗粒
调节十六烷基三甲基溴化铵水溶液的ph值至11.0~11.5,在搅拌下加热至78~82℃,滴加正硅酸乙酯,在78~82℃反应2~3h,将反应产物分散在浓盐酸-无水乙醇溶液中,在78~82℃回流3~5h,将所得产物用乙醇和水洗涤,干燥,得到介孔二氧化硅纳米颗粒;
(2)制备氨基修饰的介孔二氧化硅纳米颗粒
将介孔二氧化硅纳米颗粒分散于异丙醇或乙醇中形成分散液a,将分散液a与3-氨丙基三乙氧基硅烷混合,在78~82℃搅拌反应12~16h,将反应产物用乙醇和水洗涤,干燥,得到氨基修饰的介孔二氧化硅纳米颗粒;
(3)制备氨基修饰的介孔二氧化硅纳米载药颗粒
将氨基修饰的介孔二氧化硅纳米颗粒分散在pbs缓冲液或水中形成分散液b,将分散液b与药物的溶液混合,反应8~10h,将所得反应产物用pbs缓冲液洗涤,冷冻干燥,即得氨基修饰的介孔二氧化硅纳米载药颗粒;
(4)制备聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒
将氨基修饰的介孔二氧化硅纳米载药颗粒分散在pbs缓冲液或水中形成分散液c,加入聚丙烯酸水溶液,搅拌反应2~4h,将反应产物用pbs缓冲液洗涤,得到聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒,将聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒分散在pbs缓冲液或水中形成分散液d;
(5)制备肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒
向分散液d中加入氨基化的聚乙二醇,超声混合,然后搅拌反应12~16h,采用超滤膜过滤,收集反应所得纳米颗粒,再用去离子水洗涤,即得肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒。
上述方法的步骤(1)中,十六烷基三甲基溴化铵水溶液的浓度优选为1.8~2.5mg/ml,正硅酸乙酯与十六烷基三甲基溴化铵水溶液的体积比优选为1:(90~150)。
上述方法的步骤(2)中,分散液a中介孔二氧化硅纳米颗粒的浓度优选为8~15mg/ml,3-氨丙基三乙氧基硅烷与分散液a的体积比优选为1:(150~200)。
上述方法的步骤(3)中,优选的分散液b与药物的溶液混合的比例应使药物与氨基修饰的介孔二氧化硅纳米颗粒的质量比为1:(1~10)。
上述方法的步骤(4)中,优选的聚丙烯酸水溶液的加入量应使聚丙烯酸与氨基修饰的介孔二氧化硅纳米载药颗粒的质量比为(2.5~25):100;所述聚丙烯酸的分子量优选为1500~2000kd,但不限于该分子量范围。
上述方法的步骤(5)中,氨基化的聚乙二醇与分散液d中的聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒的质量比优选为(2~5):100;所述氨基化的聚乙二醇的分子量优选为4~6kd,但不限于该分子量范围。
上述方法的步骤(1)中,浓盐酸-无水乙醇溶液中浓盐酸与无水乙醇的体积比优选为(1~2):(8~9)。
上述方法中,分散液b中氨基修饰的介孔二氧化硅纳米颗粒的浓度优选为2~6mg/ml,分散液c中氨基修饰的介孔二氧化硅纳米载药颗粒的浓度优选为3~5mg/ml,分散液d中聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒的浓度优选为3~5mg/ml。
上述方法中采用的pbs缓冲液的ph值为7.4。
与现有技术相比,本发明的技术方案产生了以下有益的技术效果:
1.本发明提供的肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒,由表面被聚丙烯酸和聚乙二醇修饰的介孔二氧化硅纳米颗粒和药物组成,相对于脂质体、胶束等非晶硅材料,介二氧化孔硅纳米材料具有优良生物相容性、较好的生物安全性,较大的比表面积与孔体积、均匀可控的介孔尺寸、优良的稳定性及药物装载量高等优点,同时,相对于其他通过膜融合、内吞作用和小分子扩散被细胞内吞的脂质纳米材料,硅纳米材料仅能通过细胞内吞作用被细胞内吞,使得本发明的纳米载药颗粒仅能被内吞能力强的肿瘤细胞内吞。
2.本发明提供的纳米载药颗粒中,聚丙烯酸与带正电荷的表面被氨基修饰的介孔二氧化硅纳米载药颗粒通过静电作用结合,形成聚丙烯酸膜层封堵介孔二氧化硅的孔道,聚丙烯酸是一种聚阴离子的聚合物,具有ph可控的性质,会对ph刺激产生响应,在酸性环境下(ph<5.0),聚丙烯酸发生质子化,由负电荷转变为正电荷,同时,当ph值降低时,聚丙烯酸的羧基质子化减弱其与表面被氨基修饰的介孔二氧化硅纳米载药颗粒的静电相互作用,导致聚丙烯酸壳膜层变得疏松,药物从介孔二氧化硅的孔道释放。体内正常ph环境(ph≈7.4))不足以使聚丙烯酸质子化,进而保证药物不会被提前释放。当纳米载药颗粒被细胞内吞后,细胞内溶酶体和内含体的酸性环境使聚丙烯酸质子化,纳米载药颗粒表面聚合物壳疏松,从而使药物释放。
3.本发明通过筛选表面被氨基修饰的介孔二氧化硅纳米颗粒的氨基修饰基团的量,以及聚丙烯酸的量,使得作为封堵阀门聚丙烯酸膜层的覆盖厚度适当,同时使得纳米载药颗粒的粒径适当,这些因素使得本发明提供的纳米载药颗粒具备良好的被癌细胞内吞的能力,同时确保该纳米载药颗粒不被淋巴细胞内吞,得到了仅能被肿瘤细胞内吞而不能被淋巴细胞内吞纳米载药颗粒。
4.本发明将聚乙二醇通过酰胺键与聚丙烯酸连接,聚乙二醇的引入能增加纳米颗粒的生物相容性,同时,使制备聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒所带电荷由负电荷转变为中性,可以延长纳米载药颗粒在血液中的循环时间,避免纳米载药颗粒被网状内皮细胞清除及血浆蛋白结合。由于肿瘤的epr效应,本发明提供的纳米载药颗粒可以通过“被动靶向”在肿瘤部位富集,提高了药物在肿瘤组织的利用度,降低药物在血液系统和其它组织中的毒副作用。
5.通过上述4点中提到的一系列的设计,本发明成功构建了肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒,相比传统的介孔二氧化硅药物载体和脂质纳米载体,本发明提供的纳米载药颗粒具有更高效的肿瘤和细胞靶向给药能力,可以有效提高药物利用率并降低其毒副作用。
6.本发明通过实验证实了我们提供的纳米载药颗粒具有“被动靶向”肿瘤组织、特异靶向肿瘤细胞的能力,可以避免被淋巴细胞内吞,同时该纳米载药颗粒在体外和体内都具有保护淋巴细胞免受靶向药物损伤的能力,当所载药物为mek抑制剂azd6244时,本发明提供的载药纳米颗粒在体内具有与肿瘤免疫药物协同抗肿瘤和刺激免疫系统的能力。
7.本发明还提供了肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒的制备方法,该方法在对介孔二氧化硅纳米颗粒进行氨基化修饰,包裹聚丙烯酸膜层和接枝聚乙二醇的工艺简单,载药方式也简单易行,无须使用特殊的试剂和设备,具有容易推广应用的特点。
附图说明
图1是实施例1中纳米载药颗的制备过程及纳米载药颗粒响应ph值变化释放药物的示意图。
图2是实施例1制备的介孔二氧化硅纳米颗粒的透射电镜图。
图3是实施例1中paa、msnp、msnp-nh2、msnp-paa和msnp-paa-peg的红外光谱图。
图4是实施例1中msnp-paa-peg和msnp的氮气吸附-解吸等温曲线和表面孔径测试结果。
图5是实施例1中的msnp、msnp-nh2、msnp-paa和msnp-paa-peg的动态光散射测试结果。
图6是实施例1中的msnp、msnp-nh2、msnp-paa和msnp-paa-peg的zeta电位测试结果。
图7是实施例1制备的msnp-paa-peg的透射电镜图,其中(a)(b)(c)图分别是不同放大倍数下的透射电镜图。
图8是实施例2中msnp-paa-peg在不同ph值条件下的释药曲线。
图9是实施例3中msnp-paa-peg-fitc在体外肿瘤细胞摄取情况。
图10是实施例3中msnp-paa-peg-fitc在体外淋巴细胞摄取情况。
图11是实施例3中msnp-paa-peg-fitc在体外肿瘤和淋巴细胞摄取水平定量分析结果。
图12是实施例3中的msnp-paa-peg-fitc在小鼠各器官中的分布图。
图13是实施例4对肿瘤细胞和淋巴细胞的细胞毒性测试结果,其中,a图肿瘤细胞的的细胞存活率测试结果,b图为对淋巴细胞增殖的影响图,c图和d图为对ifn-γ和il-2的分泌的影响图。
图14是实施例5中的体内抗肿瘤效果测试结果,其中,a图是注射后肿瘤体积随时间的变化曲线,b图是注射26天后的肿瘤体积测试结果。
图15是实施例5中对肿瘤浸润的淋巴细胞的影响结果,其中,a图是肿瘤浸润的cd8+t细胞比例,b图是激活型cd8+t比例,c图是记忆型cd8+t比例,d图是肿瘤浸润的cd4+t细胞比例。
图16是实施例5中是治疗后体内安全性评价结果,其中,a图为he染色结果,b-i图为血液生化分析结果。
图17是实施例6中的体内抗肿瘤效果测试结果,其中,a图是注射后肿瘤体积随时间的变化曲线,b图是注射26天后的肿瘤体积测试结果。
图18是实施例6对肿瘤浸润的淋巴细胞的影响结果,a图是肿瘤浸润的cd8+t细胞比例,b图是激活型cd8+t比例,c图是记忆型cd8+t比例,d图是cd25hit细胞比例。
具体实施方式
以下通过实施例对本发明提供的肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒及其制备方法作进一步说明。有必要指出,以下实施例只用于对本发明作进一步说明,不能理解为对本发明保护范围的限制,所属领域技术人员根据上述发明内容,对本发明做出一些非本质的改进和调整进行具体实施,仍属于发明保护的范围。
下述各实施例中采用的pbs缓冲液的ph值为7.4,pbs缓冲液的浓度为10mmol/l。
实施例1
本实施例中,以小分子靶向药物mek抑制剂azd6244作为待装载药物,制备肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒,步骤如下:
(1)制备介孔二氧化硅纳米颗粒(msnp)
将100mg的十六烷基三甲基溴化铵(ctab)溶解于去离子水中形成浓度为2.1mg/ml的ctab水溶液,采用2mol/l的naoh溶液调节ctab水溶液的ph值至11.0,水浴搅拌加热至80℃,待温度稳定后,按照正硅酸乙酯(teos)与ctab水溶液的体积比为1:96的比例,以180μl/min的速度将正硅酸乙酯滴加至ctab水溶液中,在80℃搅拌反应3h得到白色沉淀,然后以10000rpm的转速离心15min,收集沉淀,并将所得沉淀分散在30ml的浓盐酸-无水乙醇溶液(浓盐酸与无水乙醇的体积比为1:9)中,在80℃回流3h,将所得产物用乙醇和去离子水交替洗涤3遍,再在60℃干燥12h,得到msnp。
通过teanai-10型透射电子显微镜(tem)观察msnp的形貌,结果如图2所示,图2的(a)图与(b)图是msnp在不同放大倍数下的透射电镜图,由图2可知,该步骤制备的msnp的粒径在100nm左右。
(2)制备氨基修饰的介孔二氧化硅纳米颗粒(msnp-nh2)
将200mg的msnp分散于20ml乙醇中形成分散液a,将100μl的3-氨丙基三乙氧基硅(aptes)烷加入到分散液a中,在80℃的水浴中搅拌反应12h,将所得反应液冷却至室温,以10000rpm的转速离心15min,收集沉淀,将所得沉淀用乙醇和去离子水交替洗涤3遍,然后在60℃干燥12h,得到msnp-nh2。
(3)制备氨基修饰的介孔二氧化硅纳米载药颗粒(msnp-nh2-azd)
将msnp-nh2分散在去离子水中形成msnp-nh2浓度为5mg/ml的分散液b,将azd6244用二甲基亚砜(dmso)溶解形成浓度为5mg/ml的azd6244溶液,取2ml分散液b与200μlazd6244溶液混合均匀,在室温反应8h,然后以10000rpm的转速离心15min,收集沉淀,将所得沉淀用pbs缓冲液洗涤3遍,冷冻干燥,即得msnp-nh2-azd。
(4)制备聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒(msnp-paa)
将100mg的msnp-nh2-azd分散在去离子水中形成msnp-nh2-azd浓度为2.5mg/ml的分散液c,加入3ml浓度为5mg/ml聚丙烯酸(paa,分子量为1800kd)水溶液,搅拌反应2h,以14800rpm的转速离心5min,收集沉淀,将所得沉淀用pbs缓冲液洗涤3次,得到msnp-paa,将msnp-paa重新分散在去离子水中形成msnp-paa浓度为5mg/ml的分散液d。
(5)制备肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒(msnp-paa-peg)
取200ml分散液d,向分散液d中加入50mg的分子量为5kd的氨基化的聚乙二醇(mpeg-5k-nh2),即mpeg-5k-nh2与分散液d中的msnp-paa的质量比为5:100,超声30min,然后搅拌反应12h,采用截留分子量为100kd超滤膜过滤,收集反应所得纳米颗粒,再用去离子水洗涤3次,即得msnp-paa-peg。
msnp-nh2是在msnp的表面接枝基团r得到的,
在制备msnp-paa-peg的过程中,采用傅里叶变换红外分析(ftir)和氮气吸附-解吸等温曲线对每一步反应进行监测,结果分别如图3~4所示,由图3可知,aptes、paa和peg均成功地与介孔二氧化硅纳米颗粒msnp连接。图5和图6分别为msnp、msnp-nh2、msnp-paa和msnp-paa-peg的动态光散射粒径和zeta电位测试结果,动态光散射测试结果和zeta电位的变化情况进一步表明aptes、paa和peg成功地耦联到介孔二氧化硅纳米颗粒msnp上。通过透射电镜可以测量纳米颗粒的直径和观测纳米颗粒表面的介孔结构,msnp-paa-peg的在不同放大倍数下的透射电镜照片如图7所示,由图7可知,msnp-paa-peg的粒径在100nm,边缘较粗糙有褶皱。
对比例1
本对比例中,制备负载异硫氰酸荧光素(fitc)的介孔二氧化硅纳米颗粒,步骤如下:
(1)按照实施例1步骤(1)的操作制备msnp;
(2)按照实施例1步骤(2)的操作制备msnp-nh2;
(3)将实施例1步骤(3)中的azd6244替换为fitc,并按照实施例1步骤(3)的操作制备得到载fitc的msnp-nh2;
(4)将实施例1步骤(4)中的msnp-nh2-azd替换为载fitc的msnp-nh2,并按照实施例1步骤(4)的操作制备得到负载fitc的聚丙烯酸修饰的介孔二氧化硅纳米颗粒(msnp-paa-fitc)及其分散液;
(5)按照实施例1步骤(5)的操作制备得到负载fitc的介孔二氧化硅纳米颗粒,记作msnp-paa-peg-fitc。
对比例2
本对比例中,制备未负载任何物质的空载介孔二氧化硅纳米颗粒(msnpnano),本对比例的操作与实施例1的操作基本相同,不同之处仅在于去掉实施例1的步骤(3)。
实施例2
本实施例中,测试实施例1制备的msnp-paa-peg在不同ph环境中的药物释放速率。
将实施例1制备的msnp-paa-peg置于不同ph条件的pbs缓冲液中(ph=7.4、ph=6.8、ph=5.5、ph=4.5),每间隔一段时间取样离心,取上清,采用hplc测试药物释放率。hplc的测试条件为:流动相,甲醇:水=8:2(v:v),流速1ml/min,检测波长257.8nm,进样体积20μl。
msnp-paa-peg在不同ph值条件下的释药曲线如图8所示,由图8可知,在ph值为7.4、6.5、5.5和4.5的条件下,药物azd6244的释放率分别是9.9%、20.1%、49.82%和71.02%,随着ph值的降低,药物释放率显著增加。由于体内正常ph环境的ph值约为7.4,在该条件下不足以使本发明提供的纳米载药颗粒的paa膜层中的paa质子化,因而可以保证药物不会被提前释放;由于肿瘤细胞过度增殖肿瘤微环境与正常组织微环境差异较大,如ph低于正常组织,肿瘤细胞通透性增强等,当纳米载药颗粒被细胞内吞后,细胞内溶酶体和内含体的酸性环境会使paa质子化,进而使纳米载药颗粒表面聚合物壳疏松,从而实现药物释放。
实施例3
本实施例中,测试对比例1制备的负载fitc的介孔二氧化硅纳米颗粒msnp-paa-peg-fitc在体外细胞摄取水平和体内的生物分布。
分别用终浓度为50μg/ml和25μg/ml的msnp-paa-peg-fitc处理黑色素瘤细胞smm101和smm103,以及淋巴细胞el4t、jurkat和小鼠脾淋巴细胞24h、48h和72h,然后通过流式细胞仪和共聚焦显微镜检测msnp-paa-peg-fitc在肿瘤细胞和淋巴细胞中的摄取情况,结果如图9~11所示,图9是流式细胞仪检测的msnp-paa-peg-fitc在肿瘤细胞的内吞比例。图10是流式细胞仪检测的msnp-paa-peg-fitc在淋巴细胞的内吞比例。图11是msnp-paa-peg-fitc在体外肿瘤和淋巴细胞摄取水平定量分析结果。由图9~11可知,msnp-paa-peg-fitc在肿瘤细胞的摄取效率显著高于淋巴细胞,在淋巴细胞中几乎不摄取,在肿瘤细胞smm101和smm103中摄取效率分别是92.95%和66.16%,在淋巴细胞el4t、jurkat和小鼠脾淋巴细胞中的摄取效率分别是8.44%、6.54%和16.61%。以上结果表明该介孔二氧化硅纳米颗粒具有特异性的被肿瘤细胞内吞,而不被淋巴细胞内吞的能力。
给荷瘤小鼠静脉注射msnp-paa-peg-fitc,分别在注射3h、6h、12h、24h和48h后,处死小鼠,取小鼠肿瘤、心、肝、脾、肺、肾组织,检测msnp-paa-peg-fitc在各器官的分布,结果如图12所示,由图12可知,由于肿瘤组织的epr效应,使得肿瘤组织的荧光信号强度显著高于心、脾、肺、肾,表明msnp-paa-peg-fitc主要在肿瘤部位富集。
实施例4
本实施例中,测定plx4032、azd6244、实施例1制备的msnp-paa-peg(简称msnp-azd)、对比例2制备的msnpnano、plx4032+azd6244(plx4032与azd6244联合用药)、plx+msnp-azd(plx4032与msnp-azd联合用药)对肿瘤细胞和t细胞的差异毒性。
(1)对肿瘤细胞的细胞毒性
以黑色素瘤细胞smm101、smm102和smm103为模型细胞,通过甲基三氯硅烷(mts)检测以上各物质在体外对肿瘤细胞的毒性。
将肿瘤细胞以2.0×103cells/well的密度接种于96孔板内,每孔含50μl的dmem培养基。在37℃细胞培养箱中培养过夜后,分别在各孔加入50μl不同浓度(0,0.01,0.1,1和10μm)的plx4032、azd6244、msnp-azd、msnpnano、plx4032+azd6244、plx+msnp-azd处理72h,每个浓度设5个重复孔。处理72h后,每孔加入20μlmts溶液,孵育1h,在酶标仪上测定波长为490nm处的吸光度(od)值,按照下式计算细胞存活率:
(2)对淋巴细胞的细胞毒性
通过荧光染料cfse检测载药纳米颗粒对淋巴细胞增殖的影响,提取小鼠脾淋巴细胞,将细胞密度调整至5×106cells/ml,加入终浓度为2.5μm的荧光染料cfse在室温染色8min,然后用5倍体积的rpmi培养基终止染色,洗涤两遍,去除残留荧光染料cfse。将标记好的细胞以2.0×106cells/well的密度接种于cd3抗体预包被的24孔板内,然后加入不同浓度(0、0.01、0.1、1和10μm)的plx4032、azd6244、msnp-azd、plx4032+azd6244、plx+msnp-azd处理72h,72h后收集细胞,采用流式细胞仪检测淋巴细胞的增殖情况;收集上清,采用elisa检测淋巴细胞ifn-γ和il-2的分泌水平。
图13是实施例4对肿瘤细胞和淋巴细胞的细胞毒性测试结果,其中,a图肿瘤细胞的的细胞存活率测试结果,b图为对淋巴细胞增殖的影响图,c图和d图为对ifn-γ和il-2的分泌的影响图。
由图13的a图可知,msnp-nano在三株小鼠黑色素瘤细胞系中都显示出非常微弱的细胞生长抑制作用,表明本发明所述的未载药的纳米颗粒对细胞无毒性,单独的msnp-azd,以及msnp-azd与plx4032联用时,载药的纳米颗粒与与游离药一样也表现出较强的肿瘤细胞毒性。
由图13的b~d图可知:低剂量(1μm)的plx4032促进了淋巴细胞的增殖、以及ifn-γ和il-2的分泌,而高浓度的plx4032抑制了淋巴细胞的增殖、以及ifn-γ和il-2的分泌;游离的azd6244呈剂量依赖性抑制淋巴细胞的增殖、以及ifn-γ和il-2的分泌,而msnp包载azd6244后可以显著减少azd6244对淋巴细胞的毒性。以上结果表明该介孔二氧化硅纳米载药颗粒在体外具有避免靶向药物造成的淋巴细胞毒性的能力。
实施例5
本实施例中,测试saline、plx4032、azd6244、plx4032+azd6244(plx4032与azd6244联合用药)、实施例1制备的msnp-paa-peg(简称msnp-azd)和plx4032+msnp-azd(plx4032与msnp-azd联合用药)的体内抗肿瘤效果和免疫调节作用。
将小鼠smm103黑色素瘤细胞接种到c57bl/6背部皮下,建立小鼠黑色素瘤移植瘤模型,在体内评价msnp-azd抗肿瘤作用和免疫调节作用。当肿瘤体积生长到80~100mm3时,分别静脉注射saline、plx4032、azd6244、plx4032+azd6244、msnp-azd和plx4032+msnp-azd,给药剂量为5mg/kgplx4032、5mg/kgazd6244和5mg/kgmsnp-azd。给药期间每三天测量一次肿瘤体积,绘制小鼠肿瘤生长曲线。治疗7次后取小鼠肿瘤组织评价以上各物质的抗肿瘤效果和对肿瘤浸润的淋巴细胞(til)的影响,取小鼠的心、肝、脾、肺、肾和血液评价以上各物质的安全性。图14是实施例5中的体内抗肿瘤效果测试结果,其中,a图是注射后肿瘤体积随时间的变化曲线,b图是注射26天后的肿瘤体积测试结果。由图14可知,所有单药治疗和双药联合治疗组均显著抑制了小鼠肿瘤的生长。plx4032联合azd6244抑制肿瘤生长效果优于单独的plx4032和azd6244,msnp-azd单独治疗组及联合plx4032治疗组抑制肿瘤生长效果均优于游离的azd6244治疗组。
将小鼠肿瘤组织消化成单细胞悬液,采用流式细胞仪检测各治疗组对肿瘤浸润的淋巴细胞的影响。结果如图15所示,图15是实施例5中对肿瘤浸润的淋巴细胞的影响结果,其中,a图是肿瘤浸润的cd8+t细胞比例,b图是激活型cd8+t比例,c图是记忆型cd8+t比例,d图是肿瘤浸润的cd4+t细胞比例。图16是实施例5中是治疗后体内安全性评价结果,其中,a图为he染色结果,b-i图为血液生化分析结果。由图15可知,游离的azd6244抑制了肿瘤浸润的细胞毒性淋巴细胞(ctl,cd3+cd8+),其中激活型的ctls(cd8+cd44+cd69+)和记忆型ctls(cd8+cd44+cd62l-)对azd6244更加敏感,而msnp-azd对肿瘤浸润的淋巴细胞无影响,具有免疫保护的作用。以上结果表明该介孔二氧化硅纳米载药颗粒在体内具有避免靶向药物造成的淋巴细胞毒性的能力。
治疗结束后取小鼠的心、肝、脾、肺、肾进行he染色,取小鼠血液进行血生化分析,检测以上各物质的体内安全性,结果如图16所示,本发明提供的纳米载药颗粒对小鼠的心、肝、脾、肺、肾和血液均无毒性,具有较好的安全性。
实施例6
本实施例中,测试saline、anti-pd-1、plx4032+azd6244(plx4032与azd6244联合用药)、plx4032+msnp-azd(plx4032与msnp-azd联合用药)、plx4032+azd6244+anti-pd-1(plx4032、azd6244与anti-pd-1联合用药)、plx4032+msnp-azd+anti-pd-1(plx4032、msnp-azd与anti-pd-1联合用药)的体内协同抗肿瘤效果。
将小鼠smm103黑色素瘤细胞接种到c57bl/6背部皮下,建立小鼠黑色素瘤移植瘤模型,在体内评价msnp-azd与pd-1抗体协同抗肿瘤作用和免疫调节作用。当肿瘤体积生长到80~100mm3时,分别静脉注射saline、anti-pd-1、plx4032+azd6244、plx4032+msnp-azd、plx4032+azd6244+anti-pd-1、plx4032+msnp-azd+anti-pd-1,给药剂量为5mg/kgplx4032、5mg/kgazd6244、5mg/kgmsnp-azd和2.5mg/kganti-pd-1。每三天测量一次肿瘤体积,绘制小鼠肿瘤生长曲线。治疗7次后,处死小鼠,取小鼠肿瘤组织称重评价药物抗肿瘤效果;用胶原酶将肿瘤组织消化为单细胞悬液,采用流式细胞仪检测药物对肿瘤浸润的淋巴细胞的影响。
结果如图17~18所示,由图17可知,mapk通路的靶向治疗和anti-pd-1免疫治疗均能显著抑制小鼠肿瘤生长;msnp-azd联合plx3032抗肿瘤效果略优于游离azd6244联合plx3032的抗肿瘤效果;plx4032+azd与anti-pd-1联合具有协同抗肿瘤的作用,当azd6244包载入msnp纳米颗粒后协同效果更明显。
由图18可知,plx4032+azd6244+anti-pd-1治疗组肿瘤cd3+淋巴细胞比例显著低于anti-pd-1单药治疗组,表明plx4032联合azd6244抑制了肿瘤浸润的淋巴细胞,而通过msnp包载azd6244可以避免azd6244对淋巴细胞的损伤。此外,在肿瘤浸润的细胞毒性淋巴细胞(ctl,cd3+cd8+)、激活型的ctls(cd8+cd44+cd69+)和记忆型ctls(cd8+cd44+cd62l-)也表现出了相同的淋巴细胞保护作用。
综上,本发明提供的介孔二氧化硅纳米载药颗粒可以避免小分子靶向药物对淋巴细胞的损伤,具有与肿瘤免疫药物协同抗肿瘤和刺激免疫系统的能力。
实施例7
本实施例中,以小分子靶向药物mtor抑制剂azd8055作为待装载药物,制备肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒,步骤如下:
(1)制备介孔二氧化硅纳米颗粒(msnp)
将十六烷基三甲基溴化铵(ctab)溶解于去离子水中形成浓度为1.8mg/ml的ctab水溶液,采用2mol/l的naoh溶液调节ctab水溶液的ph值至11,在搅拌下水浴加热至78℃,待温度稳定后,按照正硅酸乙酯(teos)与ctab水溶液的体积比为1:90的比例,以180μl/min的速度将正硅酸乙酯滴加至ctab水溶液中,在78℃搅拌反应3h得到白色沉淀,然后以10000rpm的转速离心15min,收集沉淀,并将所得沉淀分散在30ml的浓盐酸-无水乙醇溶液(浓盐酸与无水乙醇的体积比为2:8)中,在78℃回流3h,将所得产物用乙醇和去离子水交替洗涤3遍,再在60℃干燥12h,得到msnp。
(2)制备氨基修饰的介孔二氧化硅纳米颗粒(msnp-nh2)
将msnp分散于异丙醇中形成浓度为8mg/ml的分散液a,将3-氨丙基三乙氧基硅(aptes)烷加入到分散液a中,aptes与分散液a的体积比为1:180在78℃的水浴中搅拌反应16h,将所得反应液冷却至室温,以10000rpm的转速离心15min,收集沉淀,将所得沉淀用乙醇和去离子水交替洗涤3遍,然后在60℃干燥12h,得到msnp-nh2。
(3)制备氨基修饰的介孔二氧化硅纳米载药颗粒(msnp-nh2-azd8055)
将msnp-nh2分散在去离子水中形成msnp-nh2浓度为3mg/ml的分散液b,将azd8055用二甲基亚砜(dmso)溶解形成浓度为5mg/ml的azd8055溶液,将分散液b与azd8055溶液混合均匀,控制azd8055与分散液b中的msnp-nh2的质量比为1:1,在室温反应10h,然后以10000rpm的转速离心15min,收集沉淀,将所得沉淀用pbs缓冲液洗涤3遍,冷冻干燥,即得msnp-nh2-azd8055。
(4)制备聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒(msnp-paa)
将100mg的msnp-nh2-azd8055分散在去离子水中形成msnp-nh2-azd8055浓度为3mg/ml的分散液c,加入5ml浓度为5mg/ml聚丙烯酸(paa,分子量为2000kd)水溶液,搅拌反应4h,以14800rpm的转速离心5min,收集沉淀,将所得沉淀用pbs缓冲液洗涤3次,得到msnp-paa,将msnp-paa重新分散在去离子水中形成msnp-paa浓度为4mg/ml的分散液d。
(5)制备肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒(msnp-paa-peg)
向分散液d中加入分子量为6k的氨基化的聚乙二醇(mpeg-6k-nh2),使mpeg-6k-nh2与分散液d中的msnp-paa的质量比为3:100,超声20min,然后搅拌反应16h,采用截留分子量为100kd超滤膜过滤,收集反应所得纳米颗粒,再用去离子水洗涤3次,即得msnp-paa-peg。
msnp-nh2是在msnp的表面接枝基团r得到的,
实施例8
本实施例中,以小分子靶向药物pi3k/mtor抑制剂bez235作为待装载药物,制备肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒,步骤如下:
(1)制备介孔二氧化硅纳米颗粒(msnp)
将十六烷基三甲基溴化铵(ctab)溶解于去离子水中形成浓度为2.5mg/ml的ctab水溶液,采用2mol/l的naoh溶液调节ctab水溶液的ph值至11.5,在搅拌下水浴加热至82℃,待温度稳定后,按照正硅酸乙酯(teos)与ctab水溶液的体积比为1:150的比例,以180μl/min的速度将正硅酸乙酯滴加至ctab水溶液中,在82℃搅拌反应2h得到白色沉淀,然后以10000rpm的转速离心15min,收集沉淀,并将所得沉淀分散在30ml的浓盐酸-无水乙醇溶液(浓盐酸与无水乙醇的体积比为1:9)中,在82℃回流2h,将所得产物用乙醇和去离子水交替洗涤3遍,再在60℃干燥12h,得到msnp。
(2)制备氨基修饰的介孔二氧化硅纳米颗粒(msnp-nh2)
将msnp分散于乙醇中形成浓度为15mg/ml的分散液a,将3-氨丙基三乙氧基硅(aptes)烷加入到分散液a中,aptes与分散液a的体积比为1:150~200在78℃的水浴中搅拌反应16h,将所得反应液冷却至室温,以10000rpm的转速离心15min,收集沉淀,将所得沉淀用乙醇和去离子水交替洗涤3遍,然后在60℃干燥12h,得到msnp-nh2。
(3)制备氨基修饰的介孔二氧化硅纳米载药颗粒(msnp-nh2-bez235)
将msnp-nh2分散在pbs缓冲液中形成msnp-nh2浓度为6mg/ml的分散液b,将bez235用甲醇溶解形成浓度为5mg/ml的bez235溶液,将分散液b与bez235溶液混合均匀,控制bez235与分散液b中的msnp-nh2的质量比为1:2,在室温反应9h,然后以10000rpm的转速离心15min,收集沉淀,将所得沉淀用pbs缓冲液洗涤3遍,冷冻干燥,即得msnp-nh2-bez235。
(4)制备聚丙烯酸修饰的介孔二氧化硅纳米载药颗粒(msnp-paa)
将100mg的msnp-nh2-bez235分散在pbs缓冲液中形成msnp-nh2-bez235浓度为4mg/ml的分散液c,加入4ml浓度为5mg/ml聚丙烯酸(paa,分子量为1500kd)水溶液,搅拌反应3h,以14800rpm的转速离心5min,收集沉淀,将所得沉淀用pbs缓冲液洗涤3次,得到msnp-paa,将msnp-paa重新分散在去离子水中形成msnp-paa浓度为3mg/ml的分散液d。
(5)制备肿瘤组织和细胞双重靶向的介孔二氧化硅纳米载药颗粒(msnp-paa-peg)
向分散液d中加入分子量为4k的氨基化的聚乙二醇(mpeg-4k-nh2),使mpeg-4k-nh2与分散液d中的msnp-paa的质量比为2:100,超声40min,然后搅拌反应14h,采用截留分子量为100kd超滤膜过滤,收集反应所得纳米颗粒,再用去离子水洗涤3次,即得msnp-paa-peg。
msnp-nh2是在msnp的表面接枝基团r得到的,
- 还没有人留言评论。精彩留言会获得点赞!