一种基于不确定图模型侦测蛋白质复合物的方法与流程
本发明涉及计算生物信息学领域,具体涉及一种基于不确定图模型侦测蛋白质复合物的方法。
背景技术:
蛋白质复合物是一组相互作用的蛋白质,它们在同一时间和地点相互作用,形成一个单一的多分子机器。细胞过程通常由蛋白质复合物进行。识别它们对于揭示细胞组织和功能原理的尝试起着重要作用。积累的证据表明蛋白质复合物参与许多疾病机制。跟踪蛋白质复合物可以揭示模块化机制的重要见解,并提高对疾病途径的理解。
随着高通量技术的进步,已经产生了许多蛋白质-蛋白质相互作用(ppi)数据。近年来,生物网络的研究已经成为生物信息学的热点。将蛋白质结点及其相互作用采用计算机图论理论抽象成一个复杂网络,用网络的理论来研究蛋白质分子内部的相互作用是研究热点之一。传统图理论方法有基于划分的方法、基于层次聚类的方法、基于密度的局部搜索方法等。基于划分的聚类结果依赖于初始划分质量的好坏,且划分后的每个蛋白质只能属于一个功能模块,因此基于图划分的方法并不适合ppi网络的聚类分析;基于层次聚类的方法对噪声非常敏感,而且很难挖掘交叠蛋白质复合物;基于密度的方法在扩充过程中允许某个蛋白质重复出现,但无法识别ppi网络中非稠密的子图结构。由于上述侦测蛋白质复合物的方法都是基于确定图模型,边要么存在要么不存在,而这些方法都存在很明显的缺陷,比如:基于确定图模型侦测方法都忽视了邻居信息。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种基于不确定图模型侦测蛋白质复合物的方法。
为实现上述目的及其他相关目的,本发明提供一种基于不确定图模型侦测蛋白质复合物的方法,该方法包括:
将蛋白质作为不确定图的顶点,蛋白质之间的相互作用作为不确定图的边;
根据不确定图模型,定义期望稠密度,判断每个顶点是否属于核;
计算蛋白质和他邻居蛋白质的皮尔逊系数,进一步判断该蛋白质是否属于核;
根据不确定图模型,定义邻居节点相关度,判断该蛋白质是否属于附件;
对所有挖掘到的蛋白质复合物进行冗余过滤。
可选地,所述将蛋白质作为不确定图的顶点,蛋白质之间的相互作用作为不确定图的边,具体包括:
将ti时刻的蛋白质相互作用网络抽象为不确定图
定义动态不确定图ppi网络dg,dg=(ugt1,ugt2,...,ugtk),其中i=1,2,...,k。
可选地,所述根据不确定图模型,定义期望稠密度,判断每个顶点是否属于核,具体包括:
赋予ti时刻每个蛋白质的表达值;
赋予ti时刻蛋白质相互作用程度的概率值,节点vi和vj间的概率值为
定义期望稠密度为ed,
其中,
可选地,所述判断该蛋白质是否属于核的方法具体为:
将所述期望稠密度ed与第一阈值比较,若期望稠密度ed大于所述第一阈值,则该蛋白质属于核,若期望稠密度ed小于所述第一阈值,则该蛋白质不属于核。
可选地,所述计算蛋白质和他邻居蛋白质的皮尔逊系数,进一步判断该蛋白质是否属于核,具体包括:
记录x={x1,x2,...,xn}和y={y1,y2,...,yn},x和y分别表示蛋白质x和蛋白质y在n个时间点的表达值;
计算皮尔逊系数
可选地,所述进一步判断该蛋白质是否属于核,具体方法为:
若蛋白质x的皮尔逊系数值
可选地,所述根据不确定图模型,定义邻居节点相关度,判断该蛋白质是否属于附件,具体包括:
计算相关度
其中,
可选地,所述判断该蛋白质对否属于附件,具体包括:
若相关度as(va,vs)大于第三阈值,则该蛋白质属于附件,若相关度as(va,vs)小于第三阈值,则该蛋白质不属于附件,其中第三阈值表示ti时刻所有核期望稠密度的平均值。
可选地,所述对所有挖掘到的蛋白质复合物进行冗余过滤,具体包括:
计算ti时刻所有找到的蛋白质复合物的期望稠密度值,并按降序排列存入到candidatelist=(cc1,cc2,...,ccn);
对于复合物期望稠密度值cci,ccj,计算值k4=|cci∩ccj|/cci|;
通过与第四阈值进行比较判断是否冗余,若出现冗余,则去除复合物cci。
可选地,所述通过与第四阈值进行比较判断是否冗余,具体包括:
若值k4大于第四阈值,则表明出现冗余,若值k4小于第四阈值,则表明没有出现冗余。
如上所述,本发明的一种基于不确定图模型侦测蛋白质复合物的方法,具有以下有益效果:
本发明所采用的不确定图模型具有很好的表示动态ppi网络,更切合实际,从而加快了搜索效率,更能精确预言蛋白质复合物。
附图说明
为了进一步阐述本发明所描述的内容,下面结合附图对本发明的具体实施方式作进一步详细的说明。应当理解,这些附图仅作为典型示例,而不应看作是对本发明的范围的限定。
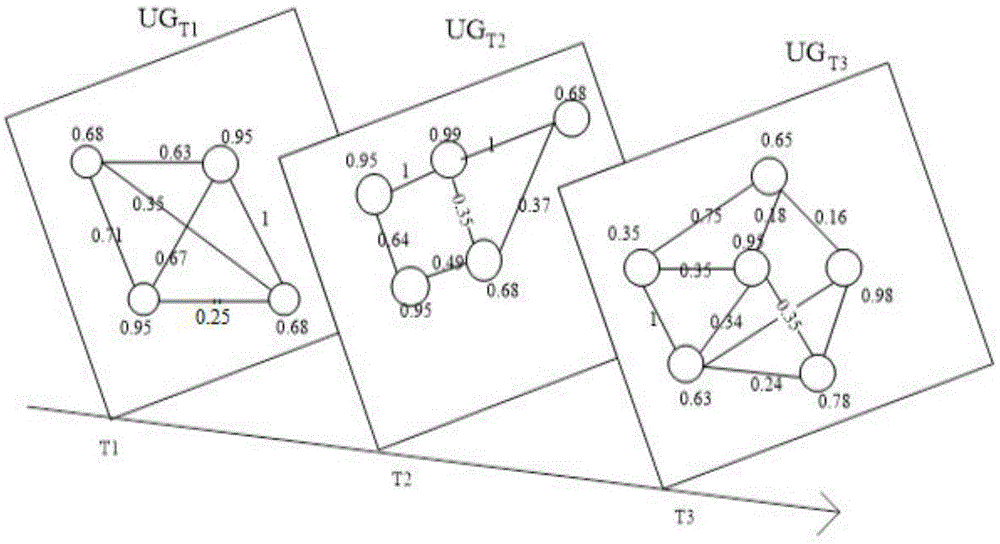
图1是本发明的t1、t2、t3时间戳的蛋白质交互网络;
图2是本发明的算法流程图。
具体实施方式
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。需说明的是,在不冲突的情况下,以下实施例及实施例中的特征可以相互组合。
需要说明的是,以下实施例中所提供的图示仅以示意方式说明本发明的基本构想,遂图式中仅显示与本发明中有关的组件而非按照实际实施时的组件数目、形状及尺寸绘制,其实际实施时各组件的型态、数量及比例可为一种随意的改变,且其组件布局型态也可能更为复杂。
如图1所示,本发明提供一种基于不确定图模型侦测蛋白质复合物的方法,包括步骤:
步骤a.蛋白质相互作用网络(ppi)中蛋白质复合物侦测问题建模,将蛋白质看成不确定图的顶点,蛋白质之间的相互作用看作不确定图的边;
步骤b.根据不确定图模型,定义期望稠密度,判断每个顶点是否属于核;
步骤c.计算蛋白质和他邻居蛋白质的皮尔逊系数,进一步判断该蛋白质是否属于核;
步骤d.根据不确定图模型,定义相关度,来判断蛋白质是否属于附件;
步骤e.对所有挖掘到的蛋白质复合物进行冗余过滤,避免挖掘到的蛋白质复合物有重复;
具体地,在步骤a中,将蛋白质看成不确定图的顶点,蛋白质之间的相互作用看作不确定图的边,具体包括:
步骤a1.将ti时刻的蛋白质相互作用网络抽象为不确定图
步骤a2.定义动态不确定图ppi网络(dupn)dg,dg=(ugt1,ugt2,...,ugtk),其中i=1,2,...,k。
在步骤b中,根据不确定图模型,定义期望稠密度,判断每个顶点是否属于核;包括步骤:
步骤b1.赋予ti时刻每个蛋白质的表达值,
步骤b2.赋予ti时刻蛋白质相互作用程度的概率值,节点vi和vj间的概率值为,
步骤b3.计算值k1,
步骤b4.计算值h1,h1=|vs|×(|vs|-1),vs表示ti蛋白质子集,
步骤b5.定义期望稠密度为ed,计算
在步骤步骤c,计算蛋白质和他邻居蛋白质的皮尔逊系数,进一步判断该蛋白质是否属于核,具体包括步骤:
步骤c1.记录x={x1,x2,...,xn}和y={y1,y2,...,yn},x和y分别表示蛋白质x和蛋白质y在n个时间点的表达值;
步骤c2.计算值k2,
步骤c3.计算值h2,
步骤c4.皮尔逊系数
具体地,计算皮尔逊系数
所述进一步判断该蛋白质是否属于核,具体方法为:
若蛋白质x的皮尔逊系数值
在步骤d中.根据不确定图模型,定义相关度,来判断蛋白质是否属于附件,包括步骤:
步骤d1.取步骤b1里面的值
步骤d2.取步骤b2里面的值p(vi,vp);
步骤d3.计算值k3,
步骤d2.计算值h3,h3=|vs|;
步骤d3.计算相关度
步骤e中,对所有挖掘到的蛋白质复合物进行冗余过滤,具体包括:
步骤e1.由前面可得到ti时刻所有复合物的集合candidate_complex=(cc1,cc2,...,ccn);
步骤e2.对于复合物cci(i∈1,2,...,n),计算所有期望稠密度ed(cci,ugti);
步骤e3.由步骤e2得到所有复合物的期望稠密度ed(cci,ugti),根据ed(cci,ugti),将所有复合物按降序排序,并存放到candidatelist=(cc1,cc2,...,ccn);
步骤e4.对于复合物cci,ccj(i∈1,2,...,n),计算值k4=|cci∩ccj|/|cci|;
步骤e5.有步骤e4得出来的值k4在比较第四阈值,从而达到去冗余的目的。具体地,若值k4大于第四阈值,则表明出现冗余,若值k4小于第四阈值,则表明没有出现冗余。
本发明所采用的不确定图模型具有很好的表示动态ppi网络,更切合实际,从而加快了搜索效率,更能精确预言蛋白质复合物。
本发明还提供一种基于不确定图模型侦测蛋白质复合物的方法,包括步骤:
步骤1.将生物蛋白质交互网络(ppi)侦测蛋白质复合物问题抽象化为图的表示形式,如图1所示,蛋白质交互网络中每个蛋白质相对应于图中的每个顶点vi,图中的边ei表示蛋白质交互网络中蛋白质之间存在相互作用。
步骤1.1.动态蛋白质交互网络生物学上共有36个时间戳,针对每个时间戳的蛋白质交互网络,本发明算法去侦测蛋白质复合物,如图1所示表示t1、t2、t3三个时间戳的蛋白质交互网络。
步骤2.动态蛋白质相互作用网络在生物学上共分为36个时间戳,其中每12个时间戳为一个周期,这里选其中t1,t2,t3三个时间戳蛋白质交互网络为例。对每个时间戳的每个蛋白质赋予表达值
步骤3.同理,对每个时间戳的每个蛋白质之间的相互作用赋予权重,该权重基于拓扑势加权方法,
步骤4.初步寻找核:
步骤4.1.ti(i∈[1,36])时间戳的蛋白质网络可以看出一个不确定图,基于不确定图模型理论,一个不确定图可以对应
步骤4.2.对于每一个可能世界vs,计算该确定图(可能世界)的期望稠密度值
步骤4.3.定义一个阈值core_thresh,对步骤4.2得到的期望稠密度值
步骤5.再次寻找核:
步骤5.1.依次计算每个蛋白质vi在36个时间戳的平均表达值,并记为vi',邻居蛋白质vj记为vj';
步骤5.2.依次遍历步骤4得到的核的邻居蛋白质,并记为nei;
步骤5.3.依次选取nei中每个蛋白质,然后根据pcc(x,y)(皮尔逊)公式计算非核候选者蛋白质的皮尔系数值,如果pcc(vi,vj)大于预定阈值tp,这里的tp值为0.3,则表示该蛋白质属于核候选者。
步骤6.将得到的所有核候选者再次进行期望稠密度计算,进一步判断核候选者内所有蛋白质是否满足条件,并并入核集(core)。
步骤7.计算核邻居结点相关度:即
步骤7.1.定义邻居集合nei,搜索核所有邻居结点并存入nei;
步骤7.2.计算结点va(va∈nei)与核所有结点的相互作用概率值
步骤7.3.计算得到蛋白质va和vj的表达值;
步骤7.4.计算核(core)内蛋白质数量为|core|;
步骤7.5.计算相关度的值as(va,core),判断是否大于预定阈值avg_score,若是,说明该蛋白质属于附件。
步骤8.去冗余,即:
步骤8.1.计算ti时刻所有找到的蛋白质复合物的期望稠密度值,并按降序排列存入candidatelist=(cc1,cc2,...,ccn);
步骤8.2.对于复合物cci,判断值
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
- 还没有人留言评论。精彩留言会获得点赞!