抗CD16A抗体与细胞因子的组合的制作方法
本发明涉及结合cd16a的抗原结合蛋白,其间歇性地与细胞因子一起施用,用于基于nk细胞的免疫疗法。在具体的实施方案中,本发明涉及具有cd16a抗原结合位点的双特异性四价串联双抗体(diabody),其用于与nk细胞结合以用于治疗癌症或(病毒)感染,其中所述串联双抗体间歇性地与细胞因子组合施用。实例是结合cd16a的串联双抗体,例如cd30/cd16a串联双抗体afm13、egfr/cd16a串联双抗体afm24和bcma/cd16a串联双抗体afm26。
背景技术:
wo2006/125668描述了抗原结合蛋白,即cd30/cd16a双特异性串联双抗体,及其用于基于nk细胞的免疫疗法的用途。
wo2016/177846描述了cd30/cd16a双特异性串联双抗体和pd-1拮抗剂的组合,其用于治疗霍奇金淋巴瘤。
自然杀伤(nk)细胞是细胞毒性的、产生ifn-γ的先天淋巴细胞,其被认为构成对抗病毒感染的细胞和癌细胞的第一道防线(cerwenkaandlanier,2001;spits,etal.,2013)。
与cd8+t细胞相反,nk细胞通过一组定义的胚系编码受体(例如抑制性kir受体和nkg2a受体以及活化性受体nkg2d、dnam-1和ncr受体)来区分异常细胞(kochetal,2013;pahlandcerwenka,2017)。nk细胞对显示出降低水平的抑制性mhci类分子或抑制性mhci类分子的不相容谱库(repertoire)的细胞起作用,使得能够识别可能已经逃脱cd8+t细胞应答的某些癌细胞。抑制性配体的低表达与靶细胞上高水平活化配体的组合导致nk细胞活化和穿孔素及颗粒酶b的释放,介导靶细胞死亡(gasserandraulet,2006;moretta,etal.,2006)。此外,nk细胞活化由增强nk细胞对靶细胞的反应性的细胞因子(例如il-2或il-15)引发(koehletal,2016)。
除了它们的直接抗肿瘤活性外,nk细胞还有助于诱导适应性抗癌响应,并可以发挥免疫调节功能(schusteretal.,2016)。越来越多的证据支持nk细胞亚群可以包含广泛的表型和功能多样性的观点(roelleandbrodin,2016;tesi,etal.,2016)。此外,nk细胞可以获得适应性免疫和免疫记忆的特性,例如特定亚群扩增和抗原特异性响应(sunetal.,2014)。在这种情况下,il-12/15/18预活化的nk细胞已被证明能增强和延长nk细胞对肿瘤细胞和细胞因子的反应性,这与ifn-γ基因座的表观遗传重塑有关(cerwenkaandlanier,2016;luetke-eversloh,etal.,2014;ni,etal.,2016;romeeetal.,2016)。
虽然根据定义,nk细胞不需要事先致敏,但未经进一步活化的新分离的(即“未致敏(naive’)”)人nk细胞只能对有限数量的肿瘤细胞(例如典型靶细胞系k562)有反应性(vivier,2011)。特别地,来自癌症患者的nk细胞显示出对自体肿瘤细胞的低反应性,除非用细胞因子离体活化(parkhurstetal.,2011,reinersetal.,2013)。这可能反映了肿瘤细胞识别不充分、免疫抑制性癌症微环境的抑制作用以及体内nk细胞反应性的连续调节。
即使存在自身mhci类,肿瘤反应性治疗性抗体也可以显著改善未致敏nk细胞对肿瘤细胞的细胞毒性(harris,2004;pahletal.,2012;parkhurstetal.,2011;reinersetal.,2013)。这种抗体介导的细胞毒性(adcc)是通过itam偶联的nk细胞活化性受体cd16a(fcγriiia)识别人源化和嵌合igg1抗体的人fc部分介导的(delandazuri,etal.,1979;lanier,etal.,1988;vivier,etal.,1991)。从携带高亲和力cd16a同种异型(158v对158f基因多态性)的患者预后更好这一观察结果可以推断出nk细胞和adcc在治疗性抗体临床反应中的作用(bibeau,etal.,2009;dall'ozzo,etal.,2004;musolino,etal.,2008)。
然而,与其他癌症患者或健康个体相比,多发性骨髓瘤患者的血清igg水平升高,并且骨髓瘤患者中cd16a亲和力的差异以及与常规治疗性抗体在这种病理生理血清浓度下对人igg的cd16a结合的潜在竞争可能妨碍adcc和nk细胞在体内发挥全部潜力(lietal.,2016)。为了改善cd16a结合(engagement),已经开发了抗体形式,其以不依赖于fc的方式以高亲和力结合cd16a(rothe,etal.,2015;wiernik,etal.,2013)。afm13是四价双特异性cd30/cd16a串联双抗体
技术实现要素:
尽管cd16a是人nk细胞上的有效活化性受体,介导nk细胞对抗体调理的癌细胞的细胞毒性,但是希望通过抗原结合蛋白的cd16a结合进一步提高活化nk细胞的细胞毒性的效率和效力。
本发明人发现,通过多特异性抗体例如双特异性cd30/cd16a串联双抗体(afm13)的cd16a结合改变了原代nk细胞的表型和功能。在最初改善nk细胞功能后,抗cd16a抗原结合蛋白暴露(通过串联双抗体)导致细胞毒性效力的瞬时选择性降低。然而,这种受损的nk细胞毒性可以通过细胞因子刺激来恢复。在实施例中,显示在没有抗原结合蛋白暴露的情况下在il-2存在下培养nk细胞一段时间后,这些经cd16a-处理的(cd16a-experienced)nk细胞在用细胞因子或其他抗性肿瘤细胞再刺激时表现出更强的细胞毒性和ifn-γ产生,表明记忆样(memory-like)功能。
因此,本文提出的发明揭示了尚未发现的cd16a触发(triggering)在引发和增强nk细胞对细胞因子和随后肿瘤细胞再刺激的应答的作用。本发明提供了抗cd16a抗原结合蛋白的间歇给药方案,例如cd30/cd16a串联双抗体(afm13)、egfr/cd16a串联双抗体afm24或bcma/cd16a串联双抗体afm26中的任一种与用活化nk细胞的细胞因子进行的治疗联合一段时间而不暴露于抗cd16a抗原结合蛋白,该方案改善nk细胞反应。
本发明揭示了cd16a在引发和增强nk细胞对细胞因子和随后肿瘤细胞再刺激的应答的新的额外作用。这意味着,在实施例中用cd30/cd16a串联双抗体刺激不仅可以杀死未经调理的cd30+肿瘤细胞,甚至可以杀死cd30-肿瘤细胞,cd30-肿瘤细胞不是cd30/cd16a串联双抗体直接靶向的。这些发现保证了抗cd16a抗原结合蛋白与细胞因子组合的间歇方案。这种间歇性组合治疗方案扩大了肿瘤反应性nk细胞的数量,并增加了它们在患者体内的功能。
即使没有共刺激信号,cd16a也是唯一触发未致敏(naive)人nk细胞细胞毒活性的活化性受体(brycesonetal.,2009;brycesonetal.,2006)。在实施例中证明,通过双特异性四价cd30/cd16a串联双抗体的cd16a活化诱发了响应于nk细胞耐受的cd30+淋巴瘤细胞的强效的nk细胞细胞毒性。重要的是,在实施例中显示cd30/cd16a串联双抗体的cd16a结合增加了nk细胞对il-15或低剂量il-2的敏感性。这导致il-15和il-2依赖性nk细胞增殖的放大,导致高功能性nk细胞的数量大大增加。在对经cd30/cd16a串联双抗体调理的肿瘤细胞的初始的优良nk细胞活性之后,cd30/cd16a串联双抗体暴露的持续时间增加导致nk细胞细胞毒性受损并减少ifn-γ的产生。然而,这种效力的降低是短暂的,因为它可以在用il-2或il-15培养这种nk细胞后恢复。值得注意的是,当通过cd16a被cd30/cd16a串联双抗体预活化时,这些细胞因子培养的nk细胞在再次刺激后对几乎所有具有耐受性的cd30+甚至cd30-淋巴瘤细胞发挥增强的细胞毒性和ifn-γ产生。可以通过随后的细胞因子处理再次刺激的这种受损的nk细胞细胞毒性也来自其他cd16a抗原结合蛋白,例如双特异性串联抗体样egfr/cd16a或bcma/cd16a。因此,本发明揭示了尚未发明的cd16a触发响应于活化细胞因子和肿瘤细胞而引发和增强nk细胞功能的作用。
对il-15和低剂量il-2的敏感性提高与cd25(高亲和力il-2受体α链)的诱导以及cd132(低亲和力γ链,为il-2和il-15受体的一部分)的上调相一致。cd30/cd16a串联双抗体的cd25诱导显著强于先前使用抗cd163g8与二抗的cd16a交联所示的(marquezetal.,2010)。类似地,已显示il-12/15/18的nk细胞活化诱导强烈的cd25表达,促进体外和荷瘤小鼠体内的il-2依赖性增殖(nietal.,2012)。因此,cd30/cd16a串联双抗体暴露后cd25的诱导使得nk细胞能够与调节性t细胞竞争少量的il-2,否则组成型的cd25表达将限制nk细胞对il-2的可用性(gasteigeretal.,2013;kimetal.,2017)。除了与cd16a结合的串联双抗体之外,fc介导的利妥昔单抗与cd16a的结合使增殖潜力增强且效力瞬时降低,表明了一种更普遍的cd16a活化的现象。然而,与利妥昔单抗相比,响应于与cd16a结合的串联双抗体的cd25和cd16的调节较不均一且在健康供体中更明显。与fc-结合的利妥昔单抗相比,与cd16a结合的串联双抗体的强大的活化潜能可归因于其与cd16a的延长的且高亲和力的二价结合,无论cd16a多态性如何(对igg抗体的fc结构域的亲和力低或高)(reuschetal.,2014)。
在暴露于cd16a结合串联双抗体之后,观察到cd16a依赖性甚至“自然”nk细胞细胞毒性的效力的瞬时和选择性降低,以及肿瘤细胞的二次应答中的脱粒和ifn-γ。cd16a依赖性活性的降低可以通过与cd16a结合后cd16表达几乎完全丧失来解释,其至少部分涉及mmp介导的切割或受体内化,如另外所述(capuanoetal.,2015;lajoieetal.,2014;motaetal.,2004;romeeetal.,2013;wierniketal.,2013)。此外,“未致敏”nk细胞效力、抗肿瘤反应性的短暂降低表明其他nk细胞活化性受体(如nkp30和nkg2d)的脱敏,所述脱敏被证明与k562细胞溶解有关(brandtetal.,2009;kuylenstiernaetal.,2011)。甚至响应于pma/离子霉素的脱粒和ifn-γ产生的减少表明效力更广泛的瞬时降低,其显然也影响pkc活化和/或ca2+动员,其直接被pma/离子霉素活化(chatilaetal.,1989)。ca2+动员与cd16a和其他活化性受体的末端信号传导密切相关(brycesonetal.,2006;cassatellaetal.,1989)。pkc活化可以介导ifn-γ的产生,并且对于k562细胞溶解是重要的,但对于adcc是非必要的,adcc反过来需要pi3k活化(bonnemaetal.,1994;haraetal.,2008)。
因此,观察到20小时的串联双抗体(例如cd30/cd16a串联双抗体)暴露后原代nk细胞效力的瞬时降低,可能超过之前发现的由短期(1.5小时)cd16a结合导致的更有限的抑制作用;据报道,这种抑制作用涉及shp-1的募集和plcγ2/vav-1/slp-76磷酸化的抑制,导致脱粒缺陷,而ifn-γ的产生和对pma/离子霉素的反应性保持不变(capuanoetal.,2015;galandrinietal.,2002)。
cd16a结合后对il-12/15/18的ifn-γ应答是保守的或甚至进一步增强,表明cd16a结合串联双抗体(例如cd30/cd16a串联双抗体)暴露后的nk细胞仅具有针对肿瘤细胞活化的ifn-γ的选择性减少。这可以通过有效诱导ifn-γ的高亲和力il-12受体和il-18受体的上调来解释。据报道,cd16a和il-12受体活化可协同促进ifn-γ的产生(kondadasulaetal.,2008)。
总之,cd16a结合串联双抗体(例如cd30/cd16a串联双抗体)暴露后nk细胞功能的短暂降低,可随后在il-2或il-15中培养后完全恢复。重要的是,这些经cd16a-处理(experienced)的、细胞因子培养的nk细胞具有记忆样功能,因为对用其它弱易感淋巴瘤细胞进行的再刺激的细胞毒活性和ifn-γ的产生显著增强。有趣的是,“cd16a诱导的记忆样nk细胞”的这种新功能可能类似于先前描述的il-12/15/18诱导的记忆样nk细胞的增强的抗肿瘤活性和ifn-γ反应(cooperetal.,2009;nietal.,2016;nietal.,2012;romeeetal.,2012;romeeetal,2016)。
这说明了包括不暴露于抗原结合蛋白的阶段的抗cd16a抗原结合蛋白(例如串联双抗体)间歇给药方案,改善了再次相遇时单个nk细胞对经抗体调理的肿瘤细胞的反应性。此外,根据本发明的cd16a结合抗原结合分子与nk细胞活化细胞因子(如il-2或il-15)的间歇施用不仅维持甚至能够增强nk细胞的抗肿瘤活性,并且增加了淋巴瘤患者中的肿瘤反应性nk细胞的数量。
以引用方式并入
本说明书中提及的所有出版物、专利和专利申请均通过引用并入本文,其程度如同每个单独的出版物、专利或专利申请被具体和单独地指出通过引用并入。
附图说明
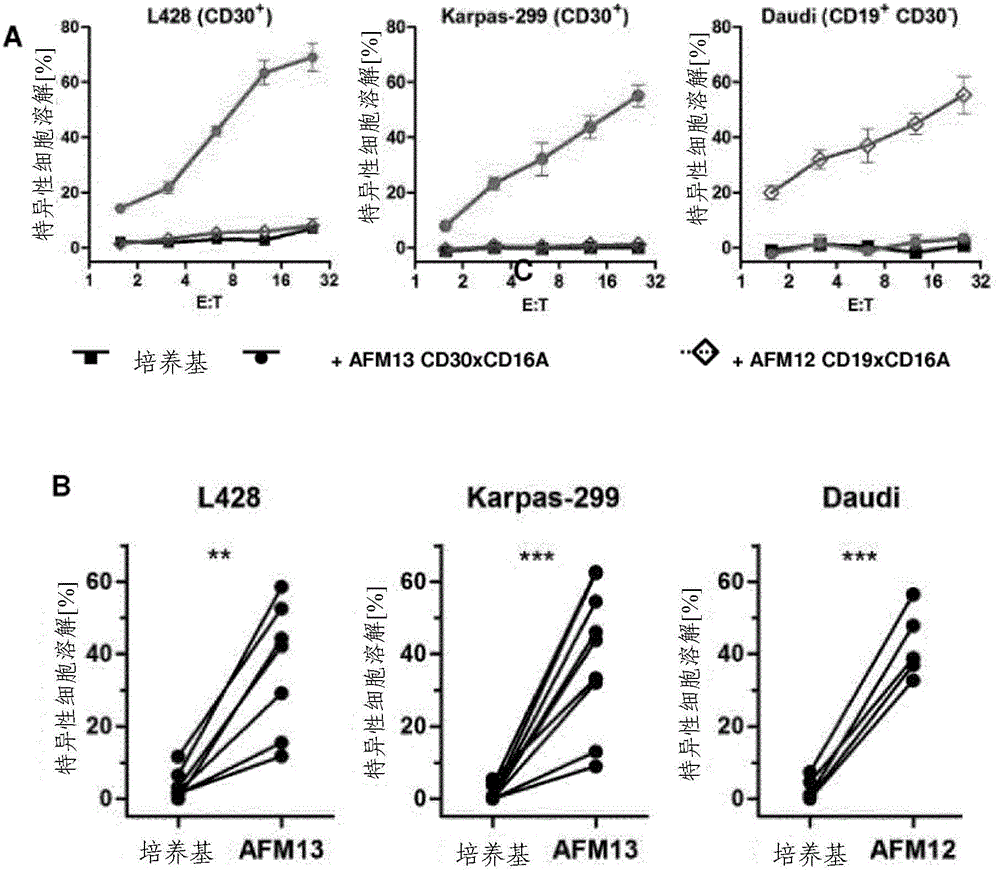
图1显示经cd30/cd16a串联双抗体调理的肿瘤细胞诱导的nk细胞的细胞毒性、ifn-γ的产生以及活化性受体:
a.在逐渐增加的效应物-靶标(e:t)比率下,在cd16axcd30串联双抗体afm13(圆圈)、cd16axcd19串联双抗体afm12(菱形)(全部为10μg/ml)存在下,与无抗体添加(正方形)相比,在4小时51cr释放测定中测量新鲜分离的原代nk细胞对cd30+cd19-karpas-299细胞和l428细胞以及cd30-cd19+daudi细胞的特异性细胞溶解;数据代表至少三次实验。b.使用/不使用afm13时karpas-299细胞、l428细胞和daudi细胞(e:t6:1)的累积特异性细胞溶解。c.将通过nk细胞的afm13诱导的karpas-299细胞和l428细胞的细胞溶解(圆圈)与亲本嵌合抗cd30igg抗体(三角形)在10-5-10μg/ml(e:t25:1)的抗体浓度下诱导的细胞溶解进行比较。afm12用作阴性对照(菱形)。d.在匹配的e:t比率下,在纯化的nk细胞(圆圈)和整个pbmc(正方形)之间比较在afm13(1μg/ml)存在下karpas-299和l428的特异性细胞溶解。用nk细胞耗尽的pbmc(pbmcδnk)(三角形)的细胞溶解进行不存在nk细胞的校正;数据代表两个实验。e.纯化的nk细胞和l428细胞(e:t1:1)在有或没有afm13(0.4μg/ml)的情况下共培养4小时后,通过流式细胞仪测量nk细胞cd107a表达(脱粒标记物)和细胞内ifn-γ表达。在共培养20小时后通过elisa定量ifn-γ的产生;四个实验的累积数据。
图2显示了通过cd30/cd16a串联双抗体经由cd16a的预活化增加了响应于il-15或低剂量il-2的nk细胞增殖和扩增:
a.实验设计方案。b.在培养基中或在包被(包被的)afm13的塑料(1μg/孔)上培养nk细胞20小时后(称为afm13-预活化),cd25、cd122和cd132的表达,描述为代表性直方图和与培养基相比的倍数变化(五个实验的累积数据)。包被的小鼠igg1用作阴性对照(包被的对照)。c.在培养基中或包被的afm13上培养后,收获cfse标记的nk细胞,重新铺板并用il-2(400u/ml)孵育3-7天后,通过流式细胞术测量cfse表达;两个实验的代表性数据(表明mfi值)。d.在afm13预活化后的il-2(400u/ml)或培养基中在培养5天后评估经历至少4次分裂(通过cfse稀释计算)的nk细胞的百分比;五个实验的累积数据。e.afm13预活化的nk细胞或对照nk细胞在逐渐升高的il-2浓度(12.5-400u/ml)下培养5天后的cfse表达和绝对nk细胞或f.afm13预活化的nk细胞或对照nk细胞在逐渐升高的il-15浓度(0.6-10ng/ml)培养5天后的cfse表达和绝对nk细胞数量;两个实验的数据。
图3显示在afm13暴露后的瞬时功能障碍后nk细胞细胞毒性的恢复:
a.实验设计方案。b.在用afm13与karpas-299细胞共培养20小时后,或在包被的afm13上(或如图2,b中的对照)培养后,测量nk细胞上的cd16表达;六个实验的代表性直方图。c.(a)暴露于包被的afm13或(b)培养基后的nk细胞上的cd16表达,或在暴露于包被的afm13或(d)未预暴露(仅il-2)后在il-2(400u/ml)中培养5天后在nk细胞上的cd16表达;九个实验的mfi数据。d.如c中所述培养的nk细胞对经afm13-调理的karpas-299细胞的细胞溶解和e脱粒;2-5次实验的累积数据(e:t2.5:1)。f.如c中所述培养的nk细胞对k562细胞的自然细胞溶解和g.脱粒。2-6次实验的累积数据(e:t2.5:1)。
图4显示在cd30/cd16a串联双抗体暴露后的瞬时选择性功能障碍后nk细胞ifn-γ的恢复:
如图3,c中所述培养的nk细胞响应于a.经afm13-调理的karpas-299细胞、b.k562细胞和c.il-12/15/18的细胞内ifnγ表达;2-4次实验的累积数据。d.高亲和力il-12rβ2和il-18rα在先前被包被的afm13活化的或在培养基中培养的nk细胞上的表达(左边两个直方图);以及在afm13暴露后在il-2中培养5天的nk细胞或在无预暴露的情况下在il-2中培养的nk细胞上的表达(右边两个直方图);4个实验的代表性数据。
图5显示cd30/cd16a串联双抗体的预活化引发nk细胞记忆样ifn-γ产生和细胞毒性:
a.由暴露于包被的afm13后在il-2中培养5天后的nk细胞,或由未经预暴露而仅在il-2中培养5天后的nk细胞响应于il-12/15、k562、l428的24小时再刺激而产生的ifn-γ;5个实验的累积数据。b.新鲜分离的nk细胞、或在暴露于包被的afm13后在il-2中培养5天的nk细胞、或未经预暴露的仅在il-2中培养的nk细胞对未调理的cd30+karpas-299细胞和hdlm-2细胞以及cd30-daudi细胞和cd30+l1236细胞的细胞溶解;2-6次实验的累积数据。
图6显示了尽管存在细胞因子刺激,再次暴露于afm13后nk细胞的靶细胞杀伤能力仍瞬时降低:
a.实验设计方案:第一次暴露于包被的afm13中20小时,随后在il-2(400u/ml)中培养5天,第二次暴露于包被的afm13中,持续2天;
b.如a中所述培养的nk细胞对afm13-调理的karpas-299细胞的细胞溶解;(e:t2.5:1)。
图7显示egfr/cd16a串联双抗体(afm24)和bcma/cd16a串联双抗体(afm26)分别响应于egfr+和bcma+靶细胞诱导nk细胞的功能性活化。显示了在egfr+a-431和bcma+mm.1s靶细胞上afm24(egfr/cd16a串联双抗体)和afm26(bcma/cd16a串联双抗体)的存在下nk细胞的细胞毒活性。用o/n培养的nk细胞作为效应细胞以2.5:1的e:t比率的4小时钙黄绿素释放。n=2个独立实验。
图8显示分别暴露于afm24(egfr/cd16a串联双抗体)和afm26(bcma/cd16a串联双抗体)后cd16a介导的nk细胞功能的恢复。
a.分别暴露于afm24和afm26后评估表型和功能性nk细胞恢复的实验方案。b.分别暴露于pbs或10μg/ml包被的afm24和afm2624小时后,nk细胞表面上的cd16的流式细胞术分析。显示了在恢复期(第1天至第6天)期间在不存在和存在指定细胞因子的情况下的cd16的细胞表面表达。在流式细胞术中将nk细胞鉴定为cd45+cd56+活细胞。c.暴露于平板结合的afm24和afm26时,定量nk细胞表面上的cd16表达。n=2个独立实验。d.在afm24靶细胞a-431和afm26靶细胞mm.1s存在下以2.5:1的e:t比率评估特异性细胞毒性nk细胞功能。将相应的串联双抗体补充至所应用的4小时钙黄绿素释放测定(c=1μg/ml)。必要时,用400u/mlil-2处理nk细胞。n=2个独立实验。
图9显示分别暴露于afm24(egfr/cd16a)和afm26(bcma/cd16a)后cd16a介导的nk细胞功能的恢复。
a.暴露于afm24和afm26后,nk细胞表面上cd16表达的定量。n=2个独立实验。b.在afm24靶细胞a-431和afm26靶细胞mm.1s存在下以2.5:1的e:t比率评估特异性细胞毒性nk细胞功能。将相应的串联双抗体补充至所应用的4小时钙黄绿素释放测定(c=1μg/ml)。必要时,用10ng/mlil-15处理nk细胞。n=2个独立实验。
发明详述
本发明提供了一种抗原结合蛋白,其包含至少一个cd16a的抗原结合位点,任选地,所述抗原结合蛋白与细胞因子组合,用于基于nk细胞的免疫疗法,其中间歇施用所述抗原结合蛋白,该间歇施用包括在两个间歇(即连续剂量之间)之间的不暴露于所述抗原结合蛋白的阶段。
基于nk细胞的免疫疗法可以是体内进行和/或包括离体步骤,例如过继性nk细胞转移或造血干细胞移植(hct),其中自体nk细胞或同种异体nk细胞可被细胞因子活化并进行离体扩增,然后转移到患者体内。
在某些实施方案中,抗原结合蛋白是多价的并且包含至少两个对cd16a特异的抗原结合位点,所述抗原结合位点导致经由cd16a受体与nk细胞二价结合,从而增加亲和力和nk细胞的细胞毒活化的效力。
在某些实施方案中,抗原结合蛋白是多特异性的,并且包含至少一个对不同于cd16a的靶抗原具有特异性的抗原结合位点,用于使nk细胞与靶标(例如肿瘤细胞或病毒)结合。
在特定实施方案中,抗原结合蛋白是四价双特异性串联双抗体,其包含cd16a的两个抗原结合位点和靶抗原的两个抗原结合位点,所述靶抗原例如肿瘤抗原或病毒抗原。
术语“组合”是指组合中的化合物(即cd16a抗原结合蛋白和细胞因子)的同时、分开或顺序给药。
如本文所用的术语“个体”包括个体,例如患者、罹患或患有可用基于nk细胞的免疫疗法治疗的病症,例如癌症或任何涉及癌症的疾病的个体。在cd30/cd16a抗原结合蛋白的实施方案中,该病症为cd30阳性恶性肿瘤,特别是cd30+淋巴瘤,例如霍奇金淋巴瘤、间变性大细胞淋巴瘤(alcl)、弥漫性大b细胞淋巴瘤(dlbcl)。
术语“抗原结合蛋白”表示具有抗原结合特性的免疫球蛋白衍生物。本发明的抗原结合蛋白通过与cd16a结合而与nk细胞结合。抗原结合蛋白可包含免疫球蛋白多肽、其片段、缀合物或包含抗原结合位点的免疫功能性免疫球蛋白部分的融合肽。结合蛋白包含至少一个抗原结合位点,该结合位点是与抗原结合的结合蛋白的区域、部分或结构域。在某些实施方案中,每个抗原结合位点由特异性结合相同抗原表位的免疫球蛋白的可变重链结构域(vh)和可变轻链结构域(vl)形成。可变重链结构域包含三个重链互补决定区(cdr):cdr1、cdr2和cdr3。可变轻链结构域包含三个互补决定区(cdr):cdr1、cdr2和cdr3。在备选实施方案中,抗原结合位点可仅由重链或轻链组成。例如,这种抗原结合位点可以衍生自仅由重链组成的纳米抗体,并且可以在不存在轻链的情况下与cd16a结合。已经描述了衍生自美洲驼(llama)或骆驼(camel)的纳米抗体。已经描述了不包含vl结构域的对cd16a具有特异性的基于vh的抗原结合位点(lietal.,2016)。结合蛋白可以是基于fv结构域的igg样或非igg样融合肽,其不具有或具有额外的恒定结构域。在本发明的某些实施方案中,结合蛋白缺乏免疫球蛋白恒定结构域。抗原结合位点的可变重链结构域和可变轻链结构域可以彼此共价连接,例如,通过肽接头,或彼此非共价结合以形成抗原结合位点。
在某些实施方案中,抗原结合蛋白包含结合cd16a但不结合cd16b的抗原结合位点。包含与cd16a结合但不结合cd16b的重链可变结构域和轻链可变结构域的抗原结合位点可由抗原结合位点提供,该抗原结合位点特异性结合cd16a的表位,所述cd16a的表位包含不存在于cd16b中的c末端序列sffppgyq(seqidno:11)的氨基酸残基和/或cd16a(seqidno:20)的残基g130和/或y141。wo2006/125668中描述了包含结合cd16a但不结合cd16b的重链可变结构域和轻链可变结构域的抗原结合位点的实例。
在一些实施方案中,抗原结合蛋白包含对cd16a特异的重链可变结构域和轻链可变结构域,其中(i)对cd16a特异的重链可变结构域(vh_cd16a)包含具有seqidno.1所示氨基酸序列的重链cdr1;具有seqidno:2或7所示氨基酸序列的重链cdr2;具有seqidno:3所示氨基酸序列的重链cdr3,以及对cd16a特异的轻链可变结构域包含具有seqidno:4所示氨基酸序列的轻链cdr1;具有seqidno:5所示的氨基酸序列的轻链cdr2;和具有seqidno:6所示氨基酸序列的轻链cdr3。在进一步的实施方案中,抗原结合蛋白包含(a)对cd16a特异的重链可变结构域(vh_cd16a),其具有seqidno:8或10所示的氨基酸序列,和/或(b)对cd16a(vl_cd16a)特异的轻链可变结构域,其具有seqidno:9所示的氨基酸序列。
在替代实施方案中,重链和轻链结构域掺入本文所述的cdr或框架序列的免疫活性同源物或变体。因此,在一些实施方案中,结合cd16a的重链或轻链结构域中的cdr序列与seqidno:1-7中描述的氨基酸序列相似但不相同。在某些情况下,与seqidno:1-7的序列相比,cdr变体序列具有99%、98%、97%、96%、95%、94%、93%、92%、91%、90%、89%、88%、87%、86%、85%、84%、83%、82%、81%或80%的序列同一性,并且具有免疫学活性。
在其他情况下,cdr变体序列包含1、2、3、4或5个保守氨基酸置换。保守置换包括用具有相似特征的另一种氨基酸置换给定氨基酸的氨基酸置换,并且还包括脂族氨基酸中丙氨酸、缬氨酸、亮氨酸和异亮氨酸的交换;羟基残基丝氨酸和苏氨酸的交换,酸性残基天冬氨酸和谷氨酸的交换,酰胺残基天冬酰胺和谷氨酰胺之间的置换,碱性残基赖氨酸和精氨酸的交换,以及芳香族残基苯丙氨酸和酪氨酸之间的取代。
在其他情况下,修饰cdr变体序列以改变非关键区域中的非关键残基或残基。可以通过已知方法鉴定不重要的氨基酸,例如亲和力成熟、cdr步行诱变(walkingmutagenesis)、定点诱变、结晶、核磁共振、光亲和标记或丙氨酸扫描诱变。
在进一步的备选实施方案中,抗原结合蛋白包含重链和轻链结构域,其是本文提供的重链和轻链结构域序列的免疫学活性同源物或变体。因此,在一些实施方案中,抗原结合蛋白包含重链或轻链结构域序列,其与seqidno:8-10中所示的氨基酸序列相似但不相同。在某些情况下,与seqidno:8-10的序列相比,变体重链或轻链结构域序列具有99%、98%、97%、96%、95%、94%、93%、92%、91%、90%、89%、88%、87%、86%、85%、84%、83%、82%、81%或80%的序列同一性,并且具有免疫学活性。
在其他情况下,变体重链或轻链结构域序列包含1、2、3、4或5个保守氨基酸置换。保守置换包括用具有相似特征的另一种氨基酸置换给定氨基酸的氨基酸置换,并且还包括脂族氨基酸中丙氨酸、缬氨酸、亮氨酸和异亮氨酸的交换;羟基残基丝氨酸和苏氨酸的交换,酸性残基天冬氨酸和谷氨酸的交换,酰胺残基天冬酰胺和谷氨酰胺之间的置换,碱性残基赖氨酸和精氨酸的交换,以及芳香族残基苯丙氨酸和酪氨酸之间的取代。
在更进一步的实例中,变体重链或轻链结构域序列包含增强cdr性质的置换,例如增加稳定性、对蛋白酶的抗性和/或对cd16a的结合亲和力。
在其他情况下,修饰变体重链或轻链结构域序列以改变非关键区域中的非关键残基或残基。可以通过已知方法鉴定不重要的氨基酸,例如亲和力成熟、cdr步行诱变、定点诱变、结晶、核磁共振、光亲和标记或丙氨酸扫描诱变。
根据本发明的抗原结合蛋白具有比单克隆抗体或使用igg等同结构域的任何其他抗体形式短的半衰期,以获得类似igg的半衰期以及采用hsa结合以延长其半衰期的构建体,特别是igg。
“半衰期”是指消除半衰期,其是抗原结合蛋白浓度达到其施用途径的人外周血中(即,血清中的游离抗体,例如但不限于静脉内注射或输注或皮下施用)测量的初始量的一半所需的时间。
在某些实施方案中,抗原结合蛋白的半衰期小于一周,特别是小于72小时、48小时、42小时、36小时、24小时、20小时、12小时或6小时。这种短的半衰期有利于采用间歇给药,该间歇给药包括根据本发明的无暴露阶段或暴露减少的阶段,因为在两个连续的间歇施用抗原结合蛋白剂量之间的间隔时间中的无暴露阶段仍然很短。
如此短的半衰期明显短于单克隆抗体的半衰期,这使得根据本发明的间歇给药方案成为可能,因为它允许在两个连续的抗原结合蛋白剂量之间在消除抗原结合蛋白后提供暴露减少的阶段或无暴露阶段。这种抗原结合蛋白的暴露减少的时间或无暴露时间对于nk细胞的恢复是重要的。如本文所用,术语“暴露减少的”是指抗原结合蛋白的量(即血浆浓度),其小于如上所述测量的抗原结合蛋白初始量的一半。因此,术语“低暴露”是指抗原结合蛋白在给药后在抗原结合蛋白的半衰期后达到的量。例如,术语“暴露减少的”可以指少于抗原结合蛋白的最初施用剂量的40%、30%、20%和15%的量,例如15-40%、15-30%或15-20%的量。
如本文所用的术语“无暴露”是指抗原结合蛋白的量(即血浆浓度),其小于在经过抗原结合蛋白的至少3个半衰期后的量,其对应于抗原结合蛋白的施用剂量的15%以下。优选地,“无暴露”是指抗原结合蛋白的量(即血浆浓度),其小于在经过抗原结合蛋白的至少4-5个半衰期后的量。优选地,“无暴露”是指抗原结合蛋白的量(即血浆浓度),其小于抗原结合蛋白施用剂量的10%、1%或0.1%。
根据本发明的抗原结合蛋白具有比单克隆抗体显著更短的半衰期,这可以通过重组抗体衍生物(与单克隆抗体相比,具有更低的分子量)提供。
根据本发明的具有短半衰期的抗原结合蛋白的实例包括:非igg样抗体片段或免疫功能性免疫球蛋白部分的融合肽,例如fab、fab'、f(ab')2、fv片段,例如单链fv、串联单链fv((scfv)2)、双特异性杀伤衔接子(engagers)(bike)、三特异性杀伤衔接子(trike)、双亲和靶向抗体(darttm)、双抗体、双抗体(db)、单链双抗体(scdb)和串联双抗体
此外,结合蛋白可以是多价的,即包含两个或更多个抗原结合位点。在某些实施方案中,抗原结合蛋白包含至少两个cd16a的抗原结合位点,即双价结合cd16a。在一些实施方案中,结合蛋白包含用于纯化的标签-氨基酸序列。
在某些实施方案中,抗原结合蛋白是多特异性的并且包含至少一个额外的抗原结合位点,其结合不同于cd16a的靶抗原,即具有第二种不同的抗原特异性。在某些实施方案中,抗原结合蛋白可包含靶抗原的至少两个抗原结合位点。
“靶抗原”通常是指与位点相关的抗原(例如细胞或病毒),nk细胞应由抗原结合蛋白引导至该位点,以引发nk细胞的细胞毒性。靶位点的实例可以是肿瘤细胞或感染因子,例如病毒或细菌性病原体或寄生虫,例如登革病毒、单纯疱疹、流感病毒、hiv或携带自身免疫靶标的细胞、自身免疫标记或自身免疫抗原。
不同于cd16a的靶抗原可选自细菌物质、病毒蛋白、自身免疫标记或肿瘤抗原。
在靶抗原是肿瘤抗原的实施方案中,肿瘤抗原可以选自肿瘤细胞表面抗原,例如特异性肿瘤标志物、或由mhc类分子展示的mhc限制性肽。如本文所用的术语“肿瘤抗原”包括肿瘤相关抗原(taa)和肿瘤特异性抗原(tsa)。术语“肿瘤细胞表面抗原”是指肿瘤细胞表面上能够被抗体识别的任何抗原或其片段。
用于肿瘤细胞的靶抗原的实例包括但不限于cd5、cd19、cd20、cd30、cd33、基质金属蛋白酶1(mmp1)、层粘连蛋白受体前体蛋白、egfr、egfrviii、bcma、ep-cam、plap、thomsen-friedenreich(tf)抗原、muc-1(粘蛋白)、igfr、il4-rα、il13-r、fcεri、ige、vgef、her2/neu、her3、psma、cea、tag-72、hpve6、hpve7、bing-4、cyclin-b1、9d7、epha3、端粒酶、间皮素、sap-1、生存素、睾丸癌抗原(bage族、cage族、gage族、mage族、sage族、xage族)、ny-eso-1/lage-1、prame、ssx-2、melan-a/mart-1、gp100/pmel17、酪氨酸酶、trp-1/-2、mc1r、β-连环蛋白、brca1/2、cdk4、cml66、mart-2、p53、ras、tgf-βrii和tcr(参见categoriesoftumorantigens,holland-freicancermedicine.6thedition,2003)。其他肿瘤抗原描述于weinberg,r.,thebiologyofcancer,2013。
在某些实施方案中,多特异性的抗原结合蛋白包含靶抗原的至少两个抗原结合位点。在特定实施方案中,抗原结合蛋白包含至少两个cd16a的抗原结合位点和至少两个靶抗原的抗原结合位点。这种抗原结合蛋白至少是四价的。
在一个具体实施方案中,提供了双特异性四价串联双抗体
在实施例中举例说明的是对cd30和cd16a(cd30/cd16a串联双抗体)具有特异性的双特异性四价cd30/cd16a串联双抗体afm13,其已描述于reusch,etal.,2014中。该cd30/cd16a串联双抗体通过仅与同种型cd16a结合而特异性地募集nk细胞。设计本文所述的cd30和cd16a双特异性串联双抗体以允许通过募集细胞毒性nk细胞而特异性靶向cd30+肿瘤细胞。在这种串联双抗体中,接头长度为使得其本身能够防止可变结构域的分子内配对,从而使分子不能自身折叠形成单链双抗体,而是被迫与另一链的互补结构域配对。还设置结构域使得相应的vh和vl结构域在该二聚化过程中配对。在由单个基因构建体表达后,两条相同的多肽链头尾相互折叠,形成约105kda的功能性非共价同源二聚体。尽管不存在分子间共价键,但同二聚体一旦形成就高度稳定,保持完整并且不会恢复为单体形式。
串联双抗体仅含有抗体可变结构域,因此预期缺乏可能与fc部分相关的副作用或非特异性相互作用。例如,fc受体存在于许多细胞类型上,例如可以与fcγ结合的白细胞或kuppfer细胞。因为串联双抗体允许与每种靶抗原(例如cd30和cd16a)二价结合,所以亲合力与igg的亲合力相同。串联双抗体的大小(约为105kda),小于igg的大小,这可以允许增强的肿瘤渗透。然而,该大小远高于首过(first-pass)肾清除的阈值,与基于抗体结合结构域或非抗体支架的较小双特异性形式相比,提供了药代动力学优势。串联双抗体(例如cd30/cd16a串联双抗体afm13)的半衰期约为19小时。串联双抗体在宿主细胞(例如哺乳动物cho细胞)中很好地表达。预期强大的上游和下游制备工艺可用于串联双抗体。
如本文所用的“间歇”施用是以间隔,特别是有规律的间隔施用抗原结合蛋白的方法。因此,施用第一剂量的抗原结合蛋白,并在施用第一剂量的抗原蛋白后施用第二剂量的抗原蛋白。优选地,对于一个治疗周期,施用第一剂量和第二剂量的间隔时间重复几次。选择两个连续的抗原结合蛋白剂量之间的间隔时间,使得基于抗原结合蛋白的药代动力学特征(例如半衰期)间歇性的暴露于抗原结合分子,从而允许在两个连续的抗原结合蛋白剂量的施用之间至少具有暴露减少的阶段,或者优选地,无暴露阶段。暴露于抗cd16a抗原结合蛋白可以瞬间降低nk细胞的效力。为了有效恢复nk细胞的细胞毒性,重要的是在恢复期期间使抗cd16a抗原结合蛋白的暴露(即浓度)保持较低或不存在。因此,选择间隔时间使得其长于抗原结合蛋白的半衰期,从而确保该间隔时间包括在消除大部分抗原结合蛋白后的暴露减少的阶段或无暴露阶段。
因此,间歇给药的间隔时间是抗原结合蛋白的半衰期的倍数。
在一些实施方案中,间隔时间可以比抗原结合蛋白的半衰期长至少一天。
在某些实施方案中,该间隔时间是抗原结合蛋白的半衰期的至少三倍。例如,间隔可以为抗原结合蛋白的半衰期的至少三倍至至少五倍,例如至少3倍、3.5倍、4倍、4.5倍或5倍。在本发明的上下文中,cd16a抗原结合蛋白在经过其半衰期的至少3倍,优选4-5倍的时间后被认为是稀释的或不存在的。
在某些实施方案中,两个连续的cd16a抗原结合蛋白剂量之间的间隔时间可以是至少1、2、3、4、5、6、7、8、9或10天,其中该间隔时间取决于抗原结合蛋白的半衰期,选择间隔时间使得其包含抗原结合蛋白的暴露减少的阶段或无暴露阶段。在特定的实施方案中,抗原结合蛋白在具有连续剂量(即第一剂量和随后的第二剂量)的给药周期中间歇性给药,其中两个连续的剂量之间的间隔时间为至少3天。
优选地,包含第一剂量的抗原结合蛋白和随后第二剂量的抗原结合蛋白的给药周期重复多次,例如2、3、4、5、6、7、8、9、10或更多次,直至完成所需的治疗周期和/或只要诊断出疾病进展和/或直至实现预定的临床终点,例如,4周或8周或6个月。
本发明提供了一种方法,包括间歇性施用如本文所述的抗cd16a抗原结合蛋白的步骤和施用至少一种细胞因子的另一个步骤。因此,本发明提供了如本文所述的抗cd16a抗原结合蛋白,其用于基于nk细胞的免疫疗法,其中间歇性施用抗cd16a抗原结合蛋白,并且还施用至少一种细胞因子。
细胞因子是nk细胞活化细胞因子,可以选自白细胞介素家族。如本文所用的术语“细胞因子”应理解为一种细胞因子或不同种类细胞因子的组合,其中细胞因子的组合可以同时、分开或顺序施用。因此,细胞因子可以在一定剂量的抗原结合蛋白之前、同时或之后施用。这包括在施用抗原结合蛋白之前或之后施用多次重复剂量的细胞因子。虽然间歇地施用抗原结合蛋白,但可以连续或间歇施用细胞因子。在后面的实施方案中,抗原结合蛋白和细胞因子可以间歇施用。在间歇施用抗原结合蛋白和细胞因子的实施方案中,抗原结合蛋白和细胞因子的施用时间和间隔时间可以不同;因此,抗原结合蛋白和细胞因子不同时施用。
因此,细胞因子可以在每次施用抗原结合蛋白之后施用,或者细胞因子的两次或更多次施用的循环可以在施用抗原结合蛋白之后施用。例如,通过在抗原结合蛋白预处理后施用低剂量的il-2可以增加活化的nk细胞的量。
例如,细胞因子可以选自白细胞介素2(il-2)、白细胞介素6(il-6)、白细胞介素12(il-12)、白细胞介素15(il-15)、白细胞介素18(il-18)、白细胞介素21(il-21)或它们的组合。在某些实施方案中,细胞因子为il-2或il-15或il-12、il-15和il-18的组合,尤其是il-2和il-15的组合。优选地,il-2以“低剂量”施用。合适的低剂量il-2是皮下约小于6.0百万单位/m2/天的量(lissonip;1993;romeeetal.,2016)。
il-2的低剂量施用降低了il-2的不期望的副作用的风险,例如tregs的活化,并且增加了活性nk细胞的数量,从而增加了患者的治疗功效。用于活化nk细胞的白细胞介素在romee,retal.,2014中进行了综述。
术语“细胞因子”还指包含用于提供增强的细胞因子介导的免疫功能的细胞因子的组合物和/或试剂。已经开发了这样的试剂以进一步减少副作用并调节细胞因子施用的分布和/或药代动力学。例如,细胞因子以具有增加的生物活性和增强的半衰期的复合物提供。这种复合物可以在类似抗体给药方案中施用,例如每周一次。可用作根据本发明的细胞因子的此类细胞因子复合物的实例为:nktr-214,一种包含与peg链结合的il-2的生物前药(charych,d.,etal.,2016)、nktr-255,一种包含与peg链结合的il-15的生物前药(nektartherapeutics,sanfrancisco,usa)、或alt-803,一种通过将il-15超级拮抗剂(il-15n72d)与细胞外il-15rαsushi结构域(il-15rαsu)融合而构建的多聚体复合物(liu,betal.,2016)。
在一个特定的实施方案中,提供了多特异性的(例如双特异性)抗原结合蛋白,其与cd16a二价结合,以用于免疫疗法,例如,基于nk细胞的免疫疗法,其中间歇地将抗原结合蛋白施用于个体。例如,抗原结合蛋白可以为串联双抗体,例如cd30/cd16a、egfr/cd16a或bcma/cd16a串联双抗体。
在进一步的实施方案中,结合cd16a的抗原结合蛋白间歇地施用于个体,并且与细胞因子组合施用。细胞因子可以与抗原结合蛋白交替施用、或者在连续施用的同时施用重复(即间歇性)剂量的抗原结合蛋白。在某些实施方案中,可以将第一剂量的抗原结合蛋白和随后的细胞因子剂量之间的时间或间隔时间调节至消除抗原结合蛋白(半衰期)。因此,在这样的实施方案中,在大部分抗原结合蛋白被消除并且在抗原结合蛋白的半衰期已经过去之后施用一定剂量的细胞因子。细胞因子的施用旨在在暴露于抗原结合蛋白后再刺激nk细胞。因此,应在抗原结合蛋白被大量消除后施用一定剂量的细胞因子。
因此,用于基于nk细胞的免疫疗法的、包含至少一个cd16a的抗原结合位点的抗原结合蛋白可以在剂量周期(dosagecycle)中间歇施用,其包括以下步骤:
(a)施用第一剂量cd16a抗原结合蛋白;和
(b)施用第二剂量cd16a抗原结合蛋白,
并且剂量周期还包括施用至少一种细胞因子,即在步骤(a)至步骤(b)的间隔期间施用一定剂量的至少一种细胞因子的步骤(c)。优选地,在步骤(a)之后,在经过抗原结合蛋白的半衰期的至少3倍时间之后施用第二剂量cd16a抗原结合蛋白。
在特定实施方案中,其中在一定剂量的抗原结合蛋白之后施用细胞因子,在第一剂量的抗原结合蛋白的至少20小时后施用细胞因子。因此,取决于抗原结合蛋白的半衰期,细胞因子可以在施用第一剂量的抗原结合蛋白至少20h、24h、30h、36h、42h、48h、54h、60h、70h、80h、90h或100h后施用。在特定实施方案中,其中抗原结合蛋白是串联双抗体并且需要无暴露期,细胞因子可以在施用第一剂量的串联双抗体至少约80-100小时后施用。在其他实施方案中,细胞因子可以在施用第一剂量的串联双抗体约20-36小时后施用,此时暴露已经减少。
例如,抗原结合蛋白和细胞因子以剂量周期给药,包括以下步骤:
(a)施用第一剂量的抗原结合蛋白;
(b)在步骤(a)之后施用至少一种细胞因子;和
(c)在步骤(b)之后施用第二剂量的抗原结合蛋白。
可以重复该剂量周期,优选至少直到诊断出疾病进展和/或实现期望的临床终点。
两次连续施用抗原结合蛋白(步骤(a)和(c))之间的间隔时间用于再刺激nk细胞的效力。在某些实施方案中,步骤(b)中施用细胞因子的时间至少在步骤(a)的第一剂量的抗原结合蛋白的半衰期之后。
根据本发明的抗原结合蛋白的短半衰期保证了间歇给药方案的应用。对于通过后续剂量的细胞因子再刺激nk细胞,有必要使先前剂量的抗原结合蛋白已被大量消除,使得nk细胞不再暴露于抗原结合蛋白。因此,使用半衰期小于48小时,尤其是小于24小时的抗原结合蛋白确保了本文所述的间歇给药方案。串联双抗体(例如本文所述的cd30/cd16a串联双抗体afm13)在人类中的半衰期约为19小时(rotheetal.,2015)。因此,根据本发明,可以在施用第一剂量cd30/cd16a串联双抗体的至少19小时(例如20小时)后施用一定剂量的细胞因子。或者,可以使用半衰期小于19小时的其他抗原结合形式,例如,抗体片段或单链或多链fv构建体。或者,细胞因子可以在一定剂量的抗原结合蛋白之前或与其同时施用,或者细胞因子可以连续施用。因此,在暴露于cd16a抗原结合蛋白期间,细胞因子可能存在于背景中。然而,为了恢复nk细胞的细胞毒性,有必要使(a)第一剂量的抗原结合蛋白和(b)第二剂量的抗原结合蛋白之间的剂量周期也包括相应的暴露减少的阶段,或者优选,基本上没有抗原结合蛋白的无暴露阶段。
在某些实施方案中,抗原结合蛋白是双特异性四价串联双抗体,即cd30/cd16a串联双抗体,例如afm13(reuschetal.,2014)。在这些实施方案中,cd30/cd16a串联双抗体用于治疗cd30+癌症,例如霍奇金淋巴瘤。
因此,本文提供了某些医学用途和方法,其中对cd30和cd16a特异性的抗原结合蛋白,例如如上所述的cd30/cd16a串联双抗体,以有效剂量给予个体,以治疗cd30+癌症,例如霍奇金淋巴瘤等。霍奇金淋巴瘤包括新诊断的、复发的、复发性或难治性霍奇金淋巴瘤。
本文所述的新的间歇剂量方案用于抗体介导的nk细胞结合的免疫治疗方法中。这种基于nk细胞的免疫疗法可用于治疗肿瘤、自身免疫疾病或病毒感染。
根据本发明的nk细胞疗法可包括离体刺激nk细胞的步骤。对于该步骤,可以从待治疗的个体收集自体nk细胞,或者可以从供体收集同种异体nk细胞。同种异体nk细胞的来源可以是外周血单核细胞(pbmc)、脐带血、干细胞分化或“现成的”谱系nk细胞系(例如nk-92细胞系及其衍生物)。
纯化后,nk细胞按照所述方案离体扩增,包括饲养细胞刺激,细胞因子组合物(cocktail),例如包括il-2、il-12、il-15、il-18、il-21及其组合。输注后,用支持性低剂量il-2、il-15(koehletal.,2016和romeeetal.,2016)或超激动剂如altor-803处理这些扩增的细胞。类似的过程适用于自体nk细胞。这种nk细胞过继转移的临床输出可能受益于靶特异性nk细胞抗原结合蛋白的肿瘤靶向。预期抗cd16a抗原结合蛋白的效果对于内源的和过继转移的(自体和同种异体)nk细胞是等同的。
通过不同的方式进行给药,例如,通过静脉内、腹膜内、皮下、肌肉内、局部或皮内给药。结合蛋白和白细胞介素的剂量将由主治医师和其他临床因素确定。任何一个个体的剂量取决于许多因素,包括患者的体型、体表面积、年龄、性别、要施用的具体化合物、施用的时间和途径、治疗的种类、一般健康状况以及其他同时施用的药物。“有效剂量”是指足以影响疾病的进程和严重程度,导致这种病理的减轻或缓解的活性成分的量。可使用已知方法确定可用于治疗和/或预防霍奇金淋巴瘤的“有效剂量”。
以下实施例进一步说明了所述实施方案,但不限制本发明的范围:
实施例1
nk细胞分离和培养
通过ficoll-hypaque密度梯度(密度1.077,biochrom,vwr)或淋巴细胞分离液(lymphoprep)密度梯度(干细胞技术(stemcelltechnologies),cat.:07861)离心从健康成人供体(blutbankmannheim,germany)的血沉棕黄层中分离pbmc。使用ls分离柱(miltenyibiotec),使用“人nk细胞分离试剂盒”(miltenyibiotec)或“mojosorttm人nk细胞分离试剂盒”(biolegend)通过阴性选择从pbmc中纯化nk细胞。为了从pbmc(pbmcδnk)中耗尽nk细胞,将cd56microbeads用于阳性选择(miltenyibiotec)。将新鲜分离的nk细胞(下文称为“未致敏的”)保持在含有10%人血清(invitrogen)的scgm培养基(cellgenix)中过夜;pbmc和pbmcδnk在rpmi完全培养基中。
或者,将分离的pbmc在补充有10%fcs(invitrogen,10270-106)的rpmi1640培养基中培养过夜。为了富集nk细胞,从过夜培养物中收获pbmc,并使用easyseptm人nk细胞富集试剂盒(干细胞技术,cat.:17955)进行一轮阴性选择。
通过串联双抗体(cd30/cd16a、egfr/cd16a或bcma/cd16a)活化nk细胞
将nk细胞与cd30+karpas-299或l428细胞以1:1的比例(各1×106个细胞)在24孔板中在0.1-1μg/ml的afm13(cd30/cd16a)或afm12(cd19/cd16a)存在下在rpmi完全培养基中共培养20小时。或者,将nk细胞在包被有0.5μg/孔(10μg/ml,每孔0.5mlpbs,包被过夜)的afm13、利妥昔单抗(mabthera;roche)、egfr/cd16a串联双抗体、bcma/cd16a串联双抗体或鼠igg1(未结合人cd16a;biolegend)的24孔板(未进行组织培养处理)中在pbs中培养20小时。需要时,nk细胞用il-2(12.5-400u/ml,nih或σ)、il-15(0.6-10ng/ml,派普泰克(peprotech))或il-12(10ng/ml,派普泰克)、il-15(20ng/ml)和il-18(100ng/ml,mbl)的组合(下文称为il-12/15/18)进行处理,并在scgm完全培养基中培养2-5天。
nk细胞增殖和数量
向nk细胞培养物加载2μmcfse(σ-aldrich),在室温下在黑暗中孵育15分钟,然后在5ml纯fcs和5mlrpmi培养基中洗涤。然后将nk细胞用单剂量il-2(12.5-400u/ml)或il-15(0.6-10ng/ml)以0.5×106/ml低细胞密度在24孔板中在scgm完全培养基中培养3-7天。通过流式细胞术测量cfse表达。通过计算经历≥4次分裂的nk细胞的百分比(由cfse稀释峰推导而来)来量化cfse稀释度。用以评估nk细胞扩增的绝对nk细胞数分别通过显微镜检查和流式细胞术(相对于计数微珠)对台盼蓝阴性细胞和生命门控(life-gated)细胞进行计数而获得。
51cr释放测定、脱粒和ifn-γ
在51cr释放测定中,在afm13、afm12、egfr/cd16a串联双抗体、bcma/cd16a串联双抗体或嵌合抗cd30igg抗体的存在下,将nk细胞与51cr标记的靶细胞共培养4小时。对于脱粒和细胞内ifn-γ表达,在没有靶细胞或nk细胞与靶细胞的比例为1:1(各为5×104细胞)的情况下,在抗-cd107a-pe(biolegend)和golgiplug(1/100v/v,bdbioscience)存在下,nk细胞在加有抗体、il-12/15/18或pma(50ng/ml)与离子霉素(1mm)的圆底96孔板中共培养4小时。通过流式细胞术测量细胞外cd107a(脱粒标记)和细胞内ifn-γ表达。
在使用人ifn-γ“elisamax”试剂盒(biolegend)将nk细胞与肿瘤细胞(1:1的比例)或il-12/15共培养24小时后,分析ifn-γ向细胞上清液中的分泌。
4小时钙黄绿素释放细胞毒性测定
对于钙黄绿素释放细胞毒性测定,从培养物中收获指定的靶细胞,用不含fcs的rpmi1640培养基洗涤,并用10μm钙黄绿素am(invitrogen/molecularprobes,cat.:c3100mp)在不含fcs的rpmi培养基中在37℃下标记30分钟。洗涤后,将标记的细胞重悬于rpmi完全培养基(补充有10%热灭活的fcs、4mml-谷氨酰胺、100u/ml青霉素g钠、100μg/ml硫酸链霉素的rpmi1640培养基)中。将1×104个靶细胞与原代人nk细胞(e:t比率为2.5:1)以及指定的抗体以总体积200μl/孔在圆底96孔微板的各孔中一起进行接种,一式两份。在每个平板上一式四份测定在不存在抗体的情况下由效应子引起的自发释放、最大释放和杀死靶标。
200g离心2分钟后,将待测物在37℃下在5%co2的潮湿气氛中孵育4小时。在额外离心5分钟后从每个孔收获100μl细胞培养物上清液,并使用荧光板读数器(ensight,perkinelmer)在520nm处测量释放的钙黄绿素的荧光。在测量的计数的基础上,根据下式计算特定细胞细胞溶解:[荧光(样品)-荧光(自发的)]/[荧光(最大)-荧光(自发的)]×100%。荧光(自发的)代表在不存在效应细胞和抗体的情况下来自靶细胞的荧光计数,荧光(最大)代表通过添加tritonx-100诱导的全部细胞溶解。
流式细胞术
使用“foxp3染色缓冲液组”(ebiosciences)和“cytofix/cytoperm”试剂盒(bdbioscience)分别在细胞外染色后进行ifn-γ和穿孔素/颗粒酶b的细胞内染色。在facscalibur或cantoii(bdbioscience)上获得样品,并用flowjo10软件(flowjollc)分析。
使用cd45percpcy5.5(bdbioscience;558411)、cd16bv421(biolegend;302038)、cd56pc7(beckmancoulter;a21692)和可固定的活力染料ef780(ebioscience;65-0864-14)测定nk细胞上的表面标志物表达。在cytoflexs(beckmancoulter)上获得样品,并使用cytexpert2.1软件进行分析。
实施例2
cd30/cd16a串联双抗体(afm13)响应于cd30+淋巴瘤细胞诱导nk细胞的功能和表型活化
在4小时51cr释放试验中,双特异性四价串联双抗体afm13(cd30/cd16a)的存在显著改善了新鲜分离的nk细胞对经典霍奇金淋巴瘤、间变性大细胞淋巴瘤和非霍奇金淋巴瘤的cd30+癌细胞系的细胞毒活性(图1,a)。这对于耐受未致敏nk细胞的cd30+肿瘤细胞尤其明显。相反,afm13串联双抗体对cd30-细胞的细胞溶解保持不变,而afm12串联双抗体(cd19/cd16a)可以增加cd19+cd30-daudi细胞的细胞溶解。afm13串联双抗体在低至10-3μg/ml的浓度下有效,并且比常规抗cd30igg1抗体强几个数量级(图1,c)。总之,在匹配的nk细胞-靶标比率下,afm13介导的细胞溶解的百分比在纯化的nk细胞和pbmc之间是相当的,而nk细胞耗尽的pbmc不能诱导肿瘤细胞细胞溶解(图1,d)。
afm13介导的nk细胞细胞毒性可以通过用il-2、il-15或il12/15/18预活化2天的nk细胞来加强,尤其是对于对细胞因子活化的nk细胞弱敏感的肿瘤细胞。有趣的是,nk细胞与afm13-调理的靶细胞的相互作用诱导了旁邻(bystander)未调理的cd30+(但不是cd30-)肿瘤细胞的细胞溶解,这并没有在与k562或经西妥昔单抗调理的靶细胞(其有效活化nk细胞)的相互作用后观察到。
因此,旁邻cd30+肿瘤细胞的细胞溶解严格需要afm13串联双抗体,并且可能是由于残留的afm13串联双抗体与nk细胞上的cd16a结合。
实施例3
通过cd16a进行的cd30/cd16a串联双抗体(afm13)的预活化增强响应于il-15或低剂量il-2的nk细胞增殖
响应于afm13-调理的肿瘤细胞,nk细胞上cd25(il-2rα)和cd132(il-2rγ)的上调暗示改善的il-2依赖性功能。因此,研究了通过afm13的预活化增强il-2依赖性nk细胞增殖。将cfse标记的nk细胞在无肿瘤系统中在包被的afm13上孵育20小时,重新接种并用il-2孵育3-7天(图2,a)。在应用的低细胞密度下,单独的细胞因子是nk细胞增殖的弱刺激物。类似于对afm13-调理的肿瘤细胞的响应,暴露于包被的afm13导致nk细胞上cd25和cd132的上调,而cd122(il-2rβ)的表达保持不变(图2,b)。值得注意的是,afm13串联双抗体预活化的nk细胞在il-2中培养后显示出明显的cfse稀释,其在第5天变得最明显并且进一步增加至第7天(图2,c)。相反,先前未暴露于afm13串联双抗体或可溶性afm13串联双抗体的nk细胞显示出相当少的cfse稀释(图2,c;数据未显示)。在afm13预活化的nk细胞中经历至少4次分裂的nk细胞的百分比显著更高,表明在暴露于afm13串联双抗体后il-2介导的增殖增强(图2,d)。在暴露于通过其人fc部分结合cd16a的利妥昔单抗后,获得了类似的结果。
接下来,评估afm13介导的预活化是否改变对低剂量il-2的敏感性。事实上,即使在50u/ml的低浓度下,afm13预活化的nk细胞也显示出与400u/ml的较高剂量相当的明显的cfse稀释度,而且增强增殖的最小浓度为25u/ml(图2,e)。此外,在低剂量和高剂量的il-2中培养后,绝对nk细胞数显著增加,导致nk细胞数量增加高达4倍。同样,afm13预活化后il-15介导的nk细胞增殖和绝对nk细胞数量增加;然而,这种效应主要在最高测试剂量的il-15(10ng/ml)下观察到(图2,f)。
因此,通过afm13串联双抗体或利妥昔单抗的对未致敏nk细胞的cd16a结合导致cd25和cd132的上调,导致nk细胞对il-15和低剂量il-2的响应性增强,其增强il-2和il-15介导的nk细胞增殖。
实施例4
在暴露于cd30/cd16a串联双抗体(afm13)之后恢复cd16a介导的nk细胞功能
伴随着活化标记物的诱导,我们观察到用afm13-调理的靶细胞或包被的afm13培养20小时后,nk细胞上的cd16表达几乎完全丧失(图3,a-c)。重要的是,这种效应是短暂的,因为当在暴露于afm13串联双抗体并随后在低剂量或高剂量的il-2或il-15中培养5天后重新接种nk细胞,cd16表达可以恢复(图3,c)。通过流式细胞术观察到的cd16下调不是由于表位包埋(masking),因为在afm13存在下抗-cd163g8对cd16的检测没有改变。相反,cd16下调至少部分依赖于金属蛋白酶介导的切割,如之前报道的由抗cd163g8、利妥昔单抗和bike导致的cd16下调(borregoetal.,1994、motaetal.,2004、romeeetal.,2013、和wierniketal.,2013)。
接下来,在随后的第二次暴露中测试暴露于afm13串联双抗体之后cd16的瞬时减少对nk细胞细胞毒性的影响。实际上,在afm13串联双抗体存在下与l428细胞共培养后,与先前未共培养的nk细胞的细胞溶解相比,新一轮afm13-调理的靶细胞的细胞溶解(第二次暴露)受损。在afm12串联双抗体存在下与l428细胞共培养后,nk细胞的细胞毒性保持不变。为了在无肿瘤细胞系统中剖析这种受损的细胞毒性功能,在暴露于包被的afm13串联双抗体后评估nk细胞的细胞毒性。nk细胞的细胞毒性、脱粒和细胞内ifn-γ表达响应于afm13-调理的karpas-299细胞,并且在包被的afm13串联双抗体上培养20小时后显著降低,然而未损失穿孔素和颗粒酶b水平(图3,de;图4,a)。同样地,afm13-调理的l428和afm12调理的daudi细胞的细胞溶解受损(图s4,c)。此外,残留的afm13介导的细胞溶解高于未调理的肿瘤细胞的细胞溶解。重要的是,随后在il2或il15中培养5天后,暴露于afm13串联双抗体之后的降低的nk细胞的细胞毒性可以完全恢复(图3,d)。类似地,包被的利妥昔单抗损害cd16表达和nk细胞细胞毒性,其可在il-2中培养后恢复。
在暴露于afm13串联双抗体后,pma/离子霉素诱导的最大脱粒水平和细胞内ifn-γ表达也降低,表明更广泛的功能障碍。事实上,在暴露于afm13串联双抗体之后,响应于经典靶细胞系k562以及hut-78细胞的cd16a非依赖性“自然”nk细胞细胞毒性、脱粒和细胞内ifn-γ表达受到干扰。(图3,f;图4,b)。这种抑制作用可以通过在il-2中后续培养5天来恢复。值得注意的是,响应于il-12/15/18的ifnγ表达并未受损,并且在暴露于afm13串联双抗体之后适度增加,这与高亲和力il-12rβ2和il-18rα受体的表达增加相一致(图4,c-d)。因此,afm13预活化的nk细胞似乎不仅对il-2和il-15更敏感,而且对il-12和il-18也更敏感。
因此,虽然nk细胞功能在直接响应afm13-调理的靶细胞的方面得到增强,但afm13串联双抗体暴露随后导致二次暴露于肿瘤细胞时存在选择性瞬时功能障碍,该功能障碍可以通过il-2或il-15刺激来挽救。
图6显示在细胞因子恢复nk细胞的细胞毒性潜力后,第二次暴露于afm13再次损害nk细胞的细胞毒性。第二次暴露于afm13后细胞毒性效力的降低与第一次暴露后的细胞毒性效力降低相当。该数据显示,在afm13存在下,细胞因子刺激不能(至少不完全)恢复由afm13诱导的nk细胞瞬时丧失的效力。因此,为了有效地通过细胞因子刺激恢复nk细胞的细胞毒性,重要的是在恢复期期间抗cd16a抗原结合蛋白的浓度保持较低或不存在抗cd16a抗原结合蛋白。
实施例5
通过cd16a进行的cd30/cd16a串联双抗体(afm13)的预活化引发有效的nk细胞ifn-γ产生
确定通过afm13串联双抗体进行的预活化是否可以在不存在afm13串联双抗体的情况下再次刺激时调节il-2培养的nk细胞的ifn-γ产生。响应于il-12/15,当il-2培养的nk细胞最初暴露于afm13时,显著改善了ifn-γ的产生(图5,a)。更引人注目的是,响应于k562或l428细胞的再刺激,额外增强了ifnγ的产生(图5,a),表明afm13串联双抗体的预活化增强了对细胞因子和淋巴瘤细胞的ifnγ应答。
通过cd16a进行的cd30/cd16a串联双抗体(afm13)的预活化引发cd30+甚至cd30-淋巴瘤细胞的细胞溶解
显示暴露于afm13串联双抗体之后在il-2中的培养物完全恢复了nk细胞的细胞毒性(图3,d+f)。因此,经afm13刺激过的nk细胞响应于第二次暴露于afm13-调理的靶标时,与仅在il-2中培养的nk细胞相比具有相同的效力。在下一步中,探讨了afm13串联双抗体的预活化是否会影响响应于第二次暴露于(非调理的)cd30+或cd30-淋巴瘤细胞的“自然”细胞溶解,该cd30+或cd30-淋巴瘤细胞几乎对5天il-2培养的nk细胞具有耐受性。值得注意的是,当il-2或il-15培养的nk细胞最初暴露于afm13时,cd30+karpas-299和hdlm-2细胞的细胞溶解被增强;这些细胞对未致敏nk细胞具有耐受性,并且几乎对il-2或il-15培养的nk细胞具有耐受性(图5,b)。更重要的是,甚至cd30-淋巴瘤细胞系l1236和daudi的细胞溶解也通过初始暴露于afm13串联双抗体而被增强(图5,b)。这种增强对于对il-2培养的nk细胞弱敏感的靶细胞特别明显,而il-2培养的nk细胞对敏感k562靶细胞的强烈细胞溶解没有进一步改善。对于afm12串联双抗体或利妥昔单抗预活化的nk细胞也观察到增强的细胞毒性功能。
因此,相对于仅由细胞因子活化的nk细胞,经afm13刺激过的细胞因子活化的nk细胞响应于cd30+甚至cd30-淋巴瘤细胞表现出增强的细胞毒性。
实施例6
暴露于egfr/cd16a(afm24)串联双抗体和bcmaxcd16a(afm26)串联双抗体后cd16a介导的nk细胞功能的恢复。
与实施例2中所示的afm13类似,egfr/cd16a串联双抗体和bcma/cd16a串联双抗体分别响应于egfr+和bcma+靶细胞而诱导nk细胞的功能性活化(图7)。
与实施例4中的afm13所示类似,通过在分别暴露于egfr/cd16a串联双抗体和bcma/cd16a串联双抗体后用il-2或il-15处理5天来实现cd16表达和cd16a介导的nk细胞功能的恢复,如实施例4(图8和图9)所述。
因此,本文所述的间歇给药方案可用于独立于靶抗原结构域的多特异性(即双特异性)cd16a抗原结合蛋白。
序列总结
参考文献
bibeau,f.,lopez-crapez,e.,di,f.f.,thezenas,s.,ychou,m.,blanchard,f.,lamy,a.,penault-llorca,f.,frebourg,t.,michel,p.,sabourin,j.c.,boissiere-michot,f.2009.impactoffc{gamma}riia-fc{gamma}riiiapolymorphismsandkrasmutationsontheclinicaloutcomeofpatientswithmetastaticcolorectalcancertreatedwithcetuximabplusirinotecan.j.clin.oncol.27,1122-1129.
bonnema,j.d.,karnitz,l.m.,schoon,r.a.,abraham,r.t.,leibson,p.j.1994.fcreceptorstimulationofphosphatidylinositol3-kinaseinnaturalkillercellsisassociatedwithproteinkinasec-independentgranulereleaseandcell-mediatedcytotoxicity.jexpmed180,1427-1435.
borrego,f.,lopez-beltran,a.,pena,j.,solana,r.1994.downregulationoffcgammareceptoriiiaalpha(cd16-ii)onnaturalkillercellsinducedbyanti-cd16mabisindependentofproteintyrosinekinasesandproteinkinasec.cellimmunol158,208-217.
brandt,c.s.,baratin,m.,yi,e.c.,kennedy,j.,gao,z.,fox,b.,haldeman,b.,ostrander,c.d.,kaifu,t.,chabannon,c.,moretta,a.,west,r.,xu,w.,vivier,e.,levin,s.d.2009.theb7familymemberb7-h6isatumorcellligandfortheactivatingnaturalkillercellreceptornkp30inhumans.jexpmed206,1495-1503.
bryceson,y.t.,ljunggren,h.g.,long,e.o.2009.minimalrequirementforinductionofnaturalcytotoxicityandintersectionofactivationsignalsbyinhibitoryreceptors.blood114,2657-2666.
bryceson,y.t.,march,m.e.,ljunggren,h.g.,long,e.o.2006.synergyamongreceptorsonrestingnkcellsfortheactivationofnaturalcytotoxicityandcytokinesecretion.blood107,159-166.
capuano,c.,romanelli,m.,pighi,c.,cimino,g.,rago,a.,molfetta,r.,paolini,r.,santoni,a.,galandrini,r.2015.anti-cd20therapyactsviafcgammariiiatodiminishresponsivenessofhumannaturalkillercells.cancerres75,4097-4108.
cassatella,m.a.,anegon,i.,cuturi,m.c.,griskey,p.,trinchieri,g.,perussia,b.1989.fcgammar(cd16)interactionwithligandinducesca2+mobilizationandphosphoinositideturnoverinhumannaturalkillercells.roleofca2+infcgammar(cd16)-inducedtranscriptionandexpressionoflymphokinegenes.jexpmed169,549-567.
cerwenka,a.,lanier,l.l.2001.naturalkillercells,virusesandcancer.nat.rev.immunol.1,41-49.
cerwenka,a.,lanier,l.l.2016.naturalkillercellmemoryininfection,inflammationandcancer.natrevimmunol16,112-123.
charychd.,khalili,s.,dixit,v.,kirk,p.,chang,t.,langowski,j.,rubas,w.,doberstein,k.,eldon,m.,hoch,u.,zalevsky,j.,2017,modelingthereceptorpharmacology,pharmacokinetics,andpharmacodynamicsofnktr-214,akinetically-controlledinterleukin-2(il2)receptoragonsitforcancerimmunotherapy,plosone12(7):e0179431.
chatila,t.,silverman,l.,miller,r.,geha,r.1989.mechanismsoftcellactivationbythecalciumionophoreionomycin.jimmunol143,1283-1289.
cooper,m.a.,elliott,j.m.,keyel,p.a.,yang,l.,carrero,j.a.,yokoyama,w.m.2009.cytokine-inducedmemory-likenaturalkillercells.procnatlacadsciusa106,1915-1919.
dall'ozzo,s.,tartas,s.,paintaud,g.,cartron,g.,colombat,p.,bardos,p.,watier,h.,thibault,g.2004.rituximab-dependentcytotoxicitybynaturalkillercells:influenceoffcgr3apolymorphismontheconcentration-effectrelationship.cancerres64,4664-4669.
delandazuri,m.o.,silva,a.,alvarez,j.,herberman,r.b.1979.evidencethatnaturalcytotoxicityandantibody-dependentcellularcytotoxicityaremediatedinhumansbythesameeffectorcellpopulations.jimmunol123,252-258.
galandrini,r.,tassi,i.,mattia,g.,lenti,l.,piccoli,m.,frati,l.,santoni,a.2002.sh2-containinginositolphosphatase(ship-1)transientlytranslocatestoraftdomainsandmodulatescd16-mediatedcytotoxicityinhumannkcells.blood100,4581-4589.
gasser,s.,raulet,d.h.2006.activationandself-toleranceofnaturalkillercells.immunolrev214,130-142.
gasteiger,g.,hemmers,s.,firth,m.a.,lefloc'h,a.,huse,m.,sun,j.c.,rudensky,a.y.2013.il-2-dependenttuningofnkcellsensitivityfortargetcellsiscontrolledbyregulatorytcells.jexpmed210,1167-1178.
hara,h.,ishihara,c.,takeuchi,a.,xue,l.,morris,s.w.,penninger,j.m.,yoshida,h.,saito,t.2008.celltype-specificregulationofitam-mediatednf-kappabactivationbytheadaptors,carma1andcard9.jimmunol181,918-930.
harris,m.2004.monoclonalantibodiesastherapeuticagentsforcancer.lancetoncol5,292-302.
kim,m.,kim,t.j.,kim,h.m.,doh,j.,lee,k.m.2017.multi-cellularnaturalkiller(nk)cellclustersenhancenkcellactivationthroughlocalizingil-2withinthecluster.scirep7,40623.
kipriyanovsm:methodsmolbiol2003;207:323-33
kipriyanovsm:methodsmol.biol.2009;562:177-93
koch,j.,steinle,a.,watzl,c.,mandelboim,o.,2013,activatingnaturalcytotoxicityreceptorsofnaturalkillercellsincancerandinfection,trends.immunol.,34(4):192-91
koehl,u.,kalberer,c.,spanholtz,j.,lee,d.a.,miller,j.s.,cooley,s.,lowdell,m.,uharek,l.,klingemann,h.,curti,a.,leung,w.andalici,e.,2016,advancesinclinicalnkcelstudies:donorselection,manufacturingandqualitycontrol,oncoimmunology,5(4)e1115178.
kondadasula,s.v.,roda,j.m.,parihar,r.,yu,j.,lehman,a.,caligiuri,m.a.,tridandapani,s.,burry,r.w.,carson,w.e.,3rd.2008.colocalizationoftheil-12receptorandfcgammariiiatonaturalkillercelllipidraftsleadstoactivationoferkandenhancedproductionofinterferon-gamma.blood111,4173-4183.
kontermann,r.e.brinkmann,u.themakingofbispecificantibodies,2017mabs,9(2):182-212).
kufedw,pollockre,weichselbaumrr.etal.2003,categoriesoftumorantigens,holland-freicancermedicine.6thedition.,editorshamilton(on):becker
kuylenstierna,c.,bjorkstrom,n.k.,andersson,s.k.,sahlstrom,p.,bosnjak,l.,paquin-proulx,d.,malmberg,k.j.,ljunggren,h.g.,moll,m.,sandberg,j.k.2011.nkg2dperformstwofunctionsininvariantnktcells:directtcr-independentactivationofnk-likecytolysisandco-stimulationofactivationbycd1d.eurjimmunol41,1913-1923.
lajoie,l.,congy-jolivet,n.,bolzec,a.,gouilleux-gruart,v.,sicard,e.,sung,h.c.,peiretti,f.,moreau,t.,vie,h.,clemenceau,b.,thibault,g.2014.adam17-mediatedsheddingoffcgammariiiaonhumannkcells:identificationofthecleavagesiteandrelationshipwithactivation.jimmunol192,741-751.
lanier,l.l.,ruitenberg,j.j.,phillips,j.h.1988.functionalandbiochemicalanalysisofcd16antigenonnaturalkillercellsandgranulocytes.jimmunol141,3478-3485.
li,w.,yang,h.,dimitrov,d.s.2016.identificationofhigh-affinityanti-cd16aallotype-independenthumanantibodydomains.expmolpathol101,281-289.
liu,b.,kong,l.,hank.,hong,h,marcus,w.d.,chen,x.,leng,e.k.,alter,s.,zhu,x.,rubinseinm.r.,shi,s.,rhode,p.r.,cai,w.wong,h.c.;2016anovelfusionofalt-803(interleukin(il)-15superagonist)withanantibodydemonstratesantigen-specificantitumorresponses,journalofbiologicalchemistry,vol.291(46),pp.23869-23881
lissonip,barnis,ardizzoiaa,olivinig,briviof,tisie,tancinig,characiejusd,kotharil.1993cancerimmunotherapywithlow-doseinterleukin-2subcutaneousadministration:potentialefficacyinmostsolidtumorhistotypesbyaconcomitanttreatmentwiththepinealhormonemelatonin.jbiolregulhomeostagents.1993oct-dec;7(4):121-5
luetke-eversloh,m.,hammer,q.,durek,p.,nordstrom,k.,gasparoni,g.,pink,m.,hamann,a.,walter,j.,chang,h.d.,dong,j.,romagnani,c.2014.humancytomegalovirusdrivesepigeneticimprintingoftheifnglocusinnkg2chinaturalkillercells.plospathog10,e1004441.
marquez,m.e.,millet,c.,stekman,h.,conesa,a.,deglesne,p.a.,toro,f.,sanctis,j.d.,blanca,i.2010.cd16cross-linkinginducesincreasedexpressionofcd56andproductionofil-12inperipheralnkcells.cellimmunol264,86-92.
moretta,l.,bottino,c.,pende,d.,castriconi,r.,mingari,m.c.,moretta,a.2006.surfacenkreceptorsandtheirligandsontumorcells.seminimmunol18,151-158.
mota,g.,moldovan,i.,calugaru,a.,hirt,m.,kozma,e.,galatiuc,c.,brasoveanu,l.,boltz-nitulescu,g.2004.interactionofhumanimmunoglobulingwithcd16onnaturalkillercells:ligandclearance,fcgammariiiaturnoverandeffectsofmetalloproteinasesonfcgammariiia-mediatedbinding,signaltransductionandkilling.scandjimmunol59,278-284.
musolino,a.,naldi,n.,bortesi,b.,pezzuolo,d.,capelletti,m.,missale,g.,laccabue,d.,zerbini,a.,camisa,r.,bisagni,g.,neri,t.m.,ardizzoni,a.2008.immunoglobulingfragmentcreceptorpolymorphismsandclinicalefficacyoftrastuzumab-basedtherapyinpatientswithher-2/neu-positivemetastaticbreastcancer.j.clin.oncol.26,1789-1796.
nakahira,m.,ahn,h.j.,park,w.r.,gao,p.,tomura,m.,park,c.s.,hamaoka,t.,ohta,t.,kurimoto,m.,fujiwara,h.2002.synergyofil-12andil-18forifn-gammageneexpression:il-12-inducedstat4contributestoifn-gammapromoteractivationbyup-regulatingthebindingactivityofil-18-inducedactivatorprotein1.jimmunol168,1146-1153.
ni,j.,holsken,o.,miller,m.,hammer,q.,luetke-eversloh,m.,romagnani,c.,cerwenka,a.2016.adoptivelytransferrednaturalkillercellsmaintainlong-termantitumoractivitybyepigeneticimprintingandcd4+tcellhelp.oncoimmunology5,e1219009.
ni,j.,miller,m.,stojanovic,a.,garbi,n.,cerwenka,a.2012.sustainedeffectorfunctionofil-12/15/18-preactivatednkcellsagainstestablishedtumors.j.exp.med.209,2351-2365.
pahl,j.,cerwenka,a.2017.trickingthebalance:nkcellsinanti-cancerimmunity.immunobiology222,11-20.
pahl,j.h.,ruslan,s.e.,buddingh,e.p.,santos,s.j.,szuhai,k.,serra,m.,gelderblom,h.,hogendoorn,p.c.,egeler,r.m.,schilham,m.w.,lankester,a.c.2012.anti-egfrantibodycetuximabenhancesthecytolyticactivityofnaturalkillercellstowardosteosarcoma.clincancerres18,432-441.
parkhurst,m.r.,riley,j.p.,dudley,m.e.,rosenberg,s.a.2011.adoptivetransferofautologousnaturalkillercellsleadstohighlevelsofcirculatingnaturalkillercellsbutdoesnotmediatetumorregression.clincancerres17,6287-6297.
reiners,k.s.,kessler,j.,sauer,m.,rothe,a.,hansen,h.p.,reusch,u.,hucke,c.,kohl,u.,durkop,h.,engert,a.,vonstrandmann,e.p.2013.rescueofimpairednkcellactivityinhodgkinlymphomawithbispecificantibodiesinvitroandinpatients.molther21,895-903.
reusch,u.,burkhardt,c.,fucek,i.,legall,f.,legall,m.,hoffmann,k.,knackmuss,s.h.,kiprijanov,s.,little,m.,zhukovsky,e.a.2014.anoveltetravalentbispecifictandab(cd30/cd16a)efficientlyrecruitsnkcellsforthelysisofcd30+tumorcells.mabs6,728-739.
rolle,a.,brodin,p.2016.immuneadaptationtoenvironmentalinfluence:thecaseofnkcellsandhcmv.trendsimmunol37,233-243.
romee,r.,foley,b.,lenvik,t.,wang,y.,zhang,b.,ankarlo,d.,luo,x.,cooley,s.,verneris,m.,walcheck,b.,miller,j.2013.nkcellcd16surfaceexpressionandfunctionisregulatedbyadisintegrinandmetalloprotease-17(adam17).blood121,3599-3608.
romee,r.,schneider,s.e.,leong,j.w.,chase,j.m.,keppel,c.r.,sullivan,r.p.,cooper,m.a.,fehniger,t.a.2012.cytokineactivationinduceshumanmemory-likenkcells.blood120,4751-4760.
romee,r,leongj.w,fehninger,t.a,2014,utilizingcytokinestofunction-enablehumannkcellsfortheimmunotherapyofcancer,hindawipublishingcorporationscientificyarticleid205796
romeer,rosariom,berrien-elliottmm,wagnerja,jewellba1,schappet,leongjw,abdel-latifs,schneiderse,willeys,nealcc,yul,ohst,leeys,muldera,claasf,cooperma,fehnigerta,2016,cytokine-inducedmemory-likenaturalkillercellsexhibitenhancedresponsesagainstmyeloidleukemiascitranslmed.sep21;8(357):357ra123.
rothe,a.,sasse,s.,topp,m.s.,eichenauer,d.a.,hummel,h.,reiners,k.s.,dietlein,m.,kuhnert,g.,kessler,j.,buerkle,c.,ravic,m.,knackmuss,s.,marschner,j.p.,poggevonstrandmann,e.,borchmann,p.,engert,a.2015.aphase1studyofthebispecificanti-cd30/cd16aantibodyconstructafm13inpatientswithrelapsedorrefractoryhodgkinlymphoma.blood125,4024-4031.
schlums,h.,cichocki,f.,tesi,b.,theorell,j.,beziat,v.,holmes,t.d.,han,h.,chiang,s.c.,foley,b.,mattsson,k.,larsson,s.,schaffer,m.,malmberg,k.j.,ljunggren,h.g.,miller,j.s.,bryceson,y.t.2015.cytomegalovirusinfectiondrivesadaptiveepigeneticdiversificationofnkcellswithalteredsignalingandeffectorfunction.immunity42,443-456.
schuster,i.s.,coudert,j.d.,andoniou,c.e.,degli-esposti,m.a.2016."naturalregulators":nkcellsasmodulatorsoftcellimmunity.frontimmunol7,235.
spiess,c.etal.,alternativemolecularformatsforbispecificantibodies.2015,mol.immunol.,67(2):95-106
spits,h.,artis,d.,colonna,m.,diefenbach,a.,disanto,j.p.,eberl,g.,koyasu,s.,locksley,r.m.,mckenzie,a.n.,mebius,r.e.,powrie,f.,vivier,e.2013.innatelymphoidcells--aproposalforuniformnomenclature.natrevimmunol13,145-149.
sun,j.c.,ugolini,s.,vivier,e.2014.immunologicalmemorywithintheinnateimmunesystem.emboj33,1295-1303.
tesi,b.,schlums,h.,cichocki,f.,bryceson,y.t.2016.epigeneticregulationofadaptivenkcelldiversification.trendsimmunol37,451-461.
vivier,e.,morin,p.,o'brien,c.,druker,b.,schlossman,s.f.,anderson,p.1991.tyrosinephosphorylationofthefcgammariii(cd16):zetacomplexinhumannaturalkillercells.inductionbyantibody-dependentcytotoxicitybutnotbynaturalkilling.jimmunol146,206-210.
weicheletal.,tandabs:potentandwell-manufacturablebi-specificantibodies;europeanpharmaceuticalreview2015,vol.20:27-32
weinberg,r.a.,thebiologyofcancer,2013,2ndedition,taylorandfrancis
wherry,e.j.,kurachi,m.2015.molecularandcellularinsightsintotcellexhaustion.natrevimmunol15,486-499.
wiernik,a.,foley,b.,zhang,b.,verneris,m.r.,warlick,e.,gleason,m.k.,ross,j.a.,luo,x.,weisdorf,d.j.,walcheck,b.,vallera,d.a.,miller,j.s.2013.targetingnaturalkillercellstoacutemyeloidleukemiainvitrowithacd16x33bispecifickillercellengagerandadam17inhibition.clincancerres19,3844-3855.
zompi,s.,colucci,f.2005.anatomyofamurder--signaltransductionpathwaysleadingtoactivationofnaturalkillercells.immunollett97,31-39.
序列表
<110>阿菲姆德股份有限公司
<120>抗cd16a抗体与细胞因子的组合
<130>a3316pct
<160>10
<170>patentinversion3.5
<210>1
<211>8
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>cdr
<400>1
glytyrthrphethrsertyrtyr
15
<210>2
<211>8
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>cdr
<400>2
ileglupromettyrglyserthr
15
<210>3
<211>13
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>cdr
<400>3
alaargglyseralatyrtyrtyraspphealaasptyr
1510
<210>4
<211>6
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>cdr
<400>4
asnileglyserlysasn
15
<210>5
<211>4
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>cdr
<400>5
glnaspasnlys
1
<210>6
<211>9
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>cdr
<400>6
glnvaltrpaspasntyrservalleu
15
<210>7
<211>8
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>cdr
<400>7
ileasnproserglyglyserthr
15
<210>8
<211>120
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>vh
<400>8
glnvalglnleuvalglnserglyalagluvallyslysproglyglu
151015
serleulysvalsercyslysalaserglytyrthrphethrsertyr
202530
tyrmethistrpvalargglnalaproglyglnglyleuglutrpmet
354045
glyalaileglupromettyrglyserthrsertyralaglnlysphe
505560
glnglyargvalthrmetthrargaspthrserthrserthrvaltyr
65707580
metgluleuserserleuargsergluaspthralavaltyrtyrcys
859095
alaargglyseralatyrtyrtyraspphealaasptyrtrpglygln
100105110
glythrleuvalthrvalserser
115120
<210>9
<211>106
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>vl
<400>9
sertyrvalleuthrglnproserservalservalalaproglygln
151015
thralathrilesercysglyglyhisasnileglyserlysasnval
202530
histrptyrglnglnargproglyglnserprovalleuvaliletyr
354045
glnaspasnlysargproserglyileprogluargpheserglyser
505560
asnserglyasnthralathrleuthrileserglythrglnalamet
65707580
aspglualaasptyrtyrcysglnvaltrpaspasntyrservalleu
859095
pheglyglyglythrlysleuthrvalleu
100105
<210>10
<211>120
<212>prt
<213>人工序列(artificalsequence)
<220>
<223>vh
<400>10
glnvalglnleuvalglnserglyalagluvallyslysproglyglu
151015
serleulysvalsercyslysalaserglytyrthrphethrsertyr
202530
tyrmethistrpvalargglnalaproglyglnglyleuglutrpmet
354045
glyileileasnproserglyglyserthrsertyralaglnlysphe
505560
glnglyargvalthrmetthrargaspthrserthrserthrvaltyr
65707580
metgluleuserserleuargsergluaspthralavaltyrtyrcys
859095
alaargglyseralatyrtyrtyraspphealaasptyrtrpglygln
100105110
glythrleuvalthrvalserser
115120
- 还没有人留言评论。精彩留言会获得点赞!