过氧麦角甾醇和EGFR靶点抗体组合物及其在头颈部鳞状细胞癌上应用的制作方法
技术领域:
:本发明涉及天然产物的应用
技术领域:
,从自然菌菇提取活性成分过氧麦角甾醇及其应用,更具体涉及圆孢蘑菇中提取过氧麦角甾醇及其过氧麦角甾醇和egfr靶点抗体的组合物在头颈部鳞状细胞癌上应用。
背景技术:
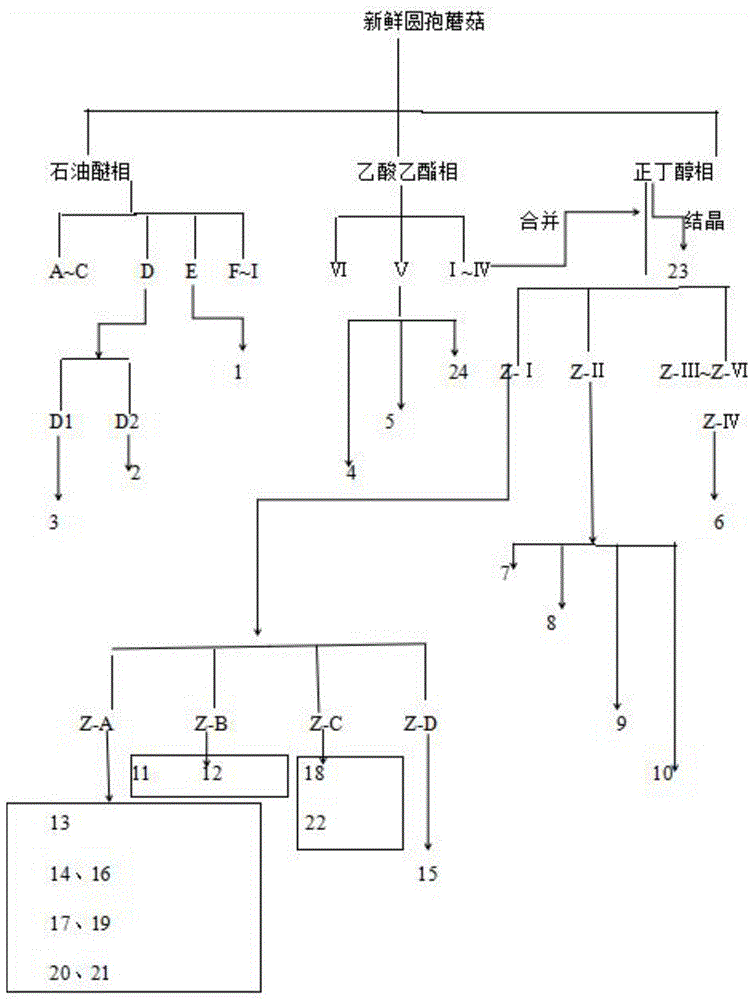
::蘑菇科(agaricaceae)系真菌伞菌目的一科,种类多,分布广泛,大多数可以食用,全世界均有分布。蘑菇科有包括蘑菇属在内的28个属。蘑菇科蘑菇属(agaricus)中常见种有圆孢蘑菇agaricusgennadii(chot.etboud)p.d.orton、双孢蘑菇agaricusbisporus(large)sing.、姬松茸agaricusblazeimurrill、紫蘑菇agaricusrubellus(gill.)sacc.、褐蘑菇agaricuscrocopeluspeck等。目前,该属的化学成分以及药理活性研究相关研究主要集中在双孢蘑菇和姬松茸两种。蘑菇科(agaricaceae)蘑菇属(agaricus)的化学成分研究从上个世纪九十年代开始,发现本属真菌的化学成分主要有挥发油、agariblazeispirol类、甾醇类、固醇类、a-1(吡咯烷酮羧酸钠)、黄酮等。可能含有神经酰胺类成分、胡萝卜苷、二萜类、三萜类、大黄素甲醚,洋芹素-7-4’-二甲醚和愈创木基丙三醇、丁烯羟酸内酯类等。圆孢蘑菇agaricusgennadii属伞菌目蘑菇科蘑菇属(agaricus),别名“圆孢托柄蘑菇”。圆孢蘑菇呈伞形,有浓郁的蘑菇香味。菌褶幼时乳白色,后渐变为粉红色至黑褐色,单个重58-350g,最大可达2-5kg。夏秋季生长于灌丛沙地、湖边芦苇丛下20-70cm的土中,在地下能开伞,单生、散生或丛生,多生长在新疆西部及西南部地区。从上世纪九十年代以来,人们已经发现同属中姬松茸的活性成分以及它的多种活性。但同属中的圆孢蘑菇agaricusgennadii的化学成分以及相关活性尚处在空白中。为开发利用这一资源,在青海省自然科学基金2018-zl-919的资助下对圆孢蘑菇的小分子化学成分进行了较系统的研究。过氧麦角甾醇是近年来从食药用真菌中发现的一个有较广泛生物活性的小分子次生代谢产物。据研究报道该化合物有促进肿瘤细胞凋亡、抗炎、抗氧化等广泛药理作用。从初步的抗菌活性实验结果来看,过氧麦角甾醇有明显的抑菌效果,尤其对金黄色葡萄球菌和苏云金芽孢杆菌抑制活性显著。过氧麦角甾醇广泛存在于食药用菌中,然而该化合物含量很低,直接从真菌中提取过氧麦角甾醇获得足够的原料目前还有很大难度。表皮生长因子受体(epidermalgrowthfactorreceptor,egfr)是一种跨膜蛋白。egfr信号转导途径在调节肿瘤细胞的生长、损伤修复和生存、新生血管生成、侵袭和转移中具有更重要的作用,同时在相当一部分的人类肿瘤中都有表达。egfr广泛存在于多种表皮来源的恶性肿瘤细胞当中:非小细胞肺癌、结直肠癌、胃癌、前列腺癌、卵巢癌、头颈部肿瘤。目前市场上以egfr为靶点的药物主要有:a)作用于受体细胞内的小分子酪氨酸激酶抑制剂(tki),如:吉非替尼、erlotinib、ekb-569、pki-166、gw-2016及ci-1033;b)作用于受体胞外区的单克隆抗体(mab)西妥昔单抗、abx-egf及emd72000等。在目前针对egfr靶点临床治疗中,一般出现单一的化疗,化疗对hnscc的治疗效果不够理想,仍有大约2/3的患者局部复发,5年生存率徘徊在30-40%。当化疗出现一定的耐药后,会有单一的单克隆抗体靶向治疗,单克隆抗体治疗hnscc病人应答率可以达到13%(7-21%),疾病控制率为45%(36-56%)与中位总存活期为5.84月。后期综合治疗以单克隆抗体联合化疗,特别是对于复发或转移或耐药的hnscc患者,当单克隆抗体同化疗联合治疗后,在临床中,表现出结果反应率有所提高,应答率可以达到16%(9-26%),疾病控制率为53%(43-63%)与中位总存活期为9.84月。但是无论哪种治疗手段,均达不到最好的治疗效果,想进一步取得好的治疗效果,化疗会带来皮疹、其他并发症或严重输液反应发生率。技术实现要素:针对上述所述问题,本发明提出了一种过氧麦角甾醇同egfr靶点抗体形成的组合物,过氧麦角甾醇和egfr靶点抗体的组合物在头颈部鳞状细胞癌上应用,该组合物对头颈部鳞状细胞癌具有良好效果。为了实现上述目的,本发明采用以下的技术方案:所述组合物为:过氧麦角甾醇及以egfr为靶点的单抗和/或双特异性抗体以及稳定剂。其中所述稳定剂选用枸橼酸。所述组合物的制备方法包括以下步骤:(1)过氧麦角甾醇的获得;(2)步骤1中的过氧麦角甾醇制备成10-200mg/ml溶液,将过氧麦角甾醇溶液与以egfr为靶点的单抗和/或双特异性抗体混合获得组合物。其中组合物中以egfr为靶点的单抗和/或双特异性抗体含量为10-1000μg/ml。其中组合物中稳定剂百分比含量0.01-0.05%。所述的抗体可以为:西妥昔单抗、abx-egf及emd72000,以及其他的以egfr为靶点的单抗和/或双特异性抗体一种或多种。过氧麦角甾醇溶液与以egfr为靶点的单抗和/或双特异性抗体在无菌条件下混合,将抗体加入过氧麦角甾醇溶液中,并在37℃水浴锅中孵育10-30min,并通过常规方法混匀该组合物,后分装该组合物于容器中,4-8℃保存。其中上述过氧麦角甾醇的制备方法如下:取新鲜、无毁坏的圆孢蘑菇子实体,切片后用90-95%乙醇浸泡一周圆孢蘑菇子实体,过滤得滤液,减压回收溶剂,滤渣加入90-95%乙醇超声提取3-5次,每次提取1-5h,减压浓缩得90-95%乙醇浸膏;用60-65%乙醇同法操作,减压浓缩得60-65%乙醇浸膏;醇提取后的圆孢蘑菇滤渣,加入蒸馏水超声提取三次,每次提取1-3h,减压浓缩得水提浸膏。其中,上述圆孢蘑菇子实体与乙醇比例为0.5-1:1-5,使乙醇完全浸泡圆孢蘑菇子实体。其中,上述减压回收条件为0.1-0.5mpa,温度25-40℃;上述超声提取条件为200-800hz,超声30-45min,温度30-50℃。将两部分醇提浸膏合并,将合并后的浸膏捏溶分散于纯水中,在分液漏斗中加入石油醚(水与石油醚比例为1:1-0.5),再将浸膏分散液加入分液漏斗中,萃取5次,萃取液合并浓缩,得石油醚部位。后再次向分液漏斗中加入乙酸乙酯(水与乙酸乙酯比例为0.5-1:1),萃取6次,萃取液合并浓缩,得乙酸乙酯部位;之后再向分液漏斗中加入正丁醇(水与正丁醇比例为0.4-1:1),萃取5次,萃取液合并浓缩,得正丁醇部位。其中,上述萃取条件为:萃取压力为20mpa~40mpa,萃取温度30℃~50℃,萃取时间为0.8h~2.5h。将上述获得的石油醚部位经中压硅胶柱快速色谱分离,石油醚-乙酸乙酯体系(100:0~0:100)梯度洗脱,洗脱液经薄层色谱检测,合并相似馏分,回收溶剂得到a~i共9个组分。组分d经中压硅胶色谱分离后,得d1和d2。d1重结晶得针状化合物3。d2经多次中压硅胶色谱与sephadexlh-20柱色谱联合分离,得白色固体化合物2。组分e经多次中压硅胶色谱与sephadexlh-20柱色谱联合分离,得黄绿色油状固体化合物1。其中,上述石油醚部位中压硅胶柱快速色谱分离条件为:以120-160μm层析硅胶为硅胶柱硅胶径高比为1:5-1:10,流速为30-90ml/min。其中,sephadexlh-20柱色谱分离条件为:以酸化甲醇为洗脱液,浓度20-40%(ph调解在3-4),流速为0.2-1.0ml/min。将上述获得的乙酸乙酯部位经中压反相c18柱快速色谱分离,水-甲醇溶剂系统(5%~100%)梯度洗脱,洗脱液按同一梯度合并,回收溶剂得到ⅰ~ⅵ共6个组分。经高效液相色谱检验后,发现ⅰ~ⅳ组分为正丁醇相中的共有组分,故合并到正丁醇相部位。ⅴ组分经中压反相c18分离后,又使用c18半制备柱分离)后得化合物4、5和24。化合物4为黄色颗粒状固体;化合物24为黄色颗粒状固体;化合物5为红色颗粒状固体。其中,上述反相c18柱色谱分离的条件为:水-甲醇溶剂系统(5%~100%)梯度洗脱;流速:1.0-3.5ml/min,柱温:25-30℃。其中,上述c18半制备柱分离条件为流动相a:1ml/l磷酸水溶液(0.2%三乙胺,磷酸调ph2.0),流动相b:乙腈;流动相a、b均需抽滤,超声10min;流动相比例:流动相a:流动相b=90:10;流速:1.0ml/min,柱温:30℃。将上述获得的正丁醇部位加入乙醇,沉淀时在容器壁上析出结晶,反复多次操作,多次重结晶得化合物23。正丁醇部位经中压反相c18分离,用水-甲醇溶液(5%~100%)梯度洗脱,洗脱液按同一梯度合并,减压回收溶剂得到z-ⅰ~z-ⅵ共6个组分。z-ⅱ组分经由mci树脂分离,用水-甲醇溶液(0%~100%)梯度洗脱,并用c18半制备柱分离纯化得化合物7、化合物8、化合物9、化合物10。z-ⅳ组分经c18半制备柱分离纯化后得化合物6。z-ⅰ组分即mci树脂纯水洗脱部位,经agilentzorbaxsb-cn全制备柱分离切段,用水-甲醇溶液(5%)等度洗脱,得z-a~z-d共4个组分。其中,上述反相c18柱色谱分离的条件为:水-甲醇溶剂系统(5%~100%)梯度洗脱;流速:1.0-3.5ml/min,柱温:25-30℃。其中,上述mci树脂分离条件为水-甲醇溶剂系统(0%~100%)梯度洗脱;流速:1.0-3.5ml/min,柱温:25-30℃。其中,上述c18半制备柱分离纯化条件为:流动相a:1ml/l磷酸水溶液(0.2%三乙胺,磷酸调ph2.0),流动相b:乙腈;流动相a、b均需抽滤,超声10min;流动相比例:流动相a:流动相b=90:10;流速:1.0-3.5ml/min,柱温:20-30℃。z-a组分经agilentzorbaxsb-cn半制备柱分离纯化,得化合物13、化合物14、化合物16、化合物17、化合物19、化合物20和21。化合物14、16、17、20和21均为白色颗粒,化合物13为白色胶状物;化合物19为纯化后结晶所得,透明针状结晶。z-b组分经agilentzorbaxsb-cn半制备柱分离纯化,得化合物11、12,化合物11、12为白色颗粒。z-c组分经agilentzorbaxsb-cn半制备柱分离纯化,得化合物18和化合物22,化合物18、22均为白色粉末。z-d组分经agilentzorbaxsb-cn半制备柱分离纯化,得化合物15,化合物15为白色粉末。其中,上述agilentzorbaxsb-cn半制备柱分离纯化条件为:流动相a:以10-30mm的na2hpo4-nah2po4缓冲溶液或k2hpo4-kh2po4缓冲溶液或tris-hcl缓冲溶液作为洗脱液a(0.2%三乙胺,磷酸调ph2.0),流动相b:乙腈;流动相a、b均需抽滤,超声10min;流动相比例:流动相a:流动相b=90:10;流速:1.0-3.5ml/min,柱温:20-30℃。采用上述的技术方案,本发明的有益效果是:1、过氧麦角甾醇同以egfr为靶点的单抗和/或双特异性抗体混合后的组合物对头颈部肿瘤hsc2杀伤作用强,且可以抑制头颈部肿瘤hsc2转移及肿瘤病灶的形成。2、稳定剂的加入可以调节抗体与组合物体系的稳定性,且过氧麦角甾醇可促进抗体挥发作用;3、组合物共同作用于受体,相较于单独疗法可大幅度降低治疗成本,获得有效的治疗效果,简化了在临床治疗过程;4、组合物对egfr靶点抗体产生耐药或者复发性和/或转移性头颈部鳞状细胞癌的患者有较好的效果,可以为临床治疗方案提供参考;5、通过该分离提取方法,快速分离圆孢蘑菇各组分,分离提取工艺简单稳定、适合工业化连续生产、产品收率较高,对于各种活性成分可以高效、简便的分离。附图说明:图1为圆孢蘑菇中活性成分的分离提纯流程图。具体实施方式:下面结合具体的实施例,对本发明实施例中的技术方案进行清楚、完整地描述。应理解,所描述的实施例是本发明一部分实施例,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施例,进一步阐述本发明。实施例1圆孢蘑菇中各活性组分粗提物制备取新鲜、无毁坏的圆孢蘑菇子实体10kg,切片后用30l95%乙醇浸泡7天(其中圆孢蘑菇子实体与乙醇比例为1:3,使乙醇完全浸泡圆孢蘑菇子实体),过滤得滤液,减压回收溶剂,滤渣加入30l95%乙醇超声提取3次,每次提取2h。减压浓缩得95%乙醇浸膏。用65%乙醇同法操作,减压浓缩得65%乙醇浸膏。醇提取后的圆孢蘑菇滤渣,加入蒸馏水30l并1800hz超声提取三次,每次提取2h。减压浓缩得水提浸膏538g。其中,上述减压回收条件为0.25mpa,温度30℃;上述超声提取条件为800hz,超声45min,温度30℃。将两部分醇提浸膏合并得84.5g,将合并后的浸膏84.5g捏溶分散于500ml纯水中,在2000ml分液漏斗中加入石油醚(水与石油醚比例为1:1),再将浸膏分散液加入分液漏斗中,萃取5次,萃取液合并浓缩,得石油醚部位10.0g。再领取2000ml分液漏斗,向分液漏斗中加入乙酸乙酯(水与乙酸乙酯比例为1:1),再将浸膏分散液加入分液漏斗中,萃取6次,萃取液合并浓缩,得乙酸乙酯部位6.0g。再领取2000ml分液漏斗,向分液漏斗中加入正丁醇(水与正丁醇比例为1:1),再将浸膏分散液加入分液漏斗中萃取5次,萃取液合并浓缩,得正丁醇部位60.0g。其中,上述萃取条件为:萃取压力为40mpa,萃取温度35℃,萃取时间为2h。所述圆孢蘑菇中活性成分的分离提纯流程图见图1。实施例2正丁醇部位组分分离纯化将上述实施例1获得的正丁醇部位60.0g加入乙醇,沉淀时在容器壁上析出结晶,反复多次操作,多次重结晶得30mg的化合物23。正丁醇部位经中压反相c18分离用水-甲醇溶液(5%~100%)梯度洗脱,洗脱液按同一梯度合并,减压回收溶剂得到z-ⅰ~z-ⅵ共6个组分。z-ⅱ组分经由mci树脂分离,用水-甲醇溶液(0%~100%)梯度洗脱,并用c18半制备柱分离纯化得4mg的化合物7、5mg的化合物8、4mg的化合物9、5mg的化合物10。z-ⅳ组分经c18半制备柱分离纯化后得5mg的化合物6。z-ⅰ组分即mci树脂纯水洗脱部位,经agilentzorbaxsb-cn全制备柱分离切段,用水-甲醇溶液(5%)等度洗脱,得z-a~z-d共4个组分。其中,上述反相c18柱色谱分离的条件为:水-甲醇溶剂系统(5%~100%)梯度洗脱;流速:1.5ml/min,柱温:30℃。其中,上述mci树脂分离条件为水-甲醇溶剂系统(0%~100%)梯度洗脱;流速:1.5ml/min,柱温:30℃。其中,上述c18半制备柱分离纯化条件为:流动相a:1ml/l磷酸水溶液(0.2%三乙胺,磷酸调ph2.0),流动相b:乙腈;流动相a、b均需抽滤,超声10min;流动相比例:流动相a:流动相b=90:10;流速:1.5ml/min,柱温:30℃。aa-a组分经agilentzorbaxsb-cn半制备柱分离纯化,得化合物4mg的13、100mg的化合物14、5mg的化合物16、4mg的化合物17、15mg的化合物19、4mg的化合物20和3mg的21。化合物14、16、17、20和21均为白色颗粒,化合物13为白色胶状物;化合物19为纯化后结晶所得,透明针状结晶。z-b组分经agilentzorbaxsb-cn半制备柱分离纯化,得化合物4mg的11、5mg的12,化合物11、12为白色颗粒。z-c组分经agilentzorbaxsb-cn半制备柱分离纯化,得5mg的化合物18和4mg的化合物22,化合物18、22均为白色粉末。z-d组分经agilentzorbaxsb-cn半制备柱分离纯化,得4mg的化合物15,化合物15为白色粉末。其中,上述agilentzorbaxsb-cn半制备柱分离纯化条件为:流动相a:以15mm的na2hpo4-nah2po4缓冲溶液或k2hpo4-kh2po4缓冲溶液或tris-hcl缓冲溶液作为洗脱液a(0.2%三乙胺,磷酸调ph2.0),流动相b:乙腈;流动相a、b均需抽滤,超声10min;流动相比例:流动相a:流动相b=90:10;流速:1.5ml/min,柱温:30℃。将分离纯化的化合物组分进行理化性质鉴定,通过常规物理检测方法:其检测方法有:eis-ms、h-nmr、c-nmr。其检测方法同药典2015版检测方法。检测结果如下:化合物6麦角甾醇(ergosterol)白色粉末;ei-msm/z=428[m-h2o],410[m-2h20],382[m-2h2o-co].1h-nmr(cd3od,500mhz):5.21(2hm,h-22,23),5.05(1h,brs,h-7),3.88(2h,m,h-3,6),1.05(3h,s,h-19),1.03(3h,d,j=6.7hz,h-21),0.93(3h,d,j=6.9hz,h-28),0.86(3h,d,j=6.8hz,h-27),0.84(3h,d,j=6.8hz,h-26),0.61(3h,s,h-18)。13c-nmr(dept)谱给出了28个碳的信号,分别为6个甲基、7个亚甲基、10个次甲基和5个季碳。其中四个碳信号在δc78.6,75.8,71.2,67.9,表明它们与氧连接。在δc143.3,137.0,133.3,122.0的四个碳信号表明有两个双键。以上数据与麦角甾醇的数据基本一致,故鉴定化合物6为麦角甾醇。化合物7过氧麦角甾醇(peroxy-ergosterol)白色粉末。1h-nmr(500mhz,cdcl3)δ:6.50(1h,d,j=8.5hz,h-7),6.24(1h,d,j=8.5hz,h-6),5.22(1h,dd,j=7.6,15.3hz,h-23),5.14(1h,dd,j=8.3,15.5hz,h-22),3.92(1h,m,h-3),1.25(3h,s,me-19),1.00(3h,d,j=6.7,me-21),0.91(3h,d,j=6.9hz,me-28),0.88(3h,s,me-18),0.83(3h,d,j=6.8hz,me-26),0.82(3h,d,j=6.8hz,me-27)。13c-nmr(125mhz,cdcl3)δ:34.7(t,c-1),30.1(t,c-2),66.4(d,c-3),36.9(t,c-4),82.2(s,c-5),135.4(d,c-6),130.7(d,c-7),79.4(s,c-8),51.1(d,c-9),36.9(s,c-10),23.4(t,c-11),39.4(t,c-12),44.6(s,c-13),51.8(d,c-14),20.6(t,c-15),28.6(t,c-16),56.2(d,c-17),12.9(q,c-18),18.2(q,c-19),39.4(d,c-20),20.9(q,c-21),135.2(d,c-22),132.3(d,c-23),42.8(d,c-24),33.1(d,c-25),20.0(q,c-26),19.6(q,c-27),17.6(q,c-28);以上数据与过氧麦角甾醇的数据一致,故鉴定化合物7为过氧麦角甾醇。化合物8macrosphelidea白色粉末,esi-msm/z:365.2[m+na]+,341.2[m-h]-,由此推断相对分子质量为342,结合氢谱和碳谱,可以推得分子式为c16h22o8,1h-nmr(cdcl3):6.88(1h,dd,j=15.6,1.5hz,h-7),6.83(1h,dd,j=15.6,1.5hz,h-13),6.02(1h,dd,j=15.6,1.5hz,h-12),6.01(1h,dd,j=15.6,1.5hz,h-6),5.35(1h,m,h-3),4.93(1h,q,j=6.3hz,h-9),4.82(1h,q,j=6.3hz,h-15),4.18(1h,m,h-8),4.09(1h,m,h-14),3.57(2h,brs,8-oh,14-oh),2.58(2h,dd,j=7.2,2.4hz,2-h2),1.40(3h,d,j=6.6hz,9-ch3),1.33(3h,d,j=6.3hz,15-ch3),1.29(3h,d,j=6.6hz,3-ch3);13c-nmr(cdcl3),170.1(c-1),165.7(c-11),164.9(c-5),146.4(13-ch),145.6(7-ch),122.5(6-ch),122.1(12-ch),74.5(9-ch),74.4(8-ch),73.6(15-ch),72.8(14-ch),67.7(3-ch),40.9(2-ch2),19.6(3-ch3),17.8(9-ch3),17.7(15-ch3);以上数据与macrosphelidea的数据一致,故鉴定化合物8为macrosphelidea。化合物9腺苷(a-denosine)白色粉末,mp.230~232℃;1h-nmr(dmso-d6,500mhz)δ:3.58(1h,d,j=10.8hz,h-5′),3.67(1h,d,j=11.4hz,h-5′),5.88(1h,d,j=9.6hz,h-1′),7-33(2h,brs,j=1.0hz,nh2),8.14(1h,s,h-8),8.34(1h,s,h-2);13cnmr(dmso-d6,125mhz)δ:152.3(c-2),149.0(c-4),119.3(c-5),156.1(c-6),139.9(c-8),87.9(c-1′),73.4(c-2′),70.6(c-3′),85,8(c-4′),61.6(c-5′),156.1(c-6′),以上波谱数据及理化特征与腺苷一致,故鉴定该化合物9为腺苷(a-denosine)。化合物10对羟基苯甲酸乙酯(4-hydroxyethylbenzoate)无色油状物;易溶于丙酮、氯仿和甲醇,难溶于水。esimsm/z=167[m+h]+;1h-nmr(me2co-d,6,500hz)δ:7.87(2h,d,j=9.0hz,h-2,6),6.90(2h,d,j=9.0hz,h-3,5),4.22(2h,q,j=7.0hz,h2-1′),1.31(3h,t,j=7.0hz,h3-2′)。以上数据与对羟基苯甲酸乙酯的数据一致,故鉴定化合物10为对羟基苯甲酸乙酯。化合物12对羟基苯甲醛(p-hydroxybenzaldehyde)无色油状物;易溶于丙酮、氯仿和甲醇,难溶于水;esimsm/z=123[m+h]+;1h-nmr(me2co-d6,500hz,)δ:9.84(1h,s,h-7),7.79(2h,d,j=9.0hz,h-2,6),7.00(2h,d,j=8.5hz,h-3,5)。以上数据与对羟基苯甲醛的数据一致,故鉴定化合物12为对羟基苯甲醛。化合物134-羟基-3-甲氧基-苄醇(4-hydroxy-3-methoxy-benzylalcohol)白色固体。易溶于丙酮、氯仿和甲醇,不溶于水;esimsm/z=155[m+h]+;1h-nmr(me2co-,d,6,500hz)δ:7.53(1h,s,4-oh),6.84(1h,d,j=1.5hz,h-2),6.85(1h,d,j=8.0hz,h-5),6.75(1h,dd,j=1.5,8.0hz,h-6),4.47(2h,d,j=6.5hz,h2-7),4.03(1h,t,j=6.5hz,7-oh),3.80(3h,s,-ome);13c-nmr(me2co-,d,6,125hz)δ:136.1(c-1),112.2(c-2),147.3(c-或c-4),147.0(c-或c-4),114.7(c-5),118.5(c-6),64.4(c-7),56.3(-ome)。以上数据与4-羟基-3-甲氧基-苄醇的数据一致,故鉴定化合物13为4-羟基-3-甲氧基-苄醇。化合物145α,6α-epoxy-(22e,24r)-ergosta-8(14),22-diene-3β,7α-diol白色粉末;[α]hresims:m/z=451.3167[m+na]+(calcdforc28h44nao3,451.3183);1h-nmr(600mhz,cdcl3)δ:5.21(2h,m,h-22,23),4.42(1h,d,j=6.3hz,h-7),3.91(1h,m,h-3),3.14(1h,d,j=3.5hz,h-6),1.02(3h,d,j=6.7hz,h-21),0.92(3h,d,j=6.8hz,h-28),0.87(6h,s,h-18,19),0.84(3h,d,j=6.8hz,h-27),0.82(3h,d,j=6.8hz,h-26);13c-nmr(150mhz,cdcl3)δ:152.75(c-14),135.41(c-22),132.44(c-23),125.38(c-8),68.87(c-3),67.92(c-5),65.25(c-7),61.49(c-6),57.02(c-17),43.15(c-13),43.01(c-24)39.78(c-20),39.39(c-4),38.92(c-9),36.78(c-12),36.01(c-10),33.27(c-25),32.38(c-1),31.31(c-2),27.31(c-16),25.13(c-15),21.40(c-21),19.82(c-27),19.17(c-26),18.24(c-11),17.77(c-18,28),16.70(c-19)。以上数据与5α,6α-epoxy-(22e,24r)-ergosta-8(14),22-diene-3β,7α-diol的数据一致,故鉴定化合物14为5α,6α-epoxy-(22e,24r)-ergosta-8(14),22-diene-3β,7α-diol。化合物15对羟基苯甲醇(p-hydroxybenzylalcohol)白色针晶(甲醇);易溶于丙酮、氯仿和甲醇,不溶于水;(-)-esimsm/z=283[m-h]-;1h-nmr(500mhz,dmso-d6)δ:9.31(1h,s,oh),7.25(2h,d,j=8.0hz,h-2,6),6.89(2h,d,j=8.0hz,h-3,5),4.76(1h,s,oh),4.36(2h,s,h-7)。以上数据与对羟基苯甲醇的数据对照基本一致,故鉴定化合物15为对羟基苯甲醇。化合物16对羟基苯甲酸(p-hydroxybenzoicacid)白色片状结晶(丙酮),易溶于氯仿和甲醇,不溶于水(-)-esimsm/z=137[m-h]-;1h-nmr(500mhz,me2co-d6)δ:7.68(2h,d,j=9.0hz,h-2,6),6.79(2h,d,j=9.0hz,h-3,5)。13c-nmr(125mhz,me2co-d6)δ:168.3(cooh),162.1(c-4),132.1(c-2,6),122.2(c-1),114.9(c-3,5)。以上数据与对羟基苯甲酸的数据一致,故鉴定化合物16为对羟基苯甲酸。化合物17对羟基苄基甲基醚(p-hydroxybenzylmethylether)白色片状结晶(丙酮),易溶于氯仿和甲醇,不溶于水。(-)-esimsm/z=137[m-h]-;1h-nmr(500mhz,dmso-d6)δ:9.25(1h,oh),7.17(2h,d,j=8.0hz,h-2,6),6.69(2h,d,j=8.0hz,h-3,5),4.19(2h,s,h-7),3.18(3h,s,ome)。以上数据与对羟基苄基甲基醚的数据一致,故鉴定化合物17为对羟基苄基甲基醚。化合物18对羟基苄基乙基醚(p-hydroxybenzylethylether)白色片状结晶(丙酮),易溶于氯仿和甲醇,不溶于水;(+)-esimsm/z=153[m+h]+;1h-nmr(300mhz,dmso-d6)δ:9.29(1h,oh),7.09(2h,d,j=8.4hz,h-2,6),6.80(2h,d,j=8.4hz,h-3,5),4.31(2h,s,h-7),3.39(2h,q,j=6.9hz,och2ch3),1.11(3h,t,j=6.9hz,och2ch3)。以上数据与对羟基苄基乙基醚的数据一致,故鉴定化合物18为对羟基苄基乙基醚。化合物19对甲氧基苯甲酸(p-hydroxybenzoicacid)白色片状结晶(丙酮),易溶于氯仿和甲醇,难溶于水。(-)-esimsm/z=151[m-h]-;1h-nmr(500mhz,dmso-d6)δ:7.87(2h,d,j=8.5hz,h-2,6),6.77(2h,d,j=8.5hz,h-3,5),3.74(3h,s,ome)。以上数据与对甲氧基苯甲酸的数据一致,故鉴定化合物19为对甲氧基苯甲酸。化合物208-甲氧基-1-萘酚(8-methoxynaphthalene-1-ol)白色粉末,esi-msm/z=175[m+h]+.1h-nmr(cdc13,500mhz)δ:9.31(1h,s,1-oh),7.41(1h,d,j=8.3hz,h-4),7.36~7.28(3h,m,h-3,h-5,h-6)6.87(1h,d,j=7.5hz,,h-7),6.78(1h,d,j=7.7hz,h-2)4.06(3h,s,8-och3);13c-nmr(cdcl3,125mhz)δ:156.2(c-8)154.5(c-1)136.8(c-4a)127.7(c-3)125.6(c-6),121.9(c-5)118.8(c-4)115.1(c-8a)110.4(c-2)103.9(c-7),56.1(8-och3)。以上数据与8-甲氧基-1-萘酚基本一致,故鉴定化合物20为8-甲氧基-1-萘酚。化合物22没食子酸(gallicacid)白色针晶;ei-msm/z(%):171[m+h]+;1h-nmr(500mhz,cdcl3):7.15(2h,s,h-2,6),7.60(2h,s,3,5-oh),7.28(1h,s,4-oh),12.10(1h,s,1-cooh);13c-nmr(100mhz,cdcl3):123.1(s,c-1),114.3(d,c-2,6),146.0(s,c-3,5),139.4(s,c-4),170.6(s,cooh)。数据与过氧麦角甾醇的基本一致,故鉴定化合物22为没食子酸。化合物233,4,5-三羟基-苯甲醛(3,4,5-trihydroxybenzaldehyde)白色无定型粉末(甲醇)。1h-nmr(cd3od,500mhz)δ(ppm):9.55(1h,s,-cho),7.27(2h,s,h-2,6);13c-nmr(cd3od,100mhz)δ(ppm):192.8(-cho),149.9(c-3,5),145.2(c-4),128.0(c-1),108.3(c-2,6)。以上数据与3,4,5-三羟基-苯甲醛一致,故鉴定化合物23为3,4,5-三羟基-苯甲醛。实施例3组合物对头颈部肿瘤hsc2细胞的杀伤活性细胞组合物的制备:将实施例2分离纯化提取的过氧麦角甾醇制备成50mg/ml的溶液,后将以egfr为靶点的单抗和/或双特异性抗体,优选西妥昔单抗溶液,缓慢加入过氧麦角甾醇溶液中,无菌密封环境下将混合物在37℃水浴中孵育20min,同时混合物按照常规方法均匀混合,必须保证无菌操作。其中西妥昔单抗溶液含有稳定剂枸橼酸,且使西妥昔单抗、枸橼酸混合于过氧麦角甾醇溶液中的终浓度或含量分别为500μg/ml、0.02%。实验组别:空白对照组:生理盐水;组合物组:过氧麦角甾醇与西妥昔单抗及稳定剂(500μg/ml西妥昔单抗、0.02%稳定剂);单抗组:西妥昔单抗及稳定剂(500μg/ml西妥昔单抗、0.02%稳定剂);过氧麦角甾醇组:50mg/ml的过氧麦角甾醇溶液。实验方法:取对数生长期的头颈部肿瘤hsc2细胞,按照每孔100ul(6000-8000个细胞)接种96孔板中,待细胞贴壁后每孔分别加入空白对照组、组合物组、单抗组、过氧麦角甾醇组各100ul进行试验,37℃孵育4h和8h后,加入1μg/ml的pi染料,cfse+pi双阳性的细胞为死亡细胞,如表1所示。表1头颈部肿瘤hsc2细胞的杀伤统计结果:组别空白对照组合物组单抗组过氧麦角甾醇组杀伤率%0%78.6%33.8%2.2%除了对照组,实验各组均表现出抑制肿瘤细胞生长、杀伤细胞的作用,其中组合物组对癌细胞的抑制杀伤活性最强,可以达到近80%的杀伤率,单抗组次之,过氧麦角甾醇组有抑制杀伤癌细胞的作用,但效果较次,但其同抗体组合后能够显著达到抑制杀伤癌细胞的效果,说明过氧麦角甾醇可以促进抗体发挥作用,同抗体协同抑制杀伤癌细胞。实施例4组合物抑制头颈部肿瘤hsc2的迁移实验材料:(1)transwellchamber:24-well,8.0-μmporemembranes(corning);(2)细胞培养相关试剂:无血清培养基,10%血清培养基,pbs,0.02%edta;(3)固定液:甲醇;(4)染色液:giemsa染液;(5)封片剂:中性树胶;(6)其他:小镊子,棉棒,载玻片,盖玻片;实验组别:制备方法同实施例3组合物制备方法;空白对照组:生理盐水;组合物组:过氧麦角甾醇与西妥昔单抗及稳定剂(500μg/ml西妥昔单抗、0.02%稳定剂);单抗组:西妥昔单抗及稳定剂(500μg/ml西妥昔单抗、0.02%稳定剂);过氧麦角甾醇组:50mg/ml的过氧麦角甾醇溶液。实验过程:(1)所有细胞培养试剂和transwellchamber放在37℃温育;(2)取培养至对数生长期的颈部肿瘤hsc2细胞,消化细胞,用pbs和rmpi1640+50vt%x-vivo15的混合培养基先后洗涤一次,用无血清培养基悬浮细胞,计数,调整浓度为2×105/ml;(3)在下室(即24孔板底部)加入600-800μl含10%自体血清的rmpi1640+50vt%x-vivo15的混合培养基,上室加入100-150μl细胞悬液,同时分别加入各实验组的物质,按照1:1进行,继续在孵箱培养24小时;(4)用镊子小心取出chamber,吸干上室液体,移到预先加入约800μl甲醇的孔中,室温固定30分钟;(5)取出chamber,吸干上室固定液,移到预先加入约800μlgiemsa染液的孔中,室温染色15-30分钟;(6)轻轻用清水冲洗浸泡数次,取出chamber,吸去上室液体,用湿棉棒小心擦去上室底部膜表面上的细胞;(7)用小镊子小心揭下膜,底面朝上晾干,移至载玻片上用中性树胶封片;(8)显微镜下取9个随机视野计数,统计结果。统计结果见表1。表2细胞迁移统计数目(个)通过上述实验统计得到:在4组实验中,组合物组的头颈部肿瘤hsc2细胞迁移最少,当过氧麦角甾醇同抗体联合作用于癌细胞后,可以有效阻止癌细胞的迁移及转移,头颈部肿瘤hsc2有一部分凋亡,其余细胞迁移性降低,因此组合物抑制头颈部肿瘤hsc2细胞转移及成瘤有效,此种方法可以降低头颈部肿瘤hsc2细胞转移及成瘤风险。实施例5组合物对体内肿瘤作用组合物的制备:将实施例2分离纯化提取的过氧麦角甾醇制备成50mg/ml的溶液,后将以egfr为靶点的单抗和/或双特异性抗体,优选西妥昔单抗溶液,缓慢加入过氧麦角甾醇溶液中,无菌密封环境下将混合物在37℃水浴中孵育20min,同时混合物按照常规方法均匀混合,必须保证无菌操作。其中西妥昔单抗溶液含有稳定剂枸橼酸,且使西妥昔单抗、枸橼酸混合于过氧麦角甾醇溶液中的终浓度或含量分别为500μg/ml、0.02%。实验组别:制备方法同实施例3组合物制备方法;空白对照组:生理盐水;组合物组:过氧麦角甾醇与西妥昔单抗及稳定剂(500μg/ml西妥昔单抗、0.02%稳定剂);单抗组:西妥昔单抗及稳定剂(500μg/ml西妥昔单抗、0.02%稳定剂);过氧麦角甾醇组:50mg/ml的过氧麦角甾醇溶液。实验方法:将处于对数生长期的头颈部肿瘤hsc2制备成单细胞悬液,调整细胞浓度1*107/ml(共48只,每组12只),预备接种体积为0.2ml/只。将小鼠置于专用固定器中固定,用1ml一次性无菌注射器将细胞悬液在小鼠腹骨皮下缓慢注射进入小鼠体内。待小鼠身体状况趋于稳定后正常饲养,待肿瘤长至100mm3左右后,通过尾静脉注射给药,药物分为四组,见实验组别设计,之后每星期尾静脉注射药物1次,每次给药0.2ml,定期观察,当出现身体瘦弱、呼吸衰竭等症状时脱臼处死小鼠,未死亡小鼠在尾静脉注射给药2个月后脱臼处死解剖,观察小鼠体内肿瘤情况。具体统计结果见表2。表3小鼠统计状况其中抑瘤率计算公式如下:抑瘤率=(空白对照组瘤重-给药组瘤重)/空白对照组瘤重*100%由表2可以看出,组合物组对头颈部肿瘤hsc2肿瘤具有强有效的敏感反应,可以有效抑制肿瘤生长,抑制率可以达到69%。且在注射药物期间,小鼠并没由出现其他并发症、身体其他不适等其他反应,其中单抗组和过氧麦角甾醇组也表现出抑制肿瘤作用,但其中过氧麦角甾醇组抑制作用较小,从而从间接反映出过氧麦角甾醇组可以有效促进抗体发挥抑制肿瘤生长的作用。以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1