拓扑电子材料的判定和搜索方法与流程
本发明涉及计算物理和材料科学领域,尤其涉及一种拓扑电子材料的判定和搜索方法。
背景技术:
拓扑电子材料是近年来在凝聚态物理学中被发现的一类新型材料,包括拓扑绝缘体材料和拓扑半金属。它的主要特点是拥有非平凡的“拓扑性质”,具体体现在两个方面:1.在拓扑绝缘体材料的表面,存在着一般二维材料中无法实现的表面电子态(如没有背散射的电子态等);2.在拓扑半金属的体内,存在着线性负磁阻等所谓“量子反常”的现象。这些性质使得拓扑电子材料在高容错的拓扑量子计算、超低能耗电子器件等多方面有着应用前景。也让很多计算物理学家和材料科学家积极寻找更新的更好的此类材料。
然而,发现新的拓扑电子材料并非易事。现有的判断拓扑电子材料的方法需要先对布里渊区密集采点(一般要几十上百个点)进行第一性原理计算,然后再计算“拓扑不变量”,通过拓扑不变量的数值来判断材料是否是拓扑的。因此,对于一个给定的化合物,想要预先判断它有无拓扑性质,必须计算它的“拓扑不变量”。拓扑不变量首先是一个复杂的数学概念,涉及到大多数领域内科学家所不具备的当代数学知识如“代数拓扑学”;其次,即便在数学知识完备的情况下,在第一性原理计算软件中实现拓扑不变量的计算也是非常繁琐的,时间代价和人工代价极高。因此,急需一种判定和搜索拓扑电子材料的简单易行的方法。
技术实现要素:
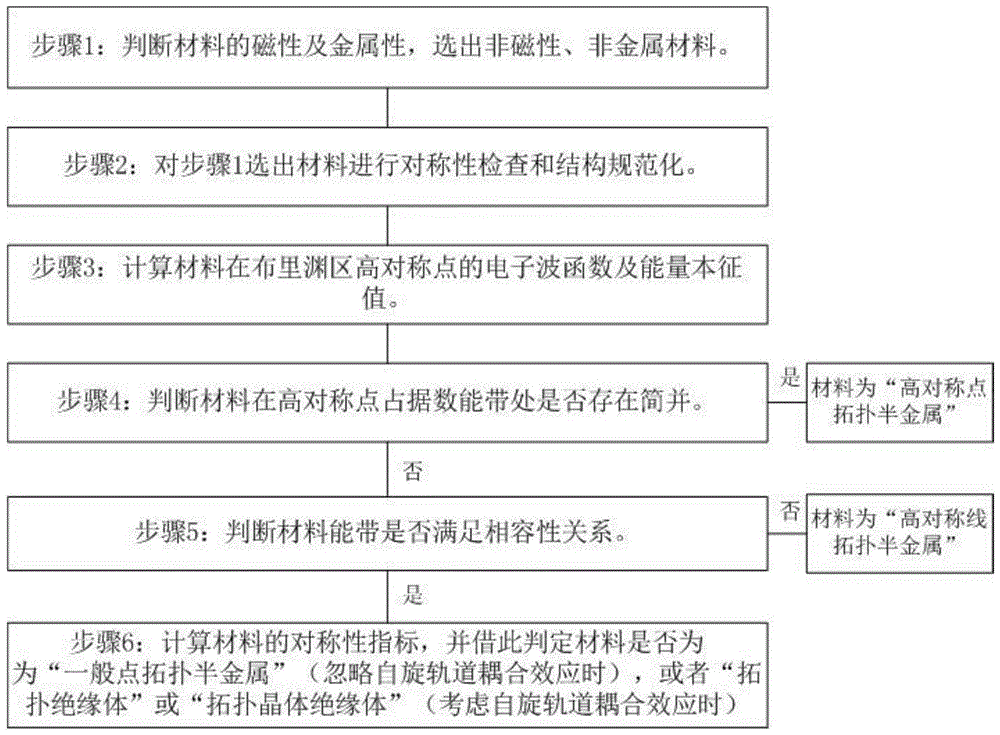
因此,本发明的目的在于克服上述现有技术的缺陷,提供一种全面、快速、自动化的拓扑电子材料的判定方法,其包括如下步骤:
步骤1:判断给定材料的磁性及金属性,选出非磁性、非金属材料;
步骤2:基于所选材料的结构文件计算在布里渊区高对称点的电子波函数及能量本征值;
步骤3:基于步骤2所得到的电子波函数和能量本征值判断材料在高对称点占据数能带处是否存在简并。
根据本发明的拓扑电子材料的判定方法,优选地,所述步骤2还包括对所选材料的结构文件进行对称性检查及结构规范化。
根据本发明的拓扑电子材料的判定方法,优选地,在步骤3中,如果材料在高对称点占据数能带处存在简并,则判定该材料属于“高对称点拓扑半金属”。
根据本发明的拓扑电子材料的判定方法,优选地,如果材料在高对称点占据数能带处不存在简并,则进行如下步骤4:
步骤4:判断材料的能带是否满足相容性关系。
根据本发明的拓扑电子材料的判定方法,优选地,在步骤3中,根据所述能量本征值获得材料的直接能隙,如果所述直接能隙大于等于2mev,则判断材料在高对称点占据数能带处不存在简并。
根据本发明的拓扑电子材料的判定方法,优选地,在步骤3中,根据所述能量本征值获得材料的直接能隙,如果所述直接能隙小于2mev,则进行简并态计算。
根据本发明的拓扑电子材料的判定方法,优选地,如果材料的能带不满足相容性关系,则该材料属于“高对称线拓扑半金属”。
根据本发明的拓扑电子材料的判定方法,优选地,如果材料的能带满足相容性关系,则进行如下步骤5:
步骤5:计算材料的对称性指标。
根据本发明的拓扑电子材料的判定方法,优选地,在步骤5中,忽略自旋轨道耦合效应时,如果所述对称性指标不全为0,则判定该材料属于“一般点拓扑半金属”。
根据本发明的拓扑电子材料的判定方法,优选地,在步骤5中,考虑自旋轨道耦合效应时,如果所述对称性指标不全为0,则判定该材料属于“拓扑绝缘体”或“拓扑晶体绝缘体”。
根据本发明的拓扑电子材料的判定方法,优选地,如果所述对称性指标里最后一位强拓扑指标为奇数,则判定该材料属于“拓扑绝缘体”;如果所述对称性指标里最后一位强拓扑指标为偶数,则判定该材料属于“拓扑晶体绝缘体”。
本发明提供的方法可以大大简化拓扑电子材料的判定过程,且通过对已有的材料数据库的全盘扫描,可以找出所有能用对称性本征值判断的拓扑材料。
附图说明
以下参照附图对本发明实施例作进一步说明,其中:
图1为根据本发明的判定拓扑电子材料的方法流程图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图通过具体实施例对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
总地来说,参见图1所示的流程图,本发明的拓扑电子材料的判定包括如下步骤:
步骤1:判断材料的磁性及金属性,选出非磁性、非金属材料;
步骤2:优选地对步骤1选出的材料进行对称性检查和结构规范化;
步骤3:计算材料在布里渊区高对称点的电子波函数及能量本征值;
步骤4:判断材料在高对称点占据数能带处是否存在简并,如果存在简并,则判断该材料为“高对称点拓扑半金属”,如果不存在简并,则进行如下步骤5,由于只有高对称点直接能隙小于2mev的材料才有可能是“高对称点拓扑半金属”,因此,首先从步骤3得到的能量本征值判断材料的直接能隙,如果其大于等于2mev,则直接判断其不存在简并并进行如下步骤5,如果其小于2mev,则进行简并判断的相关计算;
步骤5:判断材料能带是否满足相容性关系,如果不满足,则判断该材料为“高对称线拓扑半金属”,如果满足,则进行如下步骤6;
步骤6:计算材料的对称性指标,并依据该对称性指标判定材料是否为“一般点拓扑半金属”(忽略自旋轨道耦合效应时),或者“拓扑绝缘体”或“拓扑晶体绝缘体”(考虑自旋轨道耦合效应时)。
以下结合本发明的实施例,对于上述步骤进行详细解释。
步骤1:给定一个材料,从thematerialsproject网站上获取材料的磁矩,根据原胞中每种元素的个数计算材料的电子数,如果磁矩小于0.1μb则判定为非磁性材料,如果材料的电子数为奇数则为金属材料,根据此标准选出非磁性、非金属材料进行如下步骤。
步骤2:通常一个材料的对称性是已知的,但因为误差等原因导致材料用于计算的结构文件并不严格具有已知的对称性。结构文件是储存材料中每个原子所处的位置坐标的文件。为了保证计算结果的准确性,需要对材料中原子的坐标进行微小调整使其具有相应的对称性。根据本发明的一个实施例,这一步是通过phonopy软件包完成的,在确定对称性的过程中,原子坐标的移动范围从10-5埃逐步放宽到10-1埃,直到找到正确的对称性,如果精度到10-1埃时仍未找到对称性则该材料的结构文件被视为是错误的。除了检查对称性外,还将材料原胞的基矢进行规范化,即通过一个相似变换使得属于同种布拉菲格子的材料的基矢具有相同的规范。这一步之所以需要是因为不同的对称操作在不同的坐标基矢下的矩阵形式是不一样的,为了方便后续计算波函数的对称性本征值,在本发明的一个实施例中需要将材料原胞的基矢规范化,这样属于同种布拉菲格子的空间群的对称操作矩阵就是一致的。
步骤3:利用viennaabinitiosimulationpackage(vasp)软件包计算材料在布里渊区高对称点的电子波函数及能量本征值。根据songz,zhangt,fangz,etal.quantitativemappingsbetweensymmetryandtopologyinsolids[j].naturecommunications,2018,9(1):3530.和songz,zhangt,fangc.diagnosisfornonmagnetictopologicalsemimetalsintheabsenceofspin-orbitalcoupling[j].physicalreviewx,2018,8(3):031069.两篇文献,对于不同的空间群本发明只需要计算某些特定的高对称点即可得到这个空间群对应的拓扑分类。在上述文献里可知不同空间群需要计算哪些高对称点。利用vasp进行第一性计算时,先进行自洽计算产生电荷密度文件,再读取此电荷密度文件来进行非自洽的能带计算,产生能量本征值、波函数等输出文件用于下一步分析。
根据本发明的一个实施例,只需要计算几个高对称点的电子波函数即可,从而大大加快了计算速度。这一步计算对考虑自旋轨道耦合(soc-setting)和不考虑自旋轨道耦合(nsoc-setting)两种情形是分开进行的,下面的所有步骤也是对两种情形分别独立计算。实际材料都是有自旋轨道耦合的,本算法中,考虑自旋轨道耦合之后给出的拓扑分类对应材料真实的拓扑分类。另一方面,本算法也给出忽略自旋轨道耦合时的拓扑分类,虽然这并不对应材料真实的拓扑分类,但在物理上是有意义的。忽略自旋轨道耦合时的拓扑分类有助于理解考虑自旋轨道耦合时的拓扑性质是怎么形成的。此外,由于实验上观测精度有限等原因,对于一些自旋轨道耦合效应很弱的元素,忽略自旋轨道耦合时给出的拓扑分类可能与实验符合的更好。所以本发明的算法对一个材料会给出考虑自旋轨道耦合和不考虑自旋轨道耦合时的两种拓扑分类结果,以便给相关科研人员更好的参考。
步骤4:理论上,简并的能带具有相同的能量本征值,但实际计算中因为精度等原因导致简并的能带也会有微小的能量差。另一方面,每个空间群不同的不可约表示具有确定的维度,属于某个空间群的材料电子波函数具有该空间群的对称性,所以材料的每条能带,如果没有简并则构成该空间群某个一维表示的基函数,如果有简并则几条简并能带构成某个高维表示的基函数。根据这一性质,在判断占据数能带处是否存在简并时,只需要给定一个最大简并误差,在这个误差范围内计算第n条能带属于哪个不可约表示。如果属于该不可约表示的简并能带包括第n和n+1条,那么这个高对称点在占据数能带处就是存在简并的。若存在简并则该材料属于“高对称点拓扑半金属”,若不存在简并则进入下一步判断。
下面根据本发明的一个实施例介绍能带不可约表示的计算方法:
第一步:计算波函数在空间群对称操作下的对称性本征值,即特征标。vasp的波函数是以平面波基矢展开的,从vasp输出文件wavecar中可以读取波函数的平面波基矢展开系数。设布里渊区某点(设为k点)处的波函数
第二步:根据波函数在每个对称操作下的特征标来判断该波函数构成空间群哪个不可约表示的基函数。实际计算中,利用群表示的正交定理,用求出的特征标来和空间群每个不可约表示做内积,内积在归一化之后为1的表示即为所求。实际计算中由于vasp平面波截断能有限等计算误差导致内积不会严格是1,本发明设的最大误差为0.05。若在误差范围内无法求出整数个表示,则需要返回步骤3,提高精度重新计算材料的波函数。做正交定理时,需要选取某个规范下的空间群特征标表作为标准。本算法利用bilbaocrystallographicserver(bcs)上的双群不可约表示特征标表来判断不可约表示,这需要计算过程中所选取的规范和bcs是一致的,其中包括原胞基矢的选取,原胞坐标原点的选取,高对称点坐标的选取,每个三维转动矩阵对应su(2)矩阵的选取等。其中原胞坐标原点的取法决定了非简单空间群中非简单操作对应的平移部分的取法。另外,需要注意的是bcs上的双群不可约表示没有考虑时间反演对称性,而因为本发明计算的材料都是非磁性的,所以时间反演对称性总是具有的。考虑时间反演对称性之后空间群的不可约表示之间会出现额外的简并,而具体哪些表示简并可以在bcs的“双空间群能带表示和基础能带表示”模板中得到。
高维表示的情况是类似的,求特征标时只需要对简并的能带分别计算对称性本征值并求和,即可得到高维不可约表示的特征标。需要注意的是,属于同一个高维表示的能带,如果对其中某一条能带单独求表示是求不出来的,只有将每条能带的对称性本征值相加才构成高维表示的特征标。本发明的算法里需要先将能带划分成简并能带的集合,再对每个集合求表示。简并误差设为5mev和0.1*gap之间的较小者,这里gap定义为该高对称点第n和n+1条能带之间的能量差。
步骤5:在确定每个高对称点占据数能带处不存在简并之后,根据前文提到的能带不可约表示的计算方法,来计算占据数及其以下n条能带的不可约表示,这里n是材料的电子数。在算出每个高对称点的能带表示之后,判断任意两个高对称点的能带表示在每个相连这两点的高对称线上是否满足相容性关系,如果不满足,则在两个高对称点之间的高对称线上存在导带和价带的能带交叉,属于“高对称线拓扑半金属”,如果满足关系则进入下一步判断。空间群每个高对称点到每个相连的高对称线之间的不可约表示的相容性关系可以在bcs上获得,再通过匹配两个高对称点之间相同的高对称线即可获得两个高对称点之间的相容性关系,若两点之间有多条高对称线相连,则这两个点间有多个相容性关系,破坏其中一个表示相应的高对称线上存在能带交叉。
步骤6:根据songz,zhangt,fangz,etal.quantitativemappingsbetweensymmetryandtopologyinsolids[j].naturecommunications,2018,9(1):3530.和songz,zhangt,fangc.diagnosisfornonmagnetictopologicalsemimetalsintheabsenceofspin-orbitalcoupling[j].physicalreviewx,2018,8(3):031069.两篇文献,某些空间群具有由倒空间高对称点的能带表示定义的对称性指标,由对称性指标可以得到材料所有的拓扑不变量。对满足相容性关系的材料,如果材料所属的空间群没有对称性指标,则该材料属于拓扑平凡的普通绝缘体。而如果所属的空间群具有对称性指标,则将上一步骤中已经计算出的高对称点能带不可约表示带入两篇文献中的公式来计算材料的对称性指标。如果对称性指标全部为0,则该材料是拓扑平凡的普通绝缘体,如果不全为0,则在nsoc-setting中,该材料属于一般点拓扑半金属(gmsm),在soc-setting中,该材料属于拓扑绝缘体(ti)或者拓扑晶体绝缘体(tci)。在soc-setting里,如果对称性指标里最后一位强拓扑指标为奇数,则该材料属于拓扑绝缘体,如果对称性指标里最后一位强拓扑指标为偶数,则该材料属于拓扑晶体绝缘体。
在本发明算法的基础上,本发明对thematerialsproject网站上有icsd(inorganiccrystalstructuredatabase)数据库编号的39519个材料进行了计算。所有计算结果展示在网站http://materiae.iphy.ac.cn上。
下面通过具体示例说明本发明的拓扑电子材料的判定。
例1.
225号空间群的碲化锡(snte)材料,无磁性,每个原胞里共有20个价电子,在布里渊区有4个高对称点γ=[0,0,0],l=[1/2,1/2,1/2],w=[1/2,1/4,3/4],x=[1/2,0,1/2],考虑自旋轨道耦合效应时计算的能带表示如下:
经计算发现snte这个材料的能带表示满足225号空间群的相容性关系。225号空间群的对称性指标为z8,经过计算得到snte的对称性指标为z8=4,所以其拓扑分类是“拓扑晶体绝缘体”(tci)。
例2.
同样是225号空间群的snte,在忽略自旋轨道耦合效应时,其能带表示破坏了l和w两个高对称点之间的相容性关系,故其拓扑分类为“高对称线拓扑半金属”(hslsm)。
例3.
216号空间群的碲化贡(hgte)材料,无磁性,每个原胞里共有18个价电子。经过计算发现hgte在γ点的第17,18,19,20四条能带属于同一个4维表示
本发明利用对称性指标理论设计出一套快速且全自动的拓扑材料搜索方法。根据新的理论,判断一个材料是否具有拓扑性质(以及具有怎样的拓扑性质),只需要分析能带结构中数个高对称动量点的波函数的对称性(即计算波函数的不可约表示)即可。本方法可以从第一性原理计算软件中读取任意晶体材料的高对称点波函数的信息并自动完成对称性分析,利用得到的对称性数据通过计算“对称性指标”的方法来得到任意材料的拓扑性质。在原型设计中,本发明选取了viennaabinitiopackage作为第一性原理计算工具完成了全部代码,该方法很容易推广到其它计算软件,只需完成相应的数据接口即可。
本发明的方法大大简化了材料的拓扑性质计算,基于此,可以很容易地对已有的材料数据库的全盘扫描,以搜索出所有的拓扑材料。
虽然本发明已经通过优选实施例进行了描述,然而本发明并非局限于这里所描述的实施例,在不脱离本发明范围的情况下还包括所作出的各种改变以及变化。
- 还没有人留言评论。精彩留言会获得点赞!