CD47阻断疗法的制作方法
本发明涉及使用阻断cd47/sirpα相互作用的药物的方法。更具体地,本发明涉及组合地可用于改善癌症治疗的方法和手段。
背景技术:
通过与癌细胞抗原结合的抗体,以及通过fc受体与该抗体的fc部分结合的巨噬细胞的募集和活化,靶向癌细胞。癌细胞上的cd47与巨噬细胞上的sirpα之间的结合传递了“不要吃我”的信号,该信号使许多肿瘤细胞逃脱巨噬细胞的破坏。已经表明抑制cd47/sirpα相互作用(cd47阻断)将允许巨噬细胞“看到”并破坏靶cd47+癌细胞。wo2010/130053中描述了sirpα通过cd47阻断治疗癌症的用途。
在wo2014/094122中,我们描述了抑制cd47和sirpα之间相互作用的药物。该cd47阻断药物是人sirpα的一种形式,其结合了与特别有用形式的基于igg1的fc区连接的细胞外结构域的特定区域。在这种形式中,sirpαfc药物对存在cd47+表型的癌细胞的存活率显示出显著影响。特别是在急性髓性白血病(aml)细胞和许多其他类型的癌症中可以看到这种效果。在stanford的wo2013/109752中描述了具有显著改变的一级结构和增强的cd47结合亲和力的可溶形式的sirp。
其他cd47阻断药物已在文献中描述,这些包括各种cd47抗体(参见例如stanford的us8562997和inhibrx的wo2014/123580),每种抗体包含不同的抗原结合位点,但都具有与内源性sirpα竞争结合cd47的能力,从而允许与巨噬细胞相互作用,并最终增加cd47+癌细胞耗竭的速率。这些药物虽然具有cd47阻断效应,但在体内显示出与基于sirpαfc的药物所显示的活性完全不同的活性。例如,后者显示出对红细胞的可忽略的结合,而cd47抗体中的相反特性产生了需要适应施用后药物“下沉”的给药策略。
还提出了其他试剂用于阻断cd47/sirpα轴。这些包括cd47fc蛋白(参见virallogic的wo2010/083253)和sirpα抗体,如uhn的wo2013/056352、stanford的wo2016/022971、eberhard的us6913894和其他地方所述。
cd47阻断方法在抗癌药物开发中显示出巨大的希望。提供改善这些药物作用的方法和手段,特别是改善cd47阻断药物形式(尤其是并入了sirpα的那些)的作用将是有用的。
技术实现要素:
现在发现,与抑制t细胞检查点的药剂(例如抑制程序性死亡-1(pd-1)和ctla4途径的药剂)组合使用时,基于sirpα的cd47阻断药物的抗癌作用得到改善。本发明包括与该发现有关的多种方法/用途、材料、组合物、组合、试剂盒和其他制品。在实施方案中,t细胞检查点抑制剂是ctla-4抑制剂或与pd-1结合或与pd-1的结合伴侣例如pd-l1或pd-l2结合的拮抗剂。在一些实施方案中,cd47阻断药物是sirpα-fc。两种药物共同作用于癌细胞,导致癌细胞的耗竭超过仅sirpαfc所能解释的。
在一个方面,提供了一种用于治疗患有cd47+癌细胞的受试者的方法,该方法包括向该受试者施用sirpα-fc药物和t细胞检查点抑制剂,例如pd-1阻断药物和/或ctla-4抑制剂。术语sirpα-fc(或sirpαfc)是指由直接或间接连接至fc结构域的sirpα结构域组成的药物类。sirpα结构域衍生自人sirpα,并且包括足够的sirpα结构以保留sirpα的cd47结合活性特征,但是可溶并且至少缺少基因组编码的sirpα的跨膜结构域。示例性sirpα结构域包括如下所述的igv结构域。fc结构域具有抗体恒定区的特征,如下文更详细地描述。
在相关方面,本发明提供了sirpα-fc药物与t细胞检查点抑制剂例如pd-1途径抑制剂和/或ctla-4抑制剂的组合在治疗患有cd47+癌症的受试者中的用途。
在另一方面,还提供了包含sirpαfc和t细胞检查点抑制剂(例如pd-1阻断药和/或ctla-4抑制剂)的抗癌药物的药物组合,以及教导了它们在本文所述的治疗方法中的用途的说明。因此,提供了一种药物组合,其包含sirpαfc和至少一种t细胞检查点抑制剂,其中所述t细胞检查点抑制剂可以是纳武单抗(nivolumab)或伊匹单抗(ipilimumab)。在另一个相关的方面,药物组合包含sirpαfc和至少两种t细胞检查点抑制剂,它们是纳武单抗和伊匹单抗。这三种药物在增强抗肿瘤免疫反应方面起着协同作用,导致癌细胞的耗竭比仅sirpαfc所能解释的要多。
本发明进一步包括在以下编号的段落中概述的方法、用途、产品和组合物:
1.一种用于治疗具有cd47+疾病细胞的受试者的方法,包括向所述受试者施用t细胞检查点抑制剂和cd47阻断药物。
2.t细胞检查点抑制剂和cd47阻断药物治疗具有cd47+疾病细胞的受试者的用途。
3.一种t细胞检查点抑制剂,用于通过与cd47阻断药物共同施用治疗cd47+疾病细胞。
4.一种cd47阻断药物,用于通过与t细胞检查点抑制剂共同施用治疗cd47+疾病细胞。
5.t细胞检查点抑制剂和cd47阻断药物在制备用于治疗cd47+疾病细胞的药物中的用途。
6.t细胞检查点抑制剂在制备用于通过与cd47阻断药物共同施用治疗cd47+疾病细胞的药物中的用途。
7.cd47阻断药物在制备用于通过与t细胞检查点抑制剂共同施用治疗cd47+疾病细胞的药物中的用途。
8.一种产品,其包含t细胞检查点抑制剂和cd47阻断药物,作为组合制剂,用于同时、分别或依次用于治疗cd47+疾病细胞。
9.一种组合物,其包含t细胞检查点抑制剂、cd47阻断药物和药学上可接受的载体。
10.根据段落1-9中任一项所述的方法、用途、产品或组合物,其中所述cd47+疾病细胞包括cd47+癌细胞。
11.根据段落1-9中任一项所述的方法、用途、产品或组合物,其中所述t细胞检查点抑制剂包括pd-1阻断药物。
12.根据段落11所述的方法、用途、产品或组合物,其中所述pd-1阻断药物包括结合pd-1的药剂。
13.根据段落12所述的方法、用途、产品或组合物,其中所述pd-1阻断药物包括纳武单抗。
14.根据段落1-13中任一项所述的方法、用途、产品或组合物,其中所述pd-1阻断药物包括结合pd-l1或pd-l2的药剂。
15.根据段落14所述的方法、用途、产品或组合物,其中所述pd-1阻断药物包括pd-l1结合剂。
16.根据段落15所述的方法、用途、产品或组合物,其中所述pd-l1结合剂包括选自德瓦鲁单抗(durvalumab)、atezolizumab、avelumab和命名为bms-936559/mdx1105的igg4抗体的成员。
17.根据段落1-16中任一项所述的方法、用途、产品或组合物,其中所述t细胞检查点抑制剂包括ctla4抑制剂。
18.根据段落17所述的方法、用途、产品或组合物,其中所述ctla4抑制剂包括ctla4抗体。
19.根据段落18所述的方法、用途、产品或组合物,其中所述ctla4抗体包括伊匹单抗或曲美母单抗(tremelimumab)。
20.根据段落1-19中任一项所述的方法、用途、产品或组合物,其中所述cd47阻断药物包括fc融合蛋白,其包含融合到抗体的fc区的人sirpα的可溶性cd47结合区。
21.根据段落20所述的方法、用途、产品或组合物,其中包含可溶性sirpα的所述fc融合蛋白包含seqidno:8的氨基酸序列。
22.根据段落20所述的方法、用途、产品或组合物,其中包含可溶性sirpα的所述fc融合蛋白包含seqidno:9的氨基酸序列。
23.根据段落1-19中任一项所述的方法、用途、产品或组合物,其中所述cd47阻断药物包括具有选自l4v/i、v6i/l、a21v、v27i/l、i31t/s/f、e47v/l、k53r、e54q、h56p/r、s66t/g、k68r、v92i、f94v/l、v63i和f103v的一个或多个氨基酸取代的可溶性sirpα。
24.根据段落1-23中任一项所述的方法、用途、产品或组合物,其中所述t细胞检查点抑制剂包含纳武单抗和依匹单抗的组合。
25.根据段落1-24中任一项所述的方法、用途、产品或组合物,其中所述cd47+疾病细胞包括血液癌细胞或实体瘤癌细胞。
26.根据段落25所述的方法、用途、产品或组合物,其中所述cd47+疾病细胞是选自急性淋巴细胞性白血病(all);急性髓性白血病(aml);慢性淋巴细胞白血病(cll);慢性粒细胞白血病(cml);骨髓增生性疾病/肿瘤(mpds);和骨髓增生异常综合征的癌症细胞的癌症细胞类型。
27.根据段落25所述的方法、用途、产品或组合物,其中所述癌症是选自霍奇金氏淋巴瘤、惰性和侵袭性两种非霍奇金氏淋巴瘤、伯基特氏淋巴瘤、和滤泡性淋巴瘤(小细胞和大细胞滤泡性淋巴瘤)的淋巴瘤。28.根据段落25所述的方法、用途、产品或组合物,其中所述癌症是选自多发性骨髓瘤(mm)、巨细胞性骨髓瘤、重链骨髓瘤以及轻链或bence-jones骨髓瘤的骨髓瘤。
29.根据段落25所述的方法、用途、产品或组合物,其中所述癌症是黑素瘤。
30.根据段落25所述的方法、用途、产品或组合物,其中所述癌症是aml、骨髓增生异常综合征、cll、霍奇金淋巴瘤、无痛b细胞淋巴瘤、攻击性b细胞淋巴瘤、t细胞淋巴瘤、多发性骨髓瘤、骨髓增生性肿瘤或cd20+淋巴瘤。
31.根据段落25所述的方法、用途、产品或组合物,其中所述癌症选自非小细胞肺癌、肾癌、膀胱癌、头和颈鳞状细胞癌、merkel细胞皮肤癌、食道癌、胰腺癌、肝细胞癌、胶质母细胞瘤、胃癌、乳腺癌和卵巢癌。
32.根据段落25所述的方法、用途、产品或组合物,其中所述癌症选自黑色素瘤、转移性非小细胞肺癌、头颈癌、霍奇金淋巴瘤、泌尿道上皮癌和胃癌。
33.根据段落1-32中任一项所述的方法、用途、产品或组合物,其中所述t细胞检查点抑制剂和cd47阻断药物以协同有效量存在或使用。
34.抗癌剂的药物组合,其包含sirpαfc和t细胞检查点抑制剂,其有效增强cd47+疾病细胞的sirpαfc介导的耗竭。
35.根据段落34所述的组合用于治疗具有cd47+疾病细胞的受试者的用途。
36.根据段落35所述的用途,其中所述cd47+疾病细胞是cd47+癌细胞。
37.一种试剂盒,其包括sirpαfc和t细胞检查点抑制剂的至少一种,以及根据段落1-33中任一项所述的方法、用途、产品或组合物教导其使用的说明。
在本文中已被描述为方法的本发明的方面也可以被描述为“用途”,并且所有这些用途均被视为本发明的方面。同样,本文所述具有“用途”的组合物可以可替代地被描述为使用的过程或方法,其被视为本发明的方面。
同样地,本文中关于特定方法、用途、组合物或其他产品描述的本发明的细节应被理解为适用于本发明的其他方面或实施方式,包括被认为是本发明的用于检查或其他目的的不同类别的方面或实施方式。
作为附加方面,本发明包括本发明的范围在任何方面都比上述特定段落所定义的变型更窄的所有实施方式。例如,在本发明的被描述为类别或集合的某些方面的情况下,应当理解,类别或集合的每个成员分别是本发明的方面。同样,每个单独的子集都旨在作为本发明的方面。举例来说,如果将本发明的方面描述为选自1、2、3和4的成员,则每个单独的亚组(例如,选自{1,2,3}或{1,2,4}或{2,3,4}或{1,2}或{1,3}或{1,4}或{2,3}或{2,4}或{3,4}的成员)和每个单独的种类{1}或{2}或{3}或{4}被视为本发明的方面或变型。同样,如果本发明的方面被表征为范围,例如温度范围,则整数子范围被认为是本发明的方面或变型。
本文的标题是出于方便读者而并非意图为限制性的。根据具体实施方式和/或附图和/或权利要求书,本发明的其它方面、实施例和变型将显而易知。
尽管申请人发明了本文所述的本发明的全部范围,但申请人无意要求保护他人的现有技术中所述的主题。因此,如果专利局或其他实体或个人使得权利要求范围中的法定现有技术引起申请人注意,则申请人保留根据适用专利法行使修改权以重新限定该权利要求的主题,以将这种法定现有技术或法定现有技术的明显变型排除在该权利要求的范围之外。由这些修改的权利要求所限定的本发明的变型也旨在作为本发明的方面。
现在参考附图更详细地描述本发明的这些和其他方面,其中:
附图说明
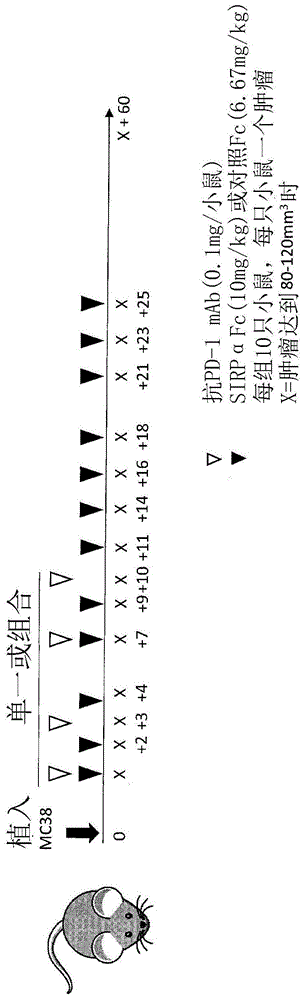
图1显示了药物组合研究设计和给药方案。
图2显示了单一疗法、组合疗法和对照给药后各个时间点(以天为单位)的肿瘤体积。
图3提供了初始治疗后60天后的生存曲线(kaplan-meier图)。抗pd-1单一疗法组和抗pd-1加sirpαfc组合疗法组中的一只动物在达到肿瘤终点之前死亡。
图4显示了纳武单抗和/或依匹单抗与sirpαfc的组合在体外调节肿瘤特异性cd8+t细胞活化和效应子功能的作用,如通过cd107a/b+和tnfα+ifnγ+的百分比所测量的。
具体实施方式
本发明提供了一种用于治疗具有癌细胞和具有cd47+表型的肿瘤的受试者的改进方法。在这种方法中,受试者接受sirpαfc(作为cd47阻断药物)和t细胞检查点抑制剂(例如pd-1阻断药物)的组合。
在一些变型中,受试者接受两种(或更多种)药剂以产生协同作用。在施用两种或多种药剂的情况下,“协同有效量”是指这样的药剂量:(i)与使用药剂的单一疗法相比产生更大的累加治疗作用;或(ii)与使用其中一种药剂的单一疗法相比,由于有效剂量较低或给药频率较低,因此至少产生了可比的治疗效果并降低了毒副作用。可以在体外研究中,例如用细胞系,在评估肿瘤细胞系杀伤的研究中,提供这种协同作用的指示。协同作用可以在临床试验中得到证实,在该试验中,对单一疗法和组合疗法的效果进行了比较和统计分析。
“阻断药物”在本文中也称为“阻断剂”。
本发明方法中使用的sirpαfc是包含抗体的fc区(或片段)和人sirpα的cd47结合区(或片段)的单链多肽的单体或同二聚体或异二聚体形式。这种一般类型的基于可溶性sirpα的药物在文献中有描述,并且包括在大学健康网的下列国际专利申请号中公开的那些:pct/ca2008/001814,公开为wo2009/046541;novartis的专利申请号pct/ep2009/067411,公开为wo2010/070047;斯坦福的专利申请第pct/us2013/021937号,公开为wo2013/109752;以及trilliumtherapeutic的专利申请号pct/ca2013/001046,公开为wo2014/094122,所有都通过引用其整体并入本文,特别是它们针对基于sirpα的构建体的描述。
在优选的实施方案中,sirpαfc具有以下讨论的性质。更具体地,药物合适地包含人sirpα蛋白,其形式为直接或间接与抗体或fc(可结晶的片段)恒定区融合。除非另有说明,否则如本文所用的术语“人sirpα”是指野生型、内源性、成熟形式的人sirpα。在人类中,sirpα蛋白以两种主要形式存在。一种形式,变体1或v1形式,具有ncbirefseqnp_542970.1所示的氨基酸序列(残基27-504构成成熟形式)。另一种形式,变体2或v2形式,相差13个氨基酸,并且具有在genbank中列出的氨基酸序列,如caa71403.1(残基30-504构成成熟形式)。这两种形式的sirpα构成人类中存在的sirpα形式的约80%,并且在本文中术语“人sirpα”包括这两种形式。术语“人sirpα”也包括其对人类是内源的次要形式,并且具有与cd47结合后触发信号转导的相同特性。本发明最特别涉及包含变体2形式或v2的药物组合。
在本发明的药物组合中,有用的sirpαfc融合蛋白包含位于人sirpα细胞外区域内的三个所谓的免疫球蛋白(ig)结构域之一。更具体地,本发明的sirpαfc蛋白掺入人sirpα(106聚体)的残基32-137,其根据目前的命名法构成并限定v2形式的igv结构域。如下所示,该sirpα序列在本文中称为seqidno:1。
eelqviqpdksvsvaagesailhctvtslipvgpiqwfrgagpareliynqkeghfprvttvsestkrenmdfsisisnitpadagtyycvkfrkgspdtefksga(seqidno:1)
在优选的实施方案中,sirpαfc融合蛋白掺入如seqidno:1所定义的igv结构域,以及在野生型人sirpα序列内邻接的另外的侧翼残基。这种优选形式的igv结构域,由人sirpα的v2形式的残基31-148表示,是具有seqidno:5的118聚体,如下所示:
eeelqviqpdksvsvaagesailhctvtslipvgpiqwfrgagpareliynqkeghfprvttvsestkrenmdfsisisnitpadagtyycvkfrkgspdtefksgagtelsvrakps
(seqidno:5)
sirpαfc融合体的fc区优选确实具有效应子功能。fc是指“可结晶的片段”并且代表主要包含重链恒定区和铰链区内组分的抗体的恒定区。因此合适的fc组分是具有效应子功能的fc组分。“具有效应子功能”的fc组分是具有至少一些效应子功能的fc组分,例如至少一些对抗体依赖性细胞毒性的贡献或某些固定补体的能力。而且,fc将至少结合一种或多种类型的fc受体。可以使用为此目的建立的测定,揭示这些性质。功能测定包括检测靶细胞裂解的标准铬释放测定。通过该定义,野生型igg1或igg4的fc区具有效应子功能,而人igg4的fc区突变以消除效应子功能,例如通过掺入包括pro233、val234、ala235的改变系列和删除gly236(eu),被认为不具有效应子功能。在优选的实施方案中,fc基于igg1同种型的人抗体。这些抗体的fc区对于本领域技术人员来说是容易识别的。在实施方案中,fc区包括下部铰链-ch2-ch3结构域。
在一个具体实施方案中,fc区基于人igg1的氨基酸序列,其在uniprotkb/swiss-prot中列为p01857,残基104-330,并且具有如下所示的并在本文中称为seqidno:2的氨基酸序列:
dkthtcppcpapellggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpapiektiskakgqprepqvytlppsrdeltknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspgk*
(seqidno:2)
因此,在实施方案中,fc区具有igg1恒定区的野生型或共有序列。在可选的实施方案中,掺入融合蛋白中的fc区衍生自具有典型效应子-活性恒定区的任何igg1抗体。此类fc区的序列可以对应于例如以下任何igg1序列的fc区(均来自genbank),例如:bag65283(残基242-473),bac04226.1(残基247-478),bac05014.1(残基240-471),cac20454.1(残基99-320),bac05016.1(残基238-469),bac85350.1(残基243-474),bac85529.1(残基244-475),和bac85429.1(残基(238-469)。
在其他实施方案中,fc区具有野生型人igg4恒定区的序列。在替代实施方案中,掺入融合蛋白中的fc区衍生自具有恒定区的任何igg4抗体,所述恒定区具有效应子活性,但是天然地,其效力明显低于igg1fc区。此类fc区的序列可以对应于例如以下任何igg4序列的fc区:来自uniprotkb/swiss-prot的p01861(残基99-327)和来自genbank的cac20457.1(残留99-327)。
在一个具体实施方案中,fc区基于人igg4的氨基酸序列,其在uniprotkb/swiss-prot中列为p01861,残基99-327,并且具有如下所示的并在本文中称为seqidno:6的氨基酸序列:
eskygppcpscpapeflggpsvflfppkpkdtlmisrtpevtcvvvdvsqedpevqfnwyvdgvevhnaktkpreeqfnstyrvvsvltvlhqdwlngkeykckvsnkglpssiektiskakgqprepqvytlppsqeemtknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflysrltvdksrwqegnvfscsvmhealhnhytqkslslslgk
(seqidno:6)
在实施方案中,fc区掺入一个或多个改变,通常不超过约5个这样的改变,包括影响某些fc特性的氨基酸取代。在一个具体和优选的实施方案中,fc区在位置228(eu编号)掺入改变,其中该位置的丝氨酸被脯氨酸(s228p)取代,从而稳定fc二聚体内的二硫键。fc区内的其他改变可包括改变糖基化的取代,例如甘氨酸或丙氨酸取代asn297;半衰期增强改变,例如t252l、t253s和t256f,如us62777375中教导,以及许多其他改变。特别有用的是增强fc特性同时在构象方面保持沉默的那些改变,例如保留fc受体结合。
在一个具体实施方案中,并且在fc组分是igg4fc的情况下,fc至少掺入s228p突变,并且具有下文列出的并在本文中称为seqidno:7的氨基酸序列:
eskygppcppcpapeflggpsvflfppkpkdtlmisrtpevtcvvvdvsqedpevqfnwyvdgvevhnaktkpreeqfnstyrvvsvltvlhqdwlngkeykckvsnkglpssiektiskakgqprepqvytlppsqeemtknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflysrltvdksrwqegnvfscsvmhealhnhytqkslslslgk
(seqidno:7)
在组合中使用的cd47阻断药物因此是可用于抑制人sirpα和人cd47之间结合,从而抑制或减少通过sirpα结合的cd47介导的信号传递的sirpαfc融合蛋白,该融合蛋白包含人sirpα组分并与之融合,fc组分,其中sirpα组分包含人sirpαv2的单个igv结构域或由其组成,并且fc组分是具有人igg的恒定区,其中所述恒定区优选地具有效应子功能。
在一个实施方案中,融合蛋白包含sirpα组分,其包含至少v2形式的野生型人sirpα的残基32-137,即seqidno:1。在优选的实施方案中,sirpα组分由人sirpα的v2形式的残基31-148组成,即seqidno:5。在另一个实施方案中,fc组分是人igg1的fc组分,命名为p01857,并且在一个具体实施方案中具有包含其下铰链-ch2-ch3区的氨基酸序列,即seqidno:2。
因此,在一个优选的实施方案中,本发明提供sirpαfc融合蛋白,作为表达的单链多肽和作为其分泌的二聚体融合体二者,其中融合蛋白掺入具有seqidno:1且优选seqidno:5的sirpα组分,以及与其融合的具有效应子功能且具有seqidno:2的fc区。当sirpα组分是seqidno:1时,该融合蛋白包含seqidno:3,如下所示:
eeelqviqpdksvsvaagesailhctvtslipvgpiqwfrgagpareliynqkeghfprvttvsestkrenmdfsisisnitpadagtyycvkfrkgspdtefksgagtelsvrakpsdkthtcppcpapellggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpapiektiskakgqprepqvytlppsrdeltknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspgk*(seqidno:3)
当sirpα组分是seqidno:5时,该融合蛋白包含seqidno:8,如下所示:
eeelqviqpdksvsvaagesailhctvtslipvgpiqwfrgagpareliynqkeghfprvttvsestkrenmdfsisisnitpadagtyycvkfrkgspdtefksgagtelsvrakpsdkthtcppcpapellggpsvflfppkpkdtlmisrtpevtcvvvdvshedpevkfnwyvdgvevhnaktkpreeqynstyrvvsvltvlhqdwlngkeykckvsnkalpapiektiskakgqprepqvytlppsrdeltknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflyskltvdksrwqqgnvfscsvmhealhnhytqkslslspgk(seqidno:8)
在替代实施方案中,融合蛋白的fc组分基于igg4,优选结合s228p突变的igg4。在融合蛋白掺入seqidno:5的优选sirpαigv结构域的情况下,所得的基于igg4的sirpα-fc蛋白具有seqidno:9,如下所示:
eeelqviqpdksvsvaagesailhctvtslipvgpiqwfrgagpareliynqkeghfprvttvsestkrenmdfsisisnitpadagtyycvkfrkgspdtefksgagtelsvrakpseskygppcppcpapeflggpsvflfppkpkdtlmisrtpevtcvvvdvsqedpevqfnwyvdgvevhnaktkpreeqfnstyrvvsvltvlhqdwlngkeykckvsnkglpssiektiskakgqprepqvytlppsqeemtknqvsltclvkgfypsdiavewesngqpennykttppvldsdgsfflysrltvdksrwqegnvfscsvmhealhnhytqkslslslgk(seqidno:9)
在优选的实施方案中,融合蛋白包含作为融合蛋白的sirpαigv结构域的序列,其为seqidno:5。优选的sirpαfc是seqidno:8。
如文献中所述,可以改变cd47阻断药物中掺入的sirpα序列。也就是说,sirpα内有用的取代包括以下的一种或多种(例如相对于seqidno:5):l4v/i、v6i/l、a21v、v27i/l、i31t/s/f、e47v/l、k53r、e54q、h56p/r、s66t/g、k68r、v92i、f94v/l、v63i和/或f103v。更其他的取代包括保守氨基酸取代,其中氨基酸被来自相同组的氨基酸取代。
在sirpαfc融合蛋白中,sirpα组分和fc组分直接或间接融合,以提供单链多肽,其最终产生为二聚体,其中单链多肽通过各个单链sirpαfc多肽的fc之间形成的二硫键偶联。连接sirpα区和fc的融合区域的性质不是关键的。融合可以在两种组分之间直接进行,其中sirp组分构成融合的n末端,fc组分构成c末端。或者,融合可以是间接的,通过由一个或多个氨基酸组成的接头,理想地是遗传编码的氨基酸,例如两个、三个、四个、五个、六个、七个、八个、九个或十个氨基酸,或5至100个氨基酸之间的任何数目氨基酸,例如5至50、5至30或5至20个氨基酸。接头可包含由构成限制性位点例如bamhi、clai、ecori、hindiii、psti、sali和xhoi位点等的dna编码的肽。
接头氨基酸通常并且理想地将提供一些灵活性以允许fc和sirpα组分采用它们的活性构象。允许这种灵活性的残基通常是gly、asn和ser,因此实际上接头内这些残基(特别是gly和ser)的任何组合都可能提供所需的连接效应。在一个示例中,这样的接头是基于所谓的g4s序列(gly-gly-gly-gly-ser,seqidno:10),其可以重复为(g4s)n(seqidno:10),其中n是1、2、3或更多,或基于(gly)n,(ser)n、(ser-gly)n或(gly-ser)n等。在另一个实施方案中,接头是gtelsvrakps(seqidno:4)。该序列构成c末端侧翼igv结构域的sirpα序列(应当理解,当与上述igv最小序列偶联时,该侧翼序列可以被认为是igv结构域的接头或不同形式)。仅需要熔融区域或连接体允许组分采用它们的活性构象,并且这可以通过本领域中有用的任何形式的连接体来实现。
sirpαfc融合可用于抑制sirpα和cd47之间的相互作用,从而阻断跨该轴的信号传导。已知cd47对巨噬细胞上的sirpα的刺激通过使肌球蛋白-ii失活以及将靶标拉入巨噬细胞所涉及的收缩细胞骨架活性来抑制巨噬细胞介导的吞噬作用。因此,该级联的激活对于cd47+疾病细胞的存活是重要的。阻断该途径可使巨噬细胞吞噬并根除cd47+疾病细胞群。
术语“cd47+”用于指目前的多肽结合靶向的细胞的表型。蛋白质cd47,也称为整联蛋白相关蛋白(iap),是由cd47基因编码的跨膜蛋白。cd47属于免疫球蛋白超家族,并与例如膜整联蛋白、血小板反应蛋白1(tsp-1)和信号调节蛋白α(sirpα)相互作用。可以使用cd47抗体作为亲和配体,通过流式细胞术鉴定cd47+细胞。适当标记的cd47抗体可商购获得用于此用途(例如,克隆b6h12可从santacruzbiotechnology获得)。检查cd47表型的细胞可以包括标准肿瘤活检样品,特别是包括取自怀疑携带内源性cd47+癌细胞的受试者的血液样品。特别感兴趣作为本融合蛋白的治疗目标的cd47病细胞是那些“过度表达”cd47。这些cd47+细胞通常是疾病细胞,并且在其表面上以一定密度存在cd47,其超过给定类型细胞的正常cd47密度。cd47过表达在不同细胞类型中会有所不同,但在本文中是指任何cd47水平,例如通过本文示例的流式细胞术或通过免疫染色或通过基因表达分析等确定,其大于在具有对于该细胞类型是正常的cd47表型的对应细胞上可测量的水平。
本发明的药物组合既包含作为cd47阻断剂的sirpαfc,又包含免疫细胞检查点抑制剂。这些包括多种负责上调基于t细胞的免疫系统的物质。关键抑制剂将阻断包括ctla4和/或pd-1的途径,并且这些抑制剂在本发明的广义范围内包括在内。
在特定的实施方案中,免疫细胞检查点抑制剂是pd-1阻断药物,并且这些药物阻断pd-1受体与配体例如pd-l1和pd-l2之间的相互作用。
根据本方法,pd-1阻断药物与sirpαfc结合使用。pd-1本身(编程性死亡-1,又名cd279)是一种淋巴细胞受体,可与称为pd-l1和pd-l2的配体相互作用。pd-1途径位于b7:cd28家族中,该家族也包含ctla4(又名cd152),并且通过控制t细胞活化参与免疫系统的体内平衡。pd-1配体1(pd-l1,cd274,b7:h1)和pd-1配体2(pd-l2,b7-dc,cd273)在癌细胞上的表达是阴性的预后因素。已经显示pd-1阻断可有效治疗多种癌症。
可以用来阻断配体诱导的pd-1刺激的pd-1阻断药物(又名pd-1途径抑制剂)种类繁多,并且包括fc融合蛋白和选择性结合pd-1的抗体及其活性片段,从而抑制与配体pd-l1和pd-l2的相互作用。特别有用的pd-1/pd-l1阻断药物包括融合蛋白和命名为以下的抗体:pidilizumab、派姆单抗(pembrolizumab)、纳武单抗、amp-224(靶向pd-1的pd-l2-igg2afc融合蛋白)、卡米单抗(camrelizumab)(shr-1210)、spartalizumab(pdr001)和regm2810,以及人源化igg4medi0680。
有用的pd-1阻断药物还包括与pd-l1或pd-l2选择性结合的药剂,其特定的pd-l1结合形式包括称为bms-936559的igg4抗体和包括以下的igg1抗体:德瓦鲁单抗(medi4736)、atezolizumab(mpdl3280a/rg7446)和avelumab(又名msb001078c)。
在一个优选的实施方案中,t细胞检查点抑制剂是为纳武单抗的pd-1阻断抗体。纳武单抗是一种结合人pd-1的批准的人igg4抗体,以商品名
sirpαfc药物组合还可以代替pd-1抑制剂或与其组合包括任何其他免疫检查点抑制剂,包括特别是ctla-4抑制剂。像pd-1一样,ctla-4负调节t细胞活化。ctla-4抑制剂是阻止ctla-4与b7配体结合的那些药剂。ctla-4抑制剂被批准用于人类,并且当与cd47阻断药物sirpαfc结合使用时这些可用于本发明。可用于本发明的特别有用的ctla-4抑制剂和药剂是结合ctla4的抗体,包括伊匹单抗或曲美母单抗以及任选的贝拉西普(belatacept)和阿巴西普(abatacept)。
在一个优选的实施方案中,ctla-4抑制剂是伊匹单抗。伊匹单抗是一种完全人重组抗体,可与表达t细胞的人ctla-4结合,商品名为
在实施方案中,本发明的药物组合包含具有seqidno:8或seqidno:9或seqidno:7的sirpαfc与称为纳武单抗的pd-1阻断抗体的组合。在一个具体的实施方案中,所述组合包含具有seqidno:8的sirpαfc和抗体纳武单抗。在另一个具体的实施方案中,所述组合包含具有seqidno:9的sirpαfc和抗体纳武单抗。在这些实施方案中,所述组合可以进一步包含ctla-4抑制剂,其为依匹单抗。
在其他实施方案中,本发明的药物组合包括具有seqidno:8或seqidno:9或seqidno:7的sirpαfc与被称为伊匹单抗的ctla-4抗体的组合。在一个具体的实施方案中,所述组合包含具有seqidno:8的sirpαfc和抗体伊匹单抗。在另一个具体的实施方案中,所述组合包含具有seqidno:9的sirpαfc和抗体伊匹单抗。在这些实施方案中,所述组合可进一步包含为纳武单抗的pd-1抗体。
组合中包含的每种药物/药剂可以单独配制以组合使用。当一种药物的作用用于增强另一种药物在两种药物的接受者中的作用时,称为这些药物“组合”使用。
在该方法中,每种药物以包含药学上可接受的载体的剂型提供,并且以治疗有效量提供。如本文所用,“药学上可接受的载体”是指在蛋白质/抗体制剂领域中生理上相容且有用的任何和所有溶剂、分散介质、包衣、抗细菌剂和抗真菌剂、等渗剂和吸收延迟剂等。药学上可接受的载体的实例包括水、盐水、磷酸盐缓冲盐水、右旋糖、甘油、乙醇等中的一种或多种,以及它们的组合。在许多情况下,组合物中将优选包括等渗剂,例如糖,多醇例如甘露醇、山梨醇或氯化钠。药学上可接受的载体可以进一步包含少量辅助物质,例如润湿剂或乳化剂,防腐剂或缓冲剂,其增强药剂的保质期或有效性。使用治疗性蛋白质制剂领域中的实践标准来配制sirpαfc融合蛋白和基于蛋白质的pd-1阻断药物。适合静脉内给药例如通过注射或输注的溶液是特别有用的。
无菌可注射溶液可以通过将所需量的活性化合物与上述成分中的一种或组合(如果需要)一起掺入适当的溶剂中,然后进行灭菌微滤来制备。通常,通过将活性化合物掺入无菌载体中来制备分散液,所述无菌载体含有基础分散介质和来自上面列举的那些的所需其它成分。在无菌粉末的情况下,制备方法是真空干燥和冷冻干燥(冻干),其产生活性成分的粉末加上来自其先前无菌过滤溶液的任何附加期望成分。
如本文所用,“有效量”是指在必要的剂量和特定时间段内有效实现所需治疗结果的量。组合中每种药物的治疗有效量可根据诸如疾病状态、年龄、性别和个体体重等因素以及药物在接受者中引发所需反应的能力而变化。治疗有效量也是其中药剂的任何毒性或有害作用超过治疗有益效果的量。
可以与载体材料组合以产生单剂型的活性成分的量将根据所治疗的受试者和具体的给药方式而变化。产生单一单位剂型所需的活性成分的量通常是产生治疗效果的组合物的量。通常,在百分之百中,该量为约0.01%至约99%的活性成分,优选约0.1%至约70%,例如约1%至约30%的活性成分,与药学上可接受的载体组合。
sirpαfc融合蛋白和pd-1阻断药物例如抗体以及ctla-4抑制剂可以通过为蛋白质递送建立的任何途径,特别是静脉内、肿瘤内、皮内和皮下注射或输液,或经鼻或肺部施用,施用给受试者。
在一些实施方案中,考虑上述多肽(或肽、蛋白质、抗体等)的变体。例如,在一些实施方案中,本发明用相对于本文所述的参考多肽或序列具有至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%或至少99%的氨基酸序列同一性。在一些实施方案中,序列仅由于保守氨基酸取代而不同。变体保留本文所述参考序列的相关生物学活性,例如cd47结合(sirpα变体)、效应子功能(对于fc变体)或检查点抑制剂活性(对于t细胞检查点抑制剂)。
本发明组合中的药物可以顺序或基本同时施用。即,可以在施用sirpαfc之前或之后给予t细胞检查点抑制剂。期望的是药物的作用在患者中重叠,并且优选的是药物或活性代谢物在接受者中同时存在。因此,接受治疗的受试者可以是已经接受了组合药物的一种如sirpαfc的受试者,然后用组合药物的另一种对该受试者进行治疗。或者,该受试者可以是已经用一种或多种t细胞检查点抑制剂治疗,然后用sirpαfc治疗的受试者。
药物组合物可以使用本领域已知的一种或多种方法,通过一种或多种施用途径施用。如本领域技术人员所理解的,施用途径和/或方式将根据所需结果而变化。本发明的融合蛋白的优选施用途径包括静脉内、肌肉内、皮内、肿瘤内、腹膜内、皮下、脊柱或其它肠胃外施用途径,例如通过注射或输注。短语“肠胃外施用”包括输注和注射,例如静脉内、肌肉内、动脉内、鞘内、囊内、眶内、心内、皮内、腹膜内、气管内、肿瘤内、皮下、皮内、关节内、包膜下、蛛网膜下、脊柱内、硬膜外和胸骨内。
或者,本发明的融合蛋白可以通过非肠胃外途径施用,例如通过经口或滴注或通过局部,表皮或粘膜施用途径施用,例如,鼻内、口服、阴道、直肠或舌下施用。
调整给药方案以提供最佳的所需反应(例如,治疗反应)。例如,可以施用单次推注的每种药物,或者可以随时间施用几个分开的剂量,或者可以按照治疗情况的指示按比例减少或增加剂量。将肠胃外组合物配制成单位剂型以便于给药和剂量均匀是特别有利的。如本文所用的“单位剂型”是指适合作为待治疗受试者的单位剂量的物理上离散的单位;每个单位含有预定量的活性化合物,经计算可与所需的药物载体一起产生所需的治疗效果。本发明的剂量单位形式的说明书由(a)活性化合物的独特特征和要实现的特定治疗效果和(b)复合这种活性化合物用于治疗个体敏感性的领域中固有的局限性决定并且直接取决于其。
药物可以组合配制,以便可以在一次给药中将组合物引入接受者,例如一次注射或一次输注袋。在另一个实施方案中,将药物分开配制以在组合疗法方案中分开施用。
对于施用,每种药物的剂量将在宿主体重的约0.0001至100mg/kg,更通常0.01至5mg/kg的范围内。例如,剂量可以是0.1mg/kg体重,0.2mg/kg体重,0.3mg/kg体重,1mg/kg体重,3mg/kg体重,5mg/kg体重或10mg/kg体重或在1-10mg/kg范围内。在单位剂型中,药物将包含1-500mg药物/剂,例如1、2、3、4、5、10、25、50、100、200、250和500mg/剂。这两种药物可以大致等摩尔量(+/-10%)施用。示例性治疗方案需要每周施用一次,每两周施用一次,每三周施用一次,每四周施用一次,每月施用一次,每3个月施用一次或每三至六个月施用一次。本发明药物组合的优选给药方案包括通过静脉内给药1mg/kg体重或3mg/kg体重,每种药物使用以下给药方案之一同时给药;(i)每四周一次,共六剂,然后每三个月一次;(ii)每三周;(iii)3mg/kg体重一次,然后每三周1mg/kg体重。在一些方法中,调节剂量以达到约1-1,000μg/ml的血浆融合蛋白浓度,并且在一些方法中约25-300μg/ml。
在实施方案中,使用给药方案治疗受试者,所述给药方案包括每周0.1mg/kg(或每周0.2mg/kg,或每周0.3mg/kg)的seqidno:8或no:9的sirpαfc药物和每2周约3mg/kg的纳武单抗。当与纳武单抗一起使用时,依匹单抗也可与批准的剂量整合。sirpαfc蛋白与红细胞的结合可忽略不计。因此,当使用药物组合给药时,不需要考虑rbc“下沉(sink)”。相对于由rbc结合的其他cd47阻断药物,估计本发明的sirpαfc融合物可以在小于成为rbc结合的药物(例如cd47抗体)所需剂量的一半的剂量下有效。此外,sirpα-fc融合蛋白是sirpα介导的信号的专用拮抗剂,因为当与其结合时其显示可忽略的cd47激动作用。因此,当建立医学上有用的单位给药方案时,不需要考虑药物诱导的任何刺激。
组合中的每种药物也可以作为持续释放制剂给药,在这种情况下需要较低频率的给药。剂量和频率取决于患者中融合蛋白的半衰期。给药的剂量和频率可以根据治疗是预防性还是治疗性而变化。在预防性应用中,相对较低的剂量在较长时间内以相对不频繁的间隔施用。一些患者在余生中继续接受治疗。在治疗应用中,有时需要以相对短的间隔施用相对高的剂量直至疾病进展减少或终止,并且优选直至患者显示疾病症状的部分或完全改善。此后,可以使用预防方案治疗患者。
该药物组合可用于“治疗”各种cd47+疾病细胞。这些尤其包括cd47+癌细胞,包括液体和实体肿瘤。可以用本发明的药物组合治疗实体瘤,以减小其大小、数量或生长速率并控制癌症干细胞的生长。此类实体肿瘤包括黑素瘤、膀胱、脑、乳腺、肺、结肠、卵巢、前列腺、肝脏、皮肤和其他组织中的cd47+肿瘤。在一个实施方案中,药物组合可用于抑制血液癌症的生长或增殖。如本文所用,“血液癌症”是指血液的癌症,并且包括白血病、淋巴瘤和骨髓瘤等。“白血病”是指血液中的癌症,其中过多的白细胞对抗感染是无效的,因此挤出构成血液的其他部分,例如血小板和红细胞。据了解,白血病病例分为急性或慢性病例。举例来说,某些形式的白血病可以是急性淋巴细胞白血病(all);急性髓性白血病(aml);慢性淋巴细胞白血病(cll);慢性粒细胞白血病(cml);骨髓增生性疾病/肿瘤(mpds);和骨髓增生异常综合征。“淋巴瘤”可以指霍奇金淋巴瘤,其中包括惰性和侵袭性非霍奇金淋巴瘤、伯基特淋巴瘤、皮肤t细胞淋巴瘤、外周t细胞淋巴瘤和滤泡性淋巴瘤(小细胞和大细胞)等。骨髓瘤可以指多发性骨髓瘤(mm)、巨细胞骨髓瘤、重链骨髓瘤和轻链或bence-jones骨髓瘤。
在一些实施方案中,用药物组合治疗的血液癌症是cd47+白血病,优选选自急性淋巴细胞白血病、急性髓性白血病、慢性淋巴细胞白血病、慢性髓性白血病和骨髓增生异常综合征,优选人急性髓性白血病。
在其他实施方案中,用sirpαfc蛋白治疗的血液学癌症是选自以下的cd47+淋巴瘤或骨髓瘤:霍奇金淋巴瘤、惰性和侵袭性两种非霍奇金淋巴瘤、伯基特氏淋巴瘤、滤泡性淋巴瘤(小细胞和大细胞)、多发性骨髓瘤(mm)、巨细胞骨髓瘤、重链骨髓瘤,轻链或bence-jones骨髓瘤以及平滑肌肉瘤(leimyosarcoma)。
在其他实施方案中,本发明的药物组合用于治疗非小细胞肺癌、肾癌、膀胱癌、头和颈鳞状细胞癌、merkel细胞皮肤癌、食道癌、胰腺癌、肝细胞癌、胃癌、乳腺癌和卵巢癌。在特定的实施方案中,当t细胞检查点抑制剂是纳武单抗时,治疗的癌症是黑色素瘤、胶质母细胞瘤、转移性非小细胞肺癌、肾细胞癌、霍奇金淋巴瘤、头颈癌、尿路上皮癌、结直肠癌和肝细胞癌。当t细胞检查点抑制剂是派姆单抗时,治疗的癌症是黑色素瘤、转移性非小细胞肺癌、头颈癌、霍奇金淋巴瘤、尿路上皮癌和胃癌之一。在另一个具体的实施方案中,当t细胞检查点抑制剂是伊匹单抗时,靶标癌症是黑素瘤。在另一个具体的实施方案中,当t细胞检查点抑制剂是pd-1抑制剂时,靶标癌症是成胶质细胞瘤。
在一个具体的实施方案中,接受治疗的受试者患有霍奇金淋巴瘤,并且所述治疗包括每周0.1-0.3mg/kg的包含seqidno:8或no:9的sirpαfc药物与每2周一次3mg/kg的纳武单抗组合。在另一个具体的实施方案中,该组合疗法用于治疗患有黑素瘤的受试者。
包含经由sirpαfc的cd47阻断和例如通过pd-1阻断和/或ctla-4阻断的t细胞检查点抑制的组合疗法也可以与用于治疗靶向适应症的任何其他药剂或方式一起利用,例如辅助治疗中的手术,或者如新辅助疗法中的其他化学疗法。
在一个特定的实施方案中,当治疗涉及依匹单抗和纳武单抗的组合以及sirpαfc时,可以同时给予药物(sirpαfc给予iv1x/周或it3x/周)。纳武单抗的推荐剂量为60分钟内以静脉输注施用1mg/kg,然后在同一天给予伊匹单抗3mg/kg,每3周一次,共4剂。推荐的纳武单抗后续推荐剂量(作为单药)为每2周60分钟静脉输注240mg,直至疾病进展或出现不可接受的毒性。
实施例1
在研究的第1天,雌性c57bl/6小鼠为八周大,体重(bw)范围为19.1至25.5克。给动物随意饲喂水(反渗透,1ppmcl),以及由18.0%的粗蛋白、5.0%的粗脂肪和5.0%的粗纤维组成的nih31modifiedandirradiatedlab
阻断药物
trilliumtherapeutics,inc.提供了预先配制的cd47阻断药物和对照,作为可溶性sirpα的小鼠形式,命名为(1)对照fc{小鼠igg2afc区(铰链-ch2-ch3)},和(2)小鼠sirpαfc{包含与野生型小鼠igg2a结构域(铰链ch2-ch3)融合的小鼠sirpa(nod菌株)的末端结构域},将其在-80℃下储存直至使用。使用小鼠蛋白是因为人sirpαfc蛋白不会与小鼠cd47靶标交叉反应。pd-1阻断药物提供为抗pd-1抗体(克隆rmp1-14,批号5792/0915,6.54mg/ml),由crdiscovery从bioxcell购买并在收到后于4℃保存。将测试药物配制在无菌pbs中,并在测试过程中使其盲目。在每个给药日,将一小瓶每种试剂解冻,以每只小鼠0.2ml(100ug)的剂量给药。通过在无菌pbs中将储备液的等分试样稀释至0.5mg/ml来制备每种抗体定量溶液。购买了配制的抗体药物。
肿瘤细胞培养
mc38鼠结肠癌细胞在含有10%胎牛血清的dmem培养基中生长至对数中期。在37℃,5%co2和95%空气的气氛中,在潮湿的培养箱中在组织培养瓶中培养肿瘤细胞。在植入肿瘤的当天,在指数生长期间收获mc38细胞,并以5×106个细胞/ml的浓度再悬浮于磷酸盐缓冲盐水(pbs)中。通过将1x106个mc38肿瘤细胞(0.1ml悬浮液)皮下植入每只测试动物的右胁腹引发肿瘤。使用卡尺以二维方式测量肿瘤,并使用以下公式计算体积:
其中,w=肿瘤宽度,l=肿瘤长度,以毫米为单位。肿瘤重量可以假设1毫克相当于1mm3肿瘤体积进行估计。
治疗
植入后,监测肿瘤,直到它们达到80-120mm3的平均尺寸。将小鼠分为五组(n=10),并开始给药。所有药剂均腹膜内(ip)递送。如图1所示,第1和2组接收200μg/动物的sirpαfc或133μg/动物的对照,每周三次,分别进行四周(tiwk×4)。第3组接受抗pd-1,为100μg/动物,每周两次,进行2周(biwkx2)。第4和5组分别接收sirpαfc或对照fc,与上述治疗方案的抗pd-1组合。
终点分析
每周两次使用卡尺测量肿瘤。分别对动物进行监测,并在肿瘤达到1500mm3的终点体积时或在最后一天(以先到者为准)对每只小鼠实施安乐死。记录因肿瘤体积终点而退出研究的动物为因肿瘤进展(tp)而安乐死,记录安乐死的日期。通过以下公式为每只小鼠计算到达终点的时间(tte),以供分析:
其中tte以天表示,终点体积以mm3表示,b是截距,m是通过对数转换的肿瘤生长数据集进行线性回归而获得的直线的斜率。数据集由超过分析中使用的终点体积的第一个观察值和在紧接达到该终点体积之前的三个连续观察值组成。计算的tte通常小于tp日期,即因肿瘤大小对动物实施安乐死的日期。具有未达到终点体积的肿瘤的动物被分配等于研究最后一天的tte值。在对数转换后计算出的tte早于到达终点的前一天或超过达到肿瘤体积终点的那一天的情况下,执行线性插值以逼近tte。如图1-3所示,单独的sirpαfc对mc38没有影响,表明没有足够的巨噬细胞浸润mc38肿瘤,或者该肿瘤生长得太快而无法控制。
实施例2
在这项研究中,人肿瘤细胞系jurkat被转染以表达人巨细胞病毒(cmv)pp65抗原。cmvpp65抗原用作替代肿瘤抗原。
巨细胞病毒(cmv)特异性cd8+t细胞被用作替代肿瘤抗原特异性t细胞的来源。这些cmv特异性cd8+t细胞是从健康的hla-a2+供体的血液中分离出来的,并根据标准方案(wolf和greenberg,natprotoc.9(4)2014),通过在il-7和il-15存在下与自体成熟cmv肽pp65脉冲的树突状细胞共培养而在14天的时间内扩增。
通过在m-csf中培养血液单核细胞9天,然后用ifnγ引发24小时来产生单核细胞衍生的巨噬细胞。随后将这些ifnγ引发的巨噬细胞与cmvpp65转染的jurkat共培养,以使吞噬作用在1umsirpαfc存在下发生。共培养24小时后,通过流式细胞术证实pp65抗原存在于巨噬细胞表面(数据未显示)。这时,将扩增的cmv特异性自体cd8+t细胞添加至巨噬细胞。
通过添加fitc结合的抗cd107a/bmab5小时并随后通过流式细胞术进行分析,评估cmv特异性cd8+t细胞的去颗粒化。通过通透化并用抗tnfα和抗ifnγmab染色,然后通过流式细胞术,评估cmv特异性cd8+t细胞产生细胞内细胞因子。通过同时四聚体染色鉴定cmv特异性cd8+t细胞。在指示的情况下,以10ug/ml加入伊匹单抗和纳武单抗。
结果总结
(a)纳武单抗和伊匹单抗与sirpαfc的三联组合与单独的sirpαfc相比在肿瘤特异性cd8t细胞活化方面具有统计学上的显著增加,这通过为去颗粒化标志物cd107a/b+的肿瘤特异性cd8+t细胞的百分比测量。
(b)与单独的sirpαfc治疗相比,纳武单抗与sirpαfc和伊匹木单抗与sirpαfc的的两种组合导致肿瘤特异性cd8t细胞的tnfα+ifnγ+百分比显著增加。与双重组合和sirpαfc单已治疗相比,三联组合纳武单抗和伊匹单抗与sirpαfc进一步增加了肿瘤特异性cd8效应子t细胞的tnfα+ifnγ+百分比。
因此表明,在以下情况下获得了sirpαfc的抗癌效果统计学上显著的改进:当治疗与pd-1抑制剂纳武单抗组合时(如图4b所示增加了50%),或与ctla-4抑制剂伊匹单抗组合时(如图4b所示增加了26%),或同时与pd-1抑制剂和ctla-4抑制剂组合时(如图4b和4a所示分别增加了136%和76%)。
总的来说,与单独的sirpαfc治疗相比,纳武单抗或伊匹单抗与sirpαfc组合以及纳武单抗和伊匹单抗与sirpαfc三联组合导致肿瘤特异性cd8+t细胞的活化和效应子功能增强。
实施例3
复发性和难治性经皮可及的实体瘤或蕈样真菌病受试者的组合疗法
以下是证明sirpαfc–检查点抑制剂组合在需要治疗的实体瘤或蕈样肉芽肿的人类受试者中疗效的方案。
将sirpαfc与程序性死亡1(pd-1)或程序性死亡配体1(pd-l1)抑制剂(例如纳武单抗、派姆单抗、德瓦鲁单抗、avelumab或atezolizumab)组合在第1天给予患癌受试者。在某些变型中,受试者的癌症诊断针对fda批准的pd-1/pd-l1抑制剂,例如黑色素瘤、头颈癌、肺癌、膀胱癌、尿路上皮癌、结直肠癌、乳腺癌和肾癌。在一些变型中,受试者患有蕈类肉牙肿(mycosisfungoide)。
选择1mg的起始剂量sirpαfc(例如,seqidno:8),一种由人sirpa的cd47结合结构域与人免疫球蛋白(igg1)的fc区连接组成的sirpαfc融合蛋白,进行肿瘤内注射。该剂量确保即使在100%的理论生物利用度下,在60kg受试者中总的全身剂量也不会超过约0.05mg/kg或3mg。肿瘤内给药最多可增加10mg,每周3次,进行2周。预期该剂量将实现非常高的局部cd47饱和度,同时大约相当于静脉注射可接受的每日剂量(60kg受试者为0.3mg/kg或18mg)。
非人类灵长类动物毒性研究还支持上述剂量和给药方案的安全性,该研究表明,皮下施用sirpαfc(单一疗法)的剂量为0.5mg/kg在2周内施用6次,以及1.5mg/kg在2周内两次施用,对皮肤无不良影响。观察到预期的血液学变化,在剂量水平之间未发现有意义的生物学差异。这项研究的最高非重度毒性剂量(hnstd)为0.5mg/kg,在2周内施用6次,以及1.5mg/kg,在2周内施用两次。
根据标准标记的剂量和方案,受试者将在第1天静脉注射sirpαfc与以下pd-1/pd-l1抑制剂之一组合:纳武单抗(
如果pd-1/pd-l1抑制剂与sirpαfc在同一天给药,则在完成pd-1/pd-l1抑制剂输注与注射sirpαfc之间应至少间隔60分钟。pd-1/pd-l1抑制剂也可以在注射sirpαfc的前一天给予。任何与pd-1/pd-l1抑制剂输注相关的反应均应为2级或更低,并已完全解决以在同一天开始sirpafc注射。
与接受单一药剂(单独使用sirpαfc或单独使用检查点抑制剂)的受试者相比,针对肿瘤体积、副作用、无进展生存期、总生存期或其他标准参数评估疗效。
在完成初始诱导治疗后有资格接受继续治疗的受试者可以根据肿瘤科医生的判断,每周接受一次额外的sirpαfc注射。组合疗法将继续使用标准剂量和给药方案(见上文)。
实施例4
sirpαfc在复发或难治性血液系统恶性肿瘤和选择的实体瘤患者中的1a/1b期剂量递增和扩展试验
sirpαfc加上纳武单抗受试者接受的起始剂量为0.1mg/kg/周的sirpαfc与纳武单抗组合,按照当前fda批准的包装说明书给药,每2周给予一次(1个周期)。对纳武单抗的毒性不可接受的受试者可以继续以单药形式接受sirpαfc。如果在与sirpαfc的同一天给予纳武单抗,则在完成纳武单抗输注和开始sirpαfc输注之间必须至少经过60分钟。在sirpαfc输注的前一天也可以给予纳武单抗。
与接受单一药剂(单独使用sirpαfc或单独使用检查点抑制剂)的受试者相比,针对癌症负担、副作用、无进展生存期、总生存期或其他标准参数评估疗效。
- 还没有人留言评论。精彩留言会获得点赞!