识别拷贝数异常的方法与流程
背景技术:
本公开总体上涉及检测一基因组中的拷贝数变化,并且更具体地涉及检测可能由于实体肿瘤组织的存在而引起的拷贝数异常。
体细胞肿瘤组织中拷贝数变化的拷贝数异常(copynumberaberrations,cnas),在许多疾病例如癌症的病因中起重要作用。cnas包括例如基因组区域的扩增及缺失。测序技术的最新进展使得能够表征包括cnas在内的多种基因组特征。这引领了从次世代测序(next-generationsequencing,ngs)数据检测cnas的生物信息学方法的发展。
然而,一个体的基因组中cnas的准确鉴定可能会与一个体中存在的其他变化混淆。例如,可能无法指示一疾病的其他拷贝数变异(copynumbervariations,cnvs),例如非肿瘤细胞中的拷贝数变化,通常可能被错误地识别为与疾病相关的cna。需要一种准确地鉴定源自一体细胞肿瘤来源的cnas,同时消除干扰因子,例如存在源自一非肿瘤来源的cnvs的方法。
技术实现要素:
本文所述的实施方式涉及一种识别在源于游离dna(cell-freedna,cfdna)的序列读数中检测到的拷贝数事件的来源的方法。一拷贝数事件的一来源可以是生殖系来源(例如,生殖系细胞中存在的一拷贝数变异);一体细胞非肿瘤来源(例如,源于一血细胞群系的细胞的一拷贝数变异);或一体细胞肿瘤来源(例如,源于实体肿瘤细胞的一拷贝数异常)。通过识别一拷贝数事件的一来源,可以筛选出并去除与肿瘤无关的拷贝数事件。这增加了一拷贝数异常辨认者(caller)的特异性,并且有利于癌症的早期检测等应用。
从一测试样本中提取游离dna以及基因组dna(genomicdna,gdna)并进行测序(例如,使用全外显子组或全基因组测序),以获得序列读数。分别对cfdna序列读数以及gdna序列读数进行分析,以识别每个对应样本中可能存在的一个或多个拷贝数事件。在此,源于cfdna的拷贝数事件的来源可以是一生殖系来源、体细胞非肿瘤来源或体细胞肿瘤来源中的任何一个。源于gdna的拷贝数事件的来源可以是一生殖系来源或一体细胞非肿瘤来源。因此,在cfdna中检测到但在gdna中未检测到的拷贝数事件很容易归因于一体细胞肿瘤来源。
所描述的方法的实施方式包括在一基因组的所有箱执行一箱级分析(例如,箱的数量为50至1000千碱基)。对于每个样本,序列读取计数被分类为所有基因组中的各个箱。每个箱中的总序列读取计数经过归一化处理,以产生由于处理条件而引起的非生物偏差。这些非生物偏差可以包括:处理偏差(例如:鸟嘌呤及胞嘧啶含量偏差以及可图谱化偏差);一箱的预期序列读取计数(例如,一些箱可能自然导致比其他箱更高的序列读取计数);一箱的预期方差(例如,一些箱可能比其他箱噪音大);以及样本的方差(例如,一些样本可能比其他样本噪音大)。通过对箱的序列读取计数进行归一化,以产生非生物偏差,具有与预期不同的归一化序列读取计数的箱指示一拷贝数事件。以下将这样的箱称为统计显着的箱。
所描述的方法的实施方式还包括对基因组中的区段进行一区段级分析。每个区段包括所有基因组中的一个或多个箱,并且被产生为使得彼此相邻的区段具有彼此显着不同的区段序列读取计数。对每个区段的区段序列读取计数进行归一化,以产生非生物偏差,因此,具有与预期不同的归一化序列读取计数的区段指示一拷贝数事件。以下将这样的区段称为统计显着的区段。
将从cfdna样本中识别出的统计显着的箱以及统计显着的区段相比较于gdna样本中的对应的箱以及区段。此比较使得能够识别拷贝数事件的一来源,所述拷贝数事件由cfdna样本中的统计显着的箱以及统计显着的区段识别出。具体地,若cfdna样本的一统计显着的箱或区段也相应地是gdna样本的一统计显着的箱或区段,则拷贝数事件很可能是源自一非肿瘤来源的一拷贝数变异。换句话说,一生殖系事件或一体细胞非肿瘤事件都可能导致在cfdna以及gdna样本中观察到拷贝数事件。相反,若自cfdna样本中的一统计显着的箱或区段与自gdna样本中的一统计显着的箱或区段不对应,则拷贝数事件很可能是一拷贝数异常。换句话说,一体细胞肿瘤事件可能导致了cfdna样本中观察到的拷贝数事件,但gdna样本中没有。
通过识别一拷贝数事件的来源,可以筛选出拷贝数变异,而拷贝数异常可以被保持,并进一步分析。因此,识别出的拷贝数异常可以进一步被分析应用,例如癌症的早期检测。
附图说明
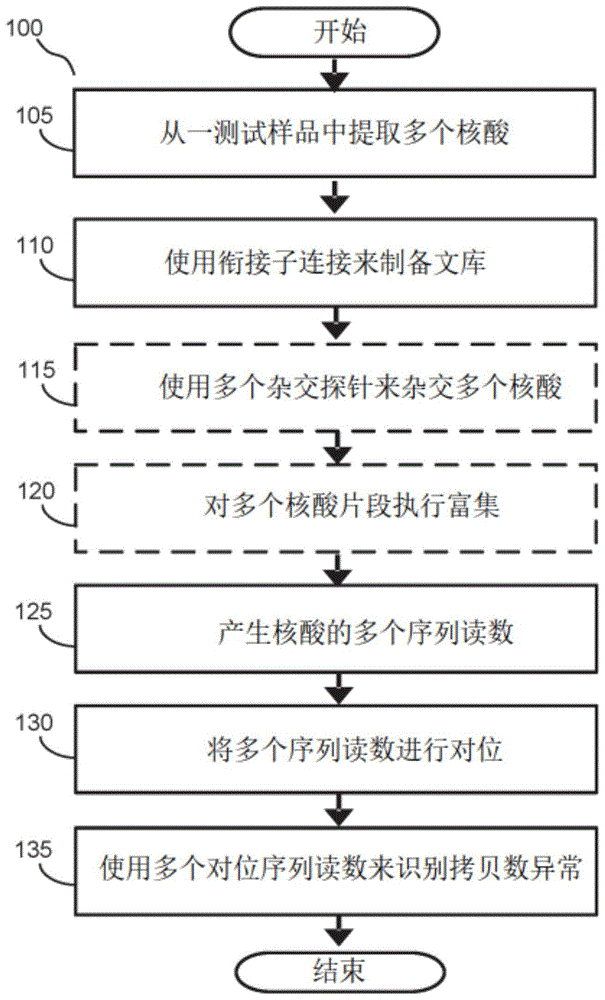
图1是根据一实施方式的用于处理从一个体获得的一测试样本以识别一拷贝数异常的一示例流程图;
图2a是根据一实施方式的用于识别在一cfdna样本中识别的一拷贝数事件的一来源的一示例流程图;
图2b是根据一实施方式的一示例流程图,其描述了用于识别源于cfdna以及gdna样本的统计显着的箱及区段的分析;
图2c描绘了根据一实施方式的一示例数据库,所述示例数据库存储用于识别一拷贝数事件的一来源的特征;
图3a是根据一实施方式的与一参考基因组的箱有关的序列读数的一示例描绘;
图3b是根据一实施方式的一示例图表,描绘了一基因组的所有不同箱的预期以及观察到的序列读取计数;
图4a及图4b描绘了分别从一乳腺癌受试者获得的一cfdna样本以及一gdna样本的一基因组的所有箱的箱得分;
图5是一图表,描绘了相对于图4a中所示的cfdna样本的对应的箱得分的图4b中所示的gdna样本的箱得分的分布;
图6a及图6b描绘了分别从一非癌症个体获得的一cfdna样本以及一gdna样本确定的一基因组的所有箱的箱得分;
图7是一图表,描绘了相对于图6a中所示的cfdna样本的对应的箱得分的图6b中所示的gdna样本的箱得分的分布;
图8a及图8b描绘了分别从一非癌症个体获得的一cfdna样本以及一gdna样本确定的一基因组的所有箱的箱得分;以及
图9是一图表,描绘了相对于图8a中所示的cfdna样本的对应的箱得分的图8b中所示的gdna样本的箱得分的分布。
具体实施方式
附图以及以下描述仅通过说明的方式涉及优选实施方式。应当注意,从下面的讨论中,本文公开的结构及方法的替代实施方式将容易地被认为是可以采用的可行替代方案,而不背离所要求保护的原理。
现在将详细参照几个实施方式,其示例在附图中示出。要注意的是,在可行的情况下,附图中可以使用相似或类似的附图标记,并且可以指示相似或类似的功能。例如,在一附图标记后的一字母,例如:“bin320a”,表示文本具体指代具有所述特定附图标记的元件。在文本中没有一随后的字母的一附图标记,例如:“bin320”,指代附图中具有所述附图标记的任何或所有元件(例如,文本中的“bin320”是指附图中的附图标记“bin320a”及/或“bin320b”)。
术语“个体”是指一人类个体。术语“健康个体”是指假定没有癌症或疾病的个体。术语“癌症受试者”是指已知患有或潜在患有癌症或疾病的个体。
术语“序列读数”是指从获自一个体的样本中读取的核苷酸序列。序列读数可通过本领域已知的各种方法获得。
术语“游离核酸”、“游离dna”或“cfdna”是指在一个体体内(例如:血流)中循环并且源于一个或多个健康细胞及/或源于一个或多个癌细胞的核酸片段。
术语“基因组核酸”、“基因组dna”或“gdna”是指包括源于一个或多个健康(例如,非肿瘤)细胞的染色体dna的核酸。在各种实施方式中,可以从源于一血细胞群系的一细胞(例如:一白细胞)提取gdna。
术语“拷贝数异常(copynumberaberrations)”或“cans”是指体肿瘤细胞中拷贝数的变化。例如,cnas可以指一实体肿瘤中的拷贝数变化。
术语“拷贝数变异(copynumbervariations)”或“cnvs”是指源于非肿瘤细胞中的生殖系细胞或体细胞的拷贝数变化。例如,cnvs可以指由于克隆性造血而引起的白细胞的拷贝数变化。
术语“拷贝数事件”是指一拷贝数异常与一拷贝数变异之一或两者。
识别拷贝数异常的一来源的方法
来自样本的生成序列读数的一般处理步骤:
图1是根据一实施方式的一示例性流程方法100,用于处理从一个体获得的一测试样本,以识别一拷贝数异常。在步骤105,从一测试样本中提取核酸。在一个实施方式中,所述测试样本可以来自已知患有或怀疑患有癌症的一癌症受试者。所述测试样本可以是选自于由血液、血浆、血清、尿液、粪便及唾液样本所组成的群组的一样本。或者,所述测试样本可以包括选自于由全血、一血液成分、一组织活检、胸膜液、心包液、脑脊髓液及腹膜液所组成的群组的一样本。根据一些实施方式,所述测试样本包含游离核酸(例如,游离dna)。在一些实施方式中,测试样本中的游离核酸源于一个或多个健康细胞及一个或多个癌细胞。根据一些实施方式,所述测试样本包括基因组dna(例如,gdna),其中所述测试样本中的gdna包括获自一个或多个健康细胞的染色体dna。在一些实施方式中,所述一个或多个健康细胞来自一健康细胞,例如一血液群系。例如,所述一个或多个健康细胞可以是白细胞。
在各种实施方式中,所述测试样本包括cfdna及gdna,因此,对所述测试样本进行处理,以提取cfdna及gdna。通常,本领域中任何已知的方法均可用于提取dna。例如,可以使用一个或多个已知的市售方案或试剂盒,例如qiaamp循环核酸试剂盒(qiaampcirculatingnucleicacidkit)(qiagen),来提取及纯化核酸。在其他实施方式中,可以通过在一管中沉淀(pelleting)及/或沉淀(precipitating)核酸来分离核酸。在一些实施方式中,对一测试样本进行处理,以获得一cfdna样本及一gdna样本,从中可以分别提取cfdna及gdna。例如,可以将一测试样本进行离心,以分离一上清液及沉淀的细胞。所述上清液可以代表一cfdna样本,而所述沉淀的细胞可以代表gdna样本。在一些实施方式中,可以将测试样本中的核酸片段化,例如可以将样本中的基因组dna(gdna)片段化(例如,剪切的gdna样本),然后进行后续处理。
提取核酸后,可以进行多种测序方法之一。例如,被提取的核酸可用于执行一靶向测序(例如,一靶向基因组(genepanel)测序)、全外显子组测序、全基因组测序或甲基化感知测序(methylation-awaresequencing)(例如,全基因组亚硫酸盐测序)之一。
在步骤110,制备一测序文库。在文库制备过程中,衔接子,例如包括一个或多个测序寡核苷酸,用于随后的丛集(cluster)生成及/或测序(例如,用于合成测序的已知p5及p7序列(sbs)(illumina,圣地亚哥,加利福尼亚)),通过衔接子连接而连接至核酸片段的末端。在一个实施方式中,在衔接子连接期间,将分子标签(uniquemolecularidentifiers,umi)添加至提取的核酸。umis是短核酸序列(例如:4至10个碱基对),其在衔接子连接期间被添加到核酸末端。在一些实施方式中,umis是简并碱基对,其作为可用于识别从核酸获得的序列读数的一独特标签。如之后所述,在扩增过程中,umis可以与连接的核酸一起进一步复制,这提供了一种在下游分析中识别源于同一原始核酸片段的序列读数的方法。
简要地参照图1,可选地执行步骤115及120。例如,执行步骤115及120以用于靶向基因组测序及全外显子组测序。然而,对于全基因组测序,不需要执行步骤115及120。
在步骤115,使用杂交探针来富集(enrich)用于一所选核酸组的一测序文库。杂交探针可被设计为以靶向并与靶向核酸序列杂交,以拉下(pulldown)及富集靶向核酸片段,其可以提供癌症(或疾病)的存在与否、癌症状态或癌症分类(例如,癌症类型或起源组织)的信息。根据此步骤,多个杂交拉下探针(hybridizationpulldownprobes)可以用于一给定的靶向序列或基因。所述探针的长度范围可为约40至约160个碱基对(bp)、约60至约120bp或约70bp至约100bp。在一实施方式中,探针覆盖靶向区域或基因的重叠部分。对于靶向基因组测序,杂交探针被设计成靶向及拉下源于基因组中包括的特定基因序列的核酸片段。对于全外显子组测序,杂交探针被设计为靶向及拉下源于一参考基因组中的外显子序列的核酸片段。
在步骤120,富集探针核酸复合物。例如,如本领域公知的,可以将一生物素部分添加到探针的5′-末端(即,生物素化),以利于使用一链霉亲和素包被的表面(例如,链霉亲和素包被的珠子)拉下靶向探针核酸复合物。可选地,一第二装置,例如聚合酶链式反应(pcr)装置,可以用于扩增靶向核酸。
在步骤125,对核酸进行测序以产生序列读数。序列读数可以通过本领域已知的手段获得。例如,许多技术及平台直接从数百万个单个核酸(例如,例如cfdna或gdna之类的dna)分子中并行地获得序列读数。此类技术可以适合于进行任何靶向测序(例如,靶向基因组测序)、全外显子组测序、全基因组测序及甲基化感知测序(例如,全基因组亚硫酸盐测序)。
在一个实施方式中,可以使用次世代测序(nextgenerationsequencing,ngs)获得来自测序文库的序列读数。次世代测序方法包括,例如,通过合成式技术(synthesistechnology)(illumina)、焦磷酸测序(454)、离子半导体技术(iontorrent测序)、单分子实时测序(pacificbiosciences)来测序;通过连接测序(solid测序)及纳米孔测序(nanoporesequencing)(oxfordnanoporetechnologies)来测序。在一些实施方式中,测序是使用具有可逆染料终端(reversibledyeterminators)的合成式测序的大规模并行测序。在其他实施方式中,测序是连接法测序(sequencing-by-ligation)。在其他实施方式中,测序是单分子测序(singlemoleculesequencing)。在其他实施方式中,测序是双边测序(paired-endsequencing)。
在步骤130,将序列读数与参考基因组对位。通常,本领域中任何已知的方法均可用于将序列读数与一参考基因组进行对位。例如,将一序列读数的核苷酸碱基与参考基因组中的核苷酸碱基进行对位,以确定序列读数的对位位置信息。对位位置信息可包括参考基因组中与序列读数的起始核苷酸碱基以及终止核苷酸碱基相对应的一区域的一起始位置及一终止位置。对位位置信息还可包括序列读数长度,其可从起始位置及终止位置被确定。在各个实施方式中,在步骤135,获得基因组的区域的对位测序读数的一bam文件,并用于分析。
在步骤135,使用对位序列读数来识别一cna。一cna指示一体细胞肿瘤事件,可以为预测癌症的存在提供信息。在一些实施方式中,使用对位序列读数来识别一cna,所述对位序列读数是从来自单个样本例如cfdna样本中提取的核酸测序而来的。在一些实施方式中,使用对位序列读数来识别一cna,所述对位序列读数是从来自多个样本(例如:cfdna样本及gdna样本)中提取的核酸测序而来的。例如,源于一gdna样本的对位序列读数可用于识别生殖系或体细胞非肿瘤事件,使得由源于一cfdna样本的对位序列读数确定的对应事件不会被错误地解释为cnas。下面参照图2a、图2b、图3a及图3b进一步详细描述用于识别cnas的方法。
识别拷贝数异常:
图2a是根据一实施方式的一示例性流程135,用于识别在一cfdna样本中识别的一拷贝数事件的一来源。具体地,图2a描绘了图1中所示的步骤135的附加步骤,用于检测一个体中的一cna。
在步骤205,获得源于一cfdna样本的对位序列读数(以下称为cfdna序列读数)以及源于一gdna样本的对位序列读数(以下称为gdna序列读数)。
在步骤210,分析对位的cfdna序列读数以及gdna序列读数,以分别识别cfdna样本及gdna样本各自在所有参考基因组的统计显着的箱及区段。一箱包括一基因组的一系列核苷酸碱基。一区段指的是一个或多个箱。因此,将每个序列读数分类在包括与所述序列读数相对应的一系列核苷酸碱基的箱及/或区段中。基因组的每个统计显着的箱或区段包括在指示一拷贝数事件的箱或区段中分类的序列读数的一总数。通常,即使考虑可能的干扰因子,一统计显着的箱或区段的序列读取计数也与箱或区段的一预期序列读取计数显着不同,所述干扰因子的示例包括处理偏差、箱或区段中的方差或样本(例如:cfdna样本或gdna样本)中的总体噪声水平。因此,一统计显着的箱及/或统计显着的区段的序列读取计数可能指示一生物学异常,例如样本中存在一拷贝数事件。
步骤210包括一箱级分析,以识别统计显着的箱;以及一区段级分析,以识别统计显着的区段。在箱及区段级执行分析可以更准确地识别可能的拷贝数事件。在一些实施方式中,仅在箱级执行分析可能不足以获得跨越多个箱的拷贝数事件。在其他实施方式中,仅在区段级执行分析可能会产生不够精细的分析结果,无法获得大小在各个箱的数量级上的拷贝数事件。
通常,cfdna序列读数的分析及gdna序列读数的分析彼此独立地进行。在各种实施方式中,并行进行cfdna序列读数及gdna序列读数的分析。在一些实施方式中,取决于何时获得序列读数(例如,何时在步骤205中获得序列读数),在分开的时间进行cfdna序列读数及gdna序列读数的分析。现在参照图2b,其是一示例性流程,其描述了根据一实施方式的用于识别源于cfdna及gdna样本的统计显着的箱及统计显着的区段的分析。具体地,图2b描绘了图2所示的步骤210中包括的步骤。因此,可以对一cfdna样本进行步骤220至260,并且类似地,可以对一gdna样本单独进行步骤220至260。
在步骤220,确定一参考基因组的每个箱的一箱序列读取计数。通常,每个箱代表所述基因组的许多连续核苷酸碱基。一基因组可以由许多箱(例如,数百或甚至数千)组成。在一些实施方式中,每个箱中的核苷酸碱基的数量在基因组中的所有箱中是恒定的。在一些实施方式中,每个箱中的核苷酸碱基的数量对于基因组中的每个箱而言是不同的。在一个实施方式中,每个箱中的核苷酸碱基的数量在25千碱基(kb)及10,000千碱基(kb)之间。在一个实施方式中,每个箱中的核苷酸碱基的数量在50千碱基(kb)至1000千碱基(kb)之间。在一个实施方式中,每个箱中的核苷酸碱基的数量在100千碱基(kb)至500kb之间。在一个实施方式中,每个箱中的核苷酸碱基的数量在50kb至100kb之间。在一个实施方式中,每个箱中的核苷酸碱基的数量在45kb至75kb之间。在一个实施方式中,每个箱中核苷酸碱基的数量为50kb。实际上,也可以使用其他箱尺寸。
一箱的箱序列读取计数表示分类在所述箱中的序列读数的一总数。若序列读数跨越了箱中包括的核苷酸碱基的阈值数量(即:对位或映射(map)到箱),则将其分类在箱中。在一个实施方式中,分类在一箱中的每个序列读数跨越所述箱中包括的至少一核苷酸碱基。现在参照图3a,其是根据一实施方式的与一参考基因组305的箱320有关的序列读数330的示例性描绘。序列读数330a、序列读数330b及序列读数330c可各自包括不同数目的核苷酸碱基,并且可跨越一个或多个箱320。
如图3a所示,序列读数330a与一箱(例如:箱320b)中的核苷酸碱基数量相比包括更少的核苷酸碱基。在此,序列读数330a被分类在箱320b中。序列读数330b跨越在箱320c及箱320d中都包括的核苷酸碱基。因此,序列读数330b被分类在箱320c及箱320d中。序列读数330c跨越包括在箱320b、箱320c及箱320d中的核苷酸碱基。因此,序列读数330c被分类在箱320b、箱320c及箱320d的每一个中。
为了确定每个箱的箱序列读取计数,对每个箱中分类的序列读数进行量化。因此,图3a中所示的箱320a的一箱序列读取计数为零;箱320b的一箱序列读取计数为2(例如,序列读数330a及序列读数330c);箱320c的一箱序列读取计数为2(例如,序列读数330b及序列读取330c);箱320d的一箱序列读取计数为2(例如,序列读数330b及序列读数330c);并且箱320e的一箱序列读取计数为1(例如,序列读数330c)。
返回图2b,在步骤225,对每个箱的箱序列读取计数进行归一化,以去除一个或多个不同的处理偏差。通常,基于先前针对同一箱确定的处理偏差,来对一箱的箱序列读取计数进行归一化。在一个实施方式中,归一化箱序列读取计数涉及将箱序列读取计数除以代表处理偏差的一值。在一个实施方式中,归一化箱序列读取计数涉及从箱序列读取计数减去代表处理偏差的一值。箱的处理偏差的示例可包括鸟嘌呤及胞嘧啶(gc)含量偏差、可图谱化性偏差或通过一主成分分析获得的其他形式的偏差。可以从图2c中所示的处理偏差存储器270中存取一箱的处理偏差。
在步骤230,通过使用箱的预期箱序列读取计数来修改箱的箱序列读取计数,来确定每个箱的一箱得分。步骤230用于归一化观察到的箱序列读取计数,使得若特定箱在所有样本一致地具有一高序列读取计数(例如,高预期箱序列读取计数),则观察到的箱序列读取计数的归一化就产生这种趋势。可以从训练特征数据库265(见图2c)中的箱预期计数存储器280存取箱的预期序列读取计数。下面进一步详细描述每个箱的预期序列读取计数的产生。
在一个实施方式中,一箱的一箱得分可以表示为所述箱的观察到的序列读取计数与所述箱的预期序列读取计数的比率的对数。例如,箱i的箱得分bi可表示为:
在其他实施方式中,箱的箱得分可以表示为箱的观察到的序列读取计数与箱的预期序列读取计数的比率(例如:
比率的广义对数转换(generalizedlogtransformation,glog)
(例如:
比率的其他方差稳定变换(variancestabilizingtransform)。
现在参照图3b,其是根据一实施方式的的一示例流程图,描绘了一参考基因组的所有不同箱的预期及观察到的序列读取计数。具体地,图3b描绘了箱的第一组370(例如,箱n、箱n+1、箱n+2)以及箱的第二组380(例如,箱m、箱m+1、箱m+2)的观察到的及预期的序列读取计数。在各种实施方式中,第一组370中的箱可来自参考基因组的一第一区段,而第二组380中的箱可来自参考基因组的第二区段。在一些实施方式中,第一组370中的箱可以来自一第一染色体,而第二组380中的箱可以来自一不同的染色体。
在此,第一组370中的箱的观察到的序列读取计数以及预期序列读取计数可能没有显着差异。然而,第二组380中箱的观察到的序列读取计数可以明显高于箱的对应的预期读取计数。因此,第二组380中的每个箱的箱得分高于第一组370中的每个箱的箱得分。第二组380中的箱的较高的箱得分表示在箱m、箱m+1及箱m+2中观察到的序列读取计数是一拷贝数事件的结果的可能性更高。
箱的第一组370以及第二组380的不同箱得分说明了通过箱的对应的预期序列读取计数来归一化每个箱的观察到的序列读取计数的好处。具体地,在图3b所示的示例中,第一组370中的观察到的箱的序列读取计数以及第二组380中的观察到的箱的序列读取计数可能没有显着差异。通过修改观察到的序列读取计数以产生预期序列读取计数,可以识别对应于箱的第二组380的一可能的拷贝数事件。
返回图2b,在步骤235,为每个箱确定一箱方差估计。在此,所述箱方差估计表示箱的一预期方差,由表示样本中方差水平的一扩大因子进一步调整。换句话说,所述箱方差估计代表从先前训练样本确定的箱预期方差与未计入箱的预期方差的当前样本(例如:cfdna或gdna样本)的一扩大因子的组合。
举个例子,一箱i的一箱方差估计(vari)可以表示为:
vari=varexpi*isample
(2)
其中varexpi表示从先前训练样本确定的箱i的预期方差,而isample表示当前样本的扩大因子。通常,通过存取图2c所示的箱预期方差存储器290来获得一箱的预期方差(例如,varexp)。
为了确定样本的扩大因子isample,样本的一偏差被确定,并与从图2c中所示的样本变异因子存储器295中取出的样本变异因子相结合。样本变异因子是先前通过对从多个训练样本得出的数据进行拟合而得出的系数值。例如,若执行一线性拟合,则样本变异因子可以包括一斜率系数以及一截距系数。若执行更高阶拟合,则样本变异因子可以包括其他系数值。
样本的偏差表示所有样本中的箱中的序列读取计数的变异性的一测量。在一个实施方式中,样本的偏差是一成对中位绝对偏差(medianabsolutepairwisedeviation,mapd),并且可以通过分析相邻的箱的序列读取计数来计算。具体而言,mapd表示所有样本中的相邻的箱的箱得分之间的绝对值差异的中值。在数学上,mapd可以表示为:
其中bi及bi+1分别是bini(箱i)及bini+1(箱i+1)的箱得分。
通过组合样本变异因子以及样本的偏差(例如,mapd)来确定扩大因子isample。例如,一样本的扩大因子isample可以表示为:
isample=斜率*σsample+截距
(4)
在此,“斜率”及“截距”系数中的每一个都是从样本变异因子存储器295存取的样本变异因子,而σsample表示样本的偏差。
在步骤240,基于箱的箱得分及箱方差估计来分析每个箱,以确定所述箱是否为统计显着的。对于每个箱i,可以将箱的箱得分(bi)以及箱方差估计(vari)相结合,以产生箱的z得分。箱i的z得分(zi)的一示例可以表示为:
为了确定一箱是否为一统计显着的箱,将所述箱的z得分与一阈值进行比较。若所述箱的z得分大于所述阈值,则将所述箱视为统计显着的箱。相反,若箱的z得分小于所述阈值,则所述箱不被视为统计显着的箱。在一个实施方式中,若一箱的z得分大于2,则将所述箱确定为统计显着的。在其他实施方式中,若一箱的z得分大于2.5、3、3.5或4,则将所述箱确定为统计显着的。在一个实施方式中,若一箱的z得分小于-2,则将所述箱确定为统计显着的。在其他实施方式中,若一箱的z得分小于-2.5、-3、-3.5或-4,则将所述箱确定为统计显着的。统计显着的箱可指示一样本(例如,cfdna或gdna样本)中存在的一个或多个拷贝数事件。
在步骤245,产生参考基因组的区段。每个区段由参考基因组的一个或多个箱组成,并具有一统计序列读取计数。一统计序列读取计数的示例可以是一平均箱序列读取计数、一中值箱序列读取计数等。通常,参考基因组的每个产生的区段具有与一相邻区段的一统计序列读取计数不同的一统计序列读取计数。因此,一第一区段可具有与一第二相邻区段的一平均箱序列读取计数显着不同的一平均箱序列读取计数。
在各个实施方式中,参考基因组的区段的产生可以包括两个分开的阶段。第一阶段可以包括基于每个区段中箱的箱序列读取计数的差异,将参考基因组初始分割成多个初始区段。第二阶段可以包括一重新分割过程,所述过程涉及将一个或多个初始区段重组为较大的区段。在此,第二阶段考虑通过初始分割过程创建的区段的长度,以结合由于在初始分割过程中发生过度分割而导致的假阳性区段。
更具体地参照初始分割方法,所述初始分割方法的一个示例包括执行一循环二元分割算法(circularbinarysegmentationalgorithm),以基于区段内箱的箱序列读取计数将参考基因组的部分递归地分解为区段。在其他实施方式中,可以使用其他算法来执行参考基因组的初始分割。作为循环二元分割方法的一示例,所述算法识别参考基因组内的一个断裂点,从而使由断裂点形成的一第一区段包括所述第一区段中的箱的一统计箱序列读取计数,其明显不同于由断裂点形成的第二区段中的箱的统计箱序列读取计数。因此,循环二元分割过程产生许多区段,其中一第一区段内的箱的统计箱序列读计数与一第二相邻区段内的箱的统计箱序列读计数显着不同。
初始分割过程可以在生成初始区段时还考虑每个箱的箱方差估计。例如,当计算一区段中的箱的一统计箱序列读取计数时,可以为每个箱i分配一权重,所述权重取决于箱的箱方差估计(例如,vari)。在一个实施方式中,分配给一箱的权重与箱的箱方差估计的大小成反比。具有一较高的箱方差估计的一箱被分配了一较低的权重,从而减少了箱的序列读取计数对区段中箱的统计箱序列读取计数的影响。相反,分配给具有一较低的箱方差估计的一箱的一权重较高,这增加了箱的序列读取计数对区段中箱的统计箱序列读取计数的影响。
现在参照重新分割过程,它分析由初始分割过程创建的区段,并识别将被重新组合的成对的错误分离的区段。重新分割过程可以产生在初始分割过程中未被考虑的区段的一特征。作为示例,一区段的一特征可以是所述区段的长度。因此,一对错误分离的区段可以指的是相邻区段,当考虑到这对区段的长度时,它们不具有显着不同的统计箱序列读取计数。通常,较长的区段与统计相序列读取计数的一较高变异相关。因此,通过考虑每个区段的长度,最初确定为每个相邻区段的统计箱序列读取计数彼此不同的相邻区段,可以被认为是一对错误分离的区段。
所述对中错误分离的区段被组合。因此,执行初始分割以及重新分割过程导致一参考基因组的生成的区段,其考虑了由每个区段的不同长度引起的差异。
在步骤250,基于区段的一观察到的区段序列读取计数以及区段的一预期区段序列读取计数,对每个区段确定一区段得分。所述区段的一观察到的区段序列读取计数代表所述区段中分类的观察到的序列读数的总数。因此,可以通过将观察到的包括在区段中的箱的箱读取计数相加,来确定所述区段的一观察到的区段读取计数。类似地,所述预期区段序列读取计数代表在所述区段中包括的所有箱的预期序列读取计数。因此,可以通过量化在所述区段中包括的箱的预期箱序列读取计数,来计算一区段的预期区段序列读取计数。可以从箱预期计数存储器280存取在区段中包括的箱的预期读取计数。
一区段的区段得分可以表示为所述区段的区段序列读取计数与预期区段序列读取计数之比率。在一个实施方式中,一区段的区段得分可以表示为所述区段的观察到的序列读取计数与所述区段的预期序列读取计数之比率的对数。区段k的区段得分sk可表示为:
在其他实施方式中,所述区段的区段得分可以表示为以下的其中一个:
比率的平方根(例如:
比率的广义对数转换(例如:
比率的其他方差稳定变换(variancestabilizingtransform)。
在步骤255,对每个区段确定一区段方差估计。通常,所述区段方差估计值表示所述区段的序列读取计数有多偏离。在一个实施方式中,可以通过使用包括在区段中的箱的箱方差估计以及通过一区段扩大因子(isegment)进一步调整箱方差估计来确定区段方差估计。举例来说,可以将一区段k的区段方差估计表示为:
vark=平均值(vari)*isegment
(7)
其中平均值(vari)表示区段k中包含的箱i的箱方差估计的平均值。可以通过存取箱期望方差存储器290来获得箱的箱方差估计。
区段扩大因子产生了区段级上偏差的增加,其通常比箱级上的偏差更高。在各种实施方式中,区段扩大因子可以根据区段的大小来缩放。例如,由大量箱组成的较大的区段可被分配一区段扩大因子,所述区段扩大因子大于分配给由较少箱组成的一较小区段的一区段扩大因子。因此,区段扩大因子产生了在较长区段中出现的更高水平的偏差。在各种实施方式中,分配给一第一样本的一区段的区段扩大因子不同于分配给一第二样本的相同区段的区段扩大因子。在各个实施方式中,可以预先根据经验确定具有一特定长度的一区段的区段扩大因子isegment。
在各个实施方式中,可以通过分析训练样本来确定每个区段的区段方差估计。例如,一旦在步骤245中产生了区段,就分析来自训练样本的序列读数,以确定每个产生的区段的一预期区段序列读取计数以及每个区段的一预期区段方差估计。
每个区段的区段方差估计可以代表为使用由样本扩大因子调整的训练样本确定的每个区段的预期区段方差估计。例如,一区段k的区段方差估计(vark)可以表示为:
其中varexpk是区段k的预期区段方差估计,而isample是上述相对于步骤235及式(4)的样本扩大因子。
在步骤260,基于区段的区段得分以及区段方差估计来分析每个区段,以确定所述区段是否为统计显着的。对于每个区段k,可以将区段的区段得分(sk)以及区段方差估计(vark)组合起来,以产生所述区段的z得分。区段k的z得分(zk)的一示例可以表示为:
为了确定一区段是否为一统计显着的区段,将所述区段的z得分与一阈值进行比较。若所述区段的z得分大于所述阈值,则将所述区段视为一统计显着的区段。相反,若所述区段的z得分小于所述阈值,则所述区段不被视为一统计显着区段。在一个实施方式中,若一区段的z得分大于2,则将所述区段确定为统计显着的。在其他实施方式中,若一区段的z得分大于2.5、3、3.5或4,则将所述区段确定为统计显着的。在一些实施方式中,若一区段的z得分小于-2,则将所述区段确定为统计显着的。在其他实施方式中,若一区段的z得分小于-2.5、-3、-3.5或-4,则将所述区段确定为统计显着的。统计显着的区段可以指示一样本(例如,cfdna或gdna样本)中存在的一个或多个拷贝数事件。
返回图2a,在步骤215,确定源于cfdna样本的统计显着的箱(例如,在步骤240确定)及/或统计显着的区段(例如,在步骤260确定)所指示的一拷贝数事件的一来源。具体地,将cfdna样本的统计显着的箱与gdna样本的对应的箱进行比较。另外,将cfdna样本的统计显着的区段与gdna样本的对应的区段进行比较。
cfdna样本的统计显着的区段及箱与gdna样本的对应的区段及箱之间的比较产生关于cfdna样本的统计显着的区段及箱是否与gdna样本的对应的区段及箱对位的一确定。如下文中所使用的,对位的区段及箱是指区段或箱在cfdna样本以及gdna样本中均统计显着的事实。相反,未对位的区段或箱是指区段或箱在一个样本(例如,cfdna样本)中是统计显着的,而在另一个样本(例如,gdna样本)中是统计不显着的。
通常,若cfdna样本的统计显着的箱以及统计显着的区段与gdna样本的对应的也是统计显着的箱及区段对位,则表明cfdna样本以及gdna样本中都存在相同的拷贝数事件。因此,拷贝数事件的来源很可能是由于非肿瘤事件(例如,一生殖系或体细胞非肿瘤事件)引起的,并且所述拷贝数事件很可能是一拷贝数变异。
相反,若cfdna样本的统计显着的箱以及统计显着的区段与对应的gdna样本的统计不显着的箱以及区段对位,则表明拷贝数事件存在于cfdna样本中,但不存在于gdna样本中。在这种情况下,cfdna样本中拷贝数事件的来源是由于体细胞肿瘤事件引起的,并且所述拷贝数事件是一拷贝数异常。
识别在cfdna样本中检测到的一拷贝数事件的来源,有利于筛选出由于一生殖系或体细胞非肿瘤事件引起的拷贝数事件。这提高了正确识别由于一实体肿瘤的存在而导致的拷贝数异常的能力。
确定训练特征:
图2c描绘了根据一实施方式的一示例数据库265,所述示例数据库265存储用于识别一拷贝数事件的一来源的特征。具体地,训练特征数据库265可以包括一处理偏差存储器270、一箱预期计数存储器280、一箱预期方差存储器290以及一样本变异因子存储器295。每个存储器270、280、290及295可以包括从训练样本中得出的特征。在各个实施方式中,训练样本获自一健康个体。在一些实施方式中,一训练样本包括一训练cfdna样本以及训练gdna样本。每个训练cfdna样本及训练gdna样本可以根据图1所示的步骤105至130进行处理,以产生对位的cfdna序列读数以及对位的gdna序列读数。如下所述,从训练样本中获取的对位的cfdna序列读数以及对位的gdna序列读数可用于确定存储在训练特征数据库265中的特征。
处理偏差存储器270包括代表参考基因组的每个箱的处理偏差的一测量的特征。在一个实施方式中,对于参考基因组的每个箱,处理偏差存储器270可以包括:(1)gc含量偏差;(2)可图谱化性偏差;以及(3)用于确定从一降维分析得出的一偏差的信息。一降维分析的一示例是主成分分析(principalcomponentanalysis,pca)。每个箱的附加处理偏差可以包括在处理偏差存储器270中。在各个实施方式中,可以将参考基因组的箱的大小可以不同,以最小化每个箱内出现的处理偏差的影响。例如,可以将参考的箱的大小调整为在多个箱之间更均匀地分配gc含量,从而最小化不同箱之间gc偏差的差异。
一箱的gc含量偏差是基于箱中的鸟嘌呤及胞嘧啶含量的水平。通常,一箱中较高的gc含量会导致一较高数量的箱序列读数。因此,处理偏差存储器270可以存储与一箱中的gc含量的量直接相关的一箱的一gc含量偏差。在部署期间,可以从处理偏差存储器270取出箱的gc含量偏差,并且可以使用箱的gc含量偏差对箱的一箱序列读取计数进行归一化。在各个实施方式中,可以使用箱的所有较小的窗口的gc含量来确定一箱的gc含量偏差。例如,一箱的一窗口可以是一定范围的核苷酸碱基(例如50、100、150个核苷酸碱基)。箱的gc含量可以是箱的所有窗口中gc含量的一平均水平。
一箱的可图谱化性偏差是基于箱的核苷酸碱基序列的可图谱化性。可以从可公开获得的数据库(例如:ucsantacruz基因组浏览器(ucsantacruzgenomebrowser))存取一箱的核苷酸碱基序列的可图谱化性。某些箱包括比其他箱具有更高的可图谱化性的核苷酸碱基序列。具有较高的可图谱化性的箱通常具有较高的箱序列读取计数。因此,处理偏差存储器270可以存储与箱的可图谱化性直接相关的一箱的一可图谱化性偏差。在部署期间,可以从处理偏差存储器270中取出箱的可图谱化性偏差,并且可以使用箱的可图谱化性偏差来归一化箱的一箱序列读取计数。在各种实施方式中,可以使用箱的所有较小的窗口的可图谱化性来确定一箱的可图谱化性,例如上述与gc含量偏差有关的窗口。箱的可图谱化性可以是箱的所有窗口的平均可图谱化性。
从一降维分析得出的偏差可以是一pca偏差。所述pca偏差表示可以由未知来源引起的一箱中的偏差。给定训练序列读数(例如,源自训练样本的cfdna序列读数及/或gdna序列读数),执行一主成分分析,以识别箱i的箱序列读取计数s(i)的主成分pcn。pca分析可以表示为:
s(i)=a+b1*pc1(i)+…+bn*pcn(i)
(10)
在此,使用从训练示例中得到的箱的箱序列读取计数来确定每个参数(a、b1...bn)以及主成分pcn。此外,参数及主成分可以存储在处理偏差存储器270中。在部署期间,可以存取箱的参数及主成分,以确定箱的一pca偏差。因此,可以通过所述箱的一pca偏差对所述箱的箱序列读取计数进行归一化。
箱预期计数存储器280保存所有基因组中每个箱的预期序列读取计数。使用训练序列读数(例如,源自一训练样本的cfdna序列读数及/或gdna序列读数)来确定所述每个箱的预期序列读取计数。具体地,将一训练样本的训练序列读数分类到参考基因组的箱中,并且针对训练样本确定箱中的训练序列读数的总数。所述箱的预期序列读取计数被计算为被分类在所有训练样本的箱中的训练序列读数的平均值。
箱预期方差存储器290保存基因组中每个箱的预期方差。通常,一箱的预期方差是所有训练样本的箱的序列读取计数的变异性的一测量。作为一个示例,一箱的预期方差可以是被分类在所有多个训练样本中的箱的训练序列读数的总数的一标准差。作为另一个示例,一箱的预期方差可以是序列读取计数的变异(例如:平均绝对偏差)的稳健测量。
样本变异因子存储器295保存可用于确定一样本的一扩大因子(例如,isample)的因子。样本变异因子存储器295中所存储的因子的示例包括通过对从训练样本得到的数据执行的一曲线拟合处理所确定的系数值。
具体地,对于每个训练样本,来自训练样本的序列读数可以用于确定参考基因组的每个箱的z得分。箱i的z得分可以表示为:
其中bi是箱i的箱得分,而vari是箱的箱方差估计。
在每个训练样本的箱z得分与z得分的理论分布之间进行第一曲线拟合。在此,z分数的一示例理论分布是一正态分布。在一个实施方式中,所述第一曲线拟合是一线性稳健回归拟合(linearrobustregressionfit),其产生一斜率值。因此,在一训练样本的箱z得分与z得分的理论分布之间执行所述第一曲线拟合可得出一斜率值。对于多个训练样本,多次执行所述第一曲线拟合以计算多个斜率值。
在训练样本的斜率值与偏差之间执行一第二曲线拟合。作为一示例,一训练样本的偏差可以是一成对中位绝对偏差(medianabsolutepairwisedeviation,mapd),其代表所有训练样本的相邻箱的箱得分之间的绝对值差的中值。在一个实施方式中,所述第二曲线拟合是一线性稳健回归拟合。在另一个实施方式中,所述第二曲线拟合可以是一高阶多项式拟合(higherorderpolynomialfit)。所述第二曲线拟合产生系数值,在第二曲线拟合是一线性稳健回归拟合的实施方式中,所述系数值包括一斜率系数及一截距系数。由第二曲线拟合产生的系数值作为样本变化因子存储在样本变异因子存储器295中。
示例
示例1:源于一癌症样本中的体细胞肿瘤来源的拷贝数异常
图4a及图4b分别描绘了从一癌症受试者获得的cfdna样本以及gdna样本的一基因组的所有多个箱的箱得分。在此,所述癌症患者已被临床诊断为乳腺癌第一期。通过对癌症患者抽血来获得一血液测试样本,并收集在一采血管中。将血样管在1600g下离心,分别提取血浆及血沉棕黄层(buffycoat)成分,并在零下20℃下保存。使用qiaamp循环核酸试剂盒(qiaampcirculatingnucleicacidkit)(qiagen,日耳曼敦(germantown),马里兰州(md))从血浆中提取cfdna并混合。使用dneasy血液及组织试剂盒(dneasybloodandtissuekit)(qiagen,日耳曼敦,马里兰州)裂解血沉棕黄层中的白细胞,并提取gdna。使用truseqnanodna试剂(illumina,圣地亚哥,加利福尼亚)从提取的cfdna样本及gdna样本中制备测序文库。文库制备后,使用hiseqx测序仪(illumina,圣地亚哥,加利福尼亚)对cfdna测序文库以及gdna测序文库进行测序,以获得来自上文相关步骤125所述的cfdna及gdna样本中的序列读数。具体而言,通过在35x的覆盖的一深度进行全基因组测序来获得cfdna序列读数以及gdna序列读数。使用图2a中所示的流程135对每个dna样本的序列读数进行对位及分析,图2a所示的流程还包括图2b中所示的相应流程210。
具体参照图4a及图4b所示的数据,在图4a及图4b的每个图表中的每个指示代表参考基因组的一箱的一箱得分。x轴上显示的选择箱代表来自癌症患者的染色体1-22号的核苷酸序列。每个箱的箱得分是相对于所述箱预期的序列读取计数的数量进行归一化,因此,没有一拷贝数事件的cfdna样本或gdna样本会描述最小偏离零的箱得分。
未对位指示(例如,在图4a及图4b中标记为“+”)是指不同于gdna样本的对应的箱及/或区段的cfdna样本的箱及/或区段。例如,若gdna样本的对应的箱为统计不显着的,则cfdna样本的一统计显着的箱在图4a中被描绘为一未对位指示。类似地,若gdna样本的对应的箱是统计显着的,则cfdna样本的一统计不显着的箱在图4a中被描绘为一未对位指示。此外,若cfdna样本的区段与gdna样本的对应的区段不同(例如,统计显着的相对于统计不显着的),则使用未对位指示来描绘cfdna样本的一区段内的所有箱。
对位箱指示(例如,在图4a及图4b中标记为“x”)是指cfdna样本及gdna样本中对位的箱。例如,若gdna样本的对应的箱也是统计显着的,则cfdna样本的一统计显着的箱被描绘为一对位箱指示。类似地,若gdna样本的对应的箱也是统计不显着的,则cfdna样本的一统计不显着的箱被描绘为一对位箱指示。
对位区段指示(例如,在图4a及图4b中标记为
参照图4a,cfdna样本包括一统计显着的区段410a,其包括箱得分高于零的箱。另外,cfdna样本包括一统计显着的区段420a,其包括箱得分低于零的箱。此外,cfdna样本包括箱430a及440a,它们是统计显着的,因为它们每一个的一箱得分都高于零。每个统计显着的区段(例如:410a及420a)以及统计显着的箱(例如:430a及440a)表示一拷贝数事件。
参照图4b,gdna样本包括区段410b及区段420b,区段410b及区段420b各自包括具有与零值无显着差异的箱得分的箱。在此,gdna样本的区段410b是cfdna样本的区段410a的对应的区段。另外,gdna样本的区段420b是cfdna样本的区段420a的对应的区段。gdna样本还包括统计显着的箱440b,其是cfdna样本的箱440a的对应的箱。
在此,cfdna样本中的统计显着的区段(例如,区段410a及420a)与gdna样本中的对应的区段(例如,区段410b及420b)不对位。具体地,cfdna样本的统计显着的区段410a与gdna样本的区段410b不对位。另外,cfdna样本的区段420a与gdna样本的区段420b不对位。这表明由统计显着的区段410a及420b中的每一个代表的拷贝数事件可能是由于一体细胞肿瘤事件。
另外,cfdna样本的箱430a未与gdna样本的对应的箱(未示)对位,而cfdna样本的箱440a与gdna样本的箱440b对位。因此,由cfdna样本的箱430a代表的拷贝数事件可能是由于一体细胞肿瘤事件,而由cfdna样本的箱430b代表的拷贝数事件可能是由于一生殖系或体细胞非肿瘤事件。
图5是一图表,描绘了相对于图4a中所示的cfdna样本的对应的箱得分的图4b中所示的gdna样本的箱得分的分布。特别地,图5描绘了一理论识别线570(例如,y=x线),其中x轴代表cfdna样本中的箱的箱得分,y轴代表gdna样本中的箱的箱得分。
如图5所示,统计显着的区段510(其表示图4a及图4b所示的区段410a及410b),统计显着的区段520(其代表图4a及图4b所示的区段420a及420b)以及统计显着的箱530(对应于图4a及图4b中所示的箱430a及430b)偏离所述识别线570。这是一种显现cfdna样本的统计显着的箱及区段与gdna样本的对应的箱及区段之间未对位的方法。
示例2:源于一非肿瘤样本中的体细胞肿瘤来源的潜在拷贝数异常
图6a及图6b描绘了分别从一非癌症个体获得的一cfdna样本以及一gdna样本确定的一基因组的所有箱的箱得分。在此,由于所述个体尚未被诊断出患有癌症,因此所述个体可以作为癌症早期检测的一候选者。通过从所述非癌症个体中抽血获得一血液测试样本,并提取cfdna及gdna。根据上述示例1中所述的方法对cfdna及gdna样本进行提取及测序,以产生用于分析的序列读数。
如图6a所示,cfdna样本包括一统计显着的区段610a,其包括箱得分高于零的箱。另外,cfdna样本包括一统计显着的箱630a,其包括高于零的一箱得分。统计显着的区段620a以及统计显着的箱630a指示拷贝数事件。如图6b所示,gdna样本包括区段620b,所述区段620b包括具有与一零值无显着差异的箱得分的箱。gdna样本的区段620b是cfdna样本的区段620a的对应的区段。另外,gdna样本还包括统计显着的箱630b,其是cfdna样本的箱630a的对应的箱。
cfdna样本的箱630a与gdna样本的箱630b对位。因此,由cfdna样本的箱630a代表的拷贝数事件可能是由于一生殖系或体细胞非肿瘤事件。cfdna样本中的统计显着的区段620a与gdna样本中的对应的区段620b不对位。这表明统计显着的区段620a代表的拷贝数事件可能是由于一体细胞肿瘤事件引起的。这表明通过使用从个体获得的cfdna及gdna样本识别可能的拷贝数异常,可以潜在地筛选一健康个体(例如,未诊断出癌症),以早期检测癌症。
图7是一图表,描绘相对于图6a中所示的cfdna样本的对应的箱得分的图6b中所示的gdna样本的箱得分的分布。特别地,图7描绘了一理论识别线770(例如,y=x线),其中x轴代表cfdna样本中的箱的箱得分,y轴代表gdna样本中的箱的箱得分。如图7所示,统计显着区段720(其代表图6a及图6b中所示的区段620a及620b)偏离所述识别线770,从而反映了cfdna样本的未对位的统计显着的区段以及gdna样本的对应的非统计显着的区段。另外,箱740(其表示图6a及图6b中的箱640a及640b)靠近识别线770。这反映出cfdna样本中箱640a的较高的箱得分与gdna样本中的箱640b的一较高的箱得分对位。
示例3:一非癌症样本中源于一生殖系或体细胞非肿瘤来源的拷贝数变异
图8a及图8b描绘了从分别从一非癌症个体获得的一cfdna样本及一gdna样本确定的一基因组的所有箱的箱得分。在此,由于所述个体尚未被诊断出患有癌症,因此所述个体可以作为癌症早期检测的一候选者。通过从非癌症个体中抽血获得一血液测试样本,并提取cfdna及gdna。根据上述示例1中所述的方法对cfdna及gdna样本进行提取及测序,以产生用于分析的序列读数。
如图8a所示,cfdna样本包括一统计显着的区段820a,所述统计显着的区段820a包括箱得分低于零的箱。另外,cfdna样本包括一统计显着的箱830a,其包括高于零的一箱得分。统计显着的区段820a以及统计显着的箱830a指示拷贝数事件。如图8b所示,gdna样本包括区段820b。gdna样本的区段820b是cfdna样本的区段820a的对应区段。在此,统计显着的区段820b包括具有不显着地偏离零的箱得分的箱的至少一子集。换句话说,区段级分析使得能够识别一统计显着的区段820b,所述区段820b包括单独不会被识别为统计学显着的箱的箱的一子集。这证明了除了执行一箱级分析外,还可以执行一区段级分析,以识别拷贝数事件的好处。gdna样本另外包括统计显着的箱830b,其是cfdna样本的箱830a的对应的箱。
在此,cfdna样本中的统计显着的区段820a与gdna样本中的对应的统计显着的区段820b对位。这表明统计显着的区段820a所代表的拷贝数事件很可能是由于一生殖系或体细胞非肿瘤事件。另外,cfdna样本的箱830a与gdna样本的箱830b对位。因此,由cfdna样本的箱830a所代表的拷贝数事件也可能是由于一生殖系或体细胞非肿瘤事件。
图9是一图表,描绘了相对于图8a所示的cfdna样本的对应的箱得分的图8b所示的gdna样本的箱得分的分布。具体而言,图9描绘了一理论识别线(identityline)970(例如,y=x线),其中x轴表示cfdna样本中的箱的箱得分,y轴表示gdna样本中的箱的箱得分。
如图9所示,箱930(其表示图8a及图8b中的箱830a及830b)靠近识别线970。这反映出cfdna样本中的箱830a的较高的箱得分与gdna样本中的箱830b的一类似的较高的箱得分对位。
另外,如图9所示,统计显着的区段920(其表示图8a及图8b中所示的区段820a及820b之间的对位)略微偏离识别线770。在此,尽管来自cfdna样本的统计显着的区段820a与gdna样本的统计显着的区段820b对位,然而区段920与识别线970的略微偏差表明,统计显着的区段820a中的箱的箱得分的偏差量与统计显着的区段820b中箱的箱得分的偏差量不同。例如,再次参照图8a及图8b,区段820a中的箱的箱得分的大小(例如,如图8a所示的大小~0.15)大于区段820b中的箱的箱得分的大小(例如,如图8b所示的大小~0.05)。这表明在区段级上,不同的样本可能具有不同的干扰因子影响每个区段中的箱得分。然而,即使考虑到区段820a及820b中的不同干扰因子,此示例仍证明了将区段820a及820b识别为统计显着的区段的能力。
其他注意事项:
实施方式的前述详细描述参照了附图,其示出了本公开的特定实施方式。具有不同结构及操作的其他实施方式不脱离本公开的范围。参照本说明书中阐述的申请人发明的许多替代方面或实施方式的某些特定示例来使用术语“本发明”等,并且其使用或不存在并不旨在限制本发明的范围。申请人的发明或权利要求的范围。本说明书被分为几个部分,以方便读者。标题不应解释为对本发明范围的限制。定义旨在作为本发明的描述的一部分。应该理解,在不脱离本发明范围的情况下,可以改变本发明的各种细节。此外,前述描述仅出于说明的目的,而非出于限制的目的。
- 还没有人留言评论。精彩留言会获得点赞!