紫杉醇和IDO1小分子抑制剂复方药物组合物及其用途的制作方法
紫杉醇和ido1小分子抑制剂复方药物组合物及其用途
技术领域
[0001]
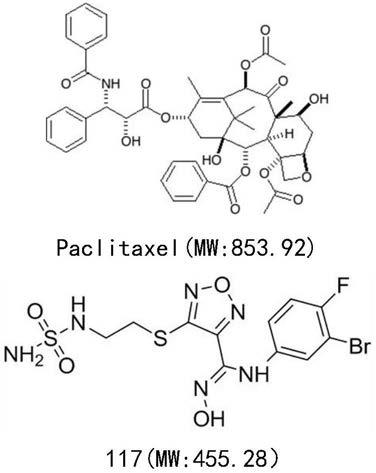
本发明属于药物技术领域,特别涉及一种紫杉醇(paclitaxel)和ido1小分子抑制剂(117)复方药物组合物及其用途。
背景技术:
[0002]
恶性肿瘤已成为严重威胁人类健康的重大疾病,在全球致死性疾病中它仅次于心血管疾病高居第二位。2018年全世界约新增癌症病例1810万,约有960万癌症患者死亡,在我国癌症已成为城市和农村居民的“头号杀手”。
[0003]
近年来,随着肿瘤免疫领域基础研究的快速发展及肿瘤免疫治疗药物在临床研究中取得的巨大成功,肿瘤免疫治疗已成为继手术、放疗、传统化疗、靶向治疗之外的重要治疗策略。与传统的癌症治疗方案主要作用于肿瘤细胞不同,肿瘤免疫治疗主要通过重新活化受抑制的或被钝化的效应细胞(主要是t细胞),重塑免疫系统的免疫监视功能,抑制肿瘤的增长。
[0004]
ido是人体肝外色氨酸代谢限速酶,它催化色氨酸分解代谢成犬尿氨酸,是引起“色氨酸耗竭”和制造“毒性代谢产物犬尿氨酸”的主要因素,也是调节肿瘤免疫应答的关键性免疫抑制酶之一。持续性“免疫抑制”下诱导出现了ido的过高表达,后者主导的肿瘤微环境中犬尿氨酸蓄积进一步诱导产生大量的treg细胞,treg细胞的大量产生进一步促进了肿瘤免疫逃逸,形成恶性循环,共同加剧和维持有利于肿瘤逃逸的抑制性免疫微环境。因此,通过抑制肿瘤微环境中犬尿氨酸/色氨酸的异常代谢重塑肿瘤免疫监视功能是肿瘤免疫治疗应用基础研究的一个重要方向。
[0005]
已完成的多项临床研究显示肿瘤免疫治疗药物与传统化疗药物联合具有更好的疗效,可显著延长肿瘤患者的生存期。
[0006]
紫杉醇是fda批准的第一个天然抗癌药物,具有广泛的抗癌活性,作为一线化疗药物应用于临床。紫杉醇单独使用时具有副作用强,易耐药抗药的缺点。有文献报道,ido是诱发紫杉醇耐药的关键蛋白,基于此,对紫杉醇和ido1小分子抑制剂(117)复方药物组合物进行了研究,证实两者联合使用较单独给药具有更好的抗肿瘤活性,是一种很有潜力的抗肿瘤复方药物。
技术实现要素:
[0007]
为了克服现有技术中存在的缺点和不足,本发明的首要目的在于提供一种紫杉醇和ido1小分子抑制剂(117)复方药物组合物;该复方药物组合物的活性成分是由紫杉醇和ido小分子抑制剂(117)组成。
[0008][0009]
一种复方药物组合物,所述复方药物组合物包括紫杉醇和ido1小分子抑制剂117;
[0010]
并且,所述紫杉醇和ido1小分子抑制剂117的摩尔比为1:1~10。优选的,所述紫杉醇和ido小分子抑制剂(117)的摩尔比为1:1~10。
[0011]
更加优选的,所述紫杉醇和ido小分子抑制剂(117)的摩尔比为1:2。
[0012]
本发明的另一目的在于提供一种上述复方药物组合物的用途。
[0013]
上述复方药物组合物制备抗肿瘤药物的应用。
[0014]
而且,药理试验证明该复方药物组合物具有协同抗肿瘤作用。
[0015]
该复方药物组合物还含有药物上可接受的载体。
[0016]
所述复方药物组合物按照常规制备工艺制备得到。
[0017]
上述的紫杉醇和ido1小分子抑制剂(117)复方药物制备抗肿瘤药物的用途。
[0018]
本发明与现有技术相比具有如下突出的优点及有益效果:
[0019]
本发明紫杉醇和ido1小分子抑制剂(117)复方药物制备抗肿瘤药物,而且,117抑制犬尿氨酸的生成,提高紫杉醇抗肿瘤效果,将紫杉醇和117组合给药较单独使用紫杉醇具有更好的生理活性。
[0020]
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在说明书、权利要求书中所特别指出的结构来实现和获得。
具体实施方式
[0021]
下面将对本发明进行更详细的描述,其中表示了本发明的优选实施例,应该理解本领域技术人员可以修改在此描述的本发明而仍然实现本发明的有益效果。因此,下列描述应当被理解为对于本领域技术人员的广泛知道,而并不作为对本发明的限制。
[0022]
下面结合实施例对本发明做进一步详细描述:
[0023]
ido1小分子抑制剂(117)合成路线如下所示:
[0024][0025]
实施例1ido小分子抑制剂(117)的合成:
[0026]
步骤1:化合物2的合成
[0027][0028]
丙二腈1(10g,151mmol)溶解于200ml水中并搅拌5分钟,冰水冷却后,将(11.8g,171mmol)亚硝酸钠和10n盐酸水溶液分别小心地加入到反应体系中,搅拌15分钟后,缓慢升温至室温并继续搅拌1.5小时。反应体系冷却至0℃后,一次性加入50%的羟胺溶液,室温搅拌1小时后,继续回流2小时,用6n的盐酸调节ph至7.0,收集析出的固体化合物,真空干燥后得到19g目标化合物2。
[0029]
步骤2:化合物3的合成
[0030]
[0031]
化合物2(20g,140mmol)加入到水/醋酸/12n盐酸(240ml/140ml/70ml)组成的混合液中,剧烈搅拌直至完全溶解。体系冷却至0℃后,顺次将氯化钠(23.7g,400mmol)和亚硝酸钠(9.5g,140mmol)缓慢加入到反应体系中,继续搅拌1.5小时后,撤去冰水浴,缓慢升温至室温,过滤得到固体化合物,水洗涤滤饼后干燥得到10.5g白色固体化合物3。
[0032]
步骤3:化合物5的合成
[0033][0034]
将化合物3(5.5g,34mmol)溶解于75ml乙醇/水(5/1,v/v)溶液中,向体系中依次加入3-溴-4-氟苯胺(6.5g,34mmol)和碳酸氢钠(2.2g,21mmol)并继续搅拌10分钟后,将体系加热到60℃直至反应完全。冷却后,加入100ml乙酸乙酯萃取、无水硫酸钠干燥并浓缩后,得到9g目标化合物5。
[0035]
步骤4:化合物6的合成
[0036][0037]
化合物5(10g,31mmol)溶解于50ml四氢呋喃中,接着,将n,n-羰基二咪唑(6g,38mmol)缓慢加入到反应体系中,加热至65℃反应2小时后,加压浓缩除去溶剂,向所得残余物中加入50ml1n盐酸溶液,并继续搅拌10分钟,过滤沉淀得到固体化合物后,用乙酸乙酯洗涤滤饼,干燥后得到8g目标化合物6。
[0038]
步骤5:化合物7的合成
[0039][0040]
将化合物6(3.4g,10mmol)溶解于200ml冰醋酸与60ml浓盐酸的混合液中,搅拌10分钟后,冷却至0℃,将10ml溶解有亚硝酸钠(2.4g,12mmol)的水溶液,缓慢滴加到反应体系中,冰水条件下继续搅拌3小时后,再将11ml溶有氯化亚铜(99mg,1.0mmol)的浓盐酸溶液,缓慢滴加到反应体系中。滴加完毕,常温反应24小时,tlc监测反应结束后,用300ml水稀释,冰水条件下继续搅拌30分钟,抽滤得到白色固体,水洗涤滤饼后,加100ml乙酸乙酯溶解固体,无水硫酸钠干燥、过滤,旋去溶剂后得2.9g白色固体化合物7。
[0041]
步骤6:化合物9的合成
[0042][0043]
将化合物7(3.6g,10mmol)溶解于40ml乙腈中,接着向体系中依次加入化合物8(1.8g,10mmol)和碳酸铯(3.5g,11mmol),常温反应1小时后,抽滤并用乙酸乙酯洗涤后,合并有机相。旋干后得到浅褐色固体,重新溶解于10ml乙酸乙酯中,并加入60ml石油醚剧烈搅拌,过滤出得到的白色固体后晾干,即得到4.5g目标化合物9。
[0044]
步骤7:化合物10的合成
[0045][0046]
将化合物9(5g,10mmol)溶解于40ml二氯甲烷中,接着向体系中加入二氧六环/浓盐酸的混合液(1/1,150ml),常温反应1小时,tlc监测反应完全后,抽滤得到固体化合物,滤饼用二氯甲烷洗涤、除杂、烘干后得到目标化合物10。
[0047]
步骤8:化合物11的合成
[0048][0049]
将氯磺酰异氰酸酯(1.4g,10mmol)溶剂于20ml干燥二氯甲烷中,冰水冷却至0℃,将5ml溶有叔丁醇(1.0g,15mmol)的二氯甲烷溶液小心地滴加到反应体系中,继续反应1小时后备用。在另一个单口烧瓶中,将化合物10(4.4g,10mmol)溶解于30ml二氯甲烷中,分批加入n,n-二异丙基乙基胺(5.2g,40mmol)后,将第一步制备得到的溶液滴加到反应体系中,并继续搅拌0.5小时。反应完全后,加入50ml水,分相,有机相用0.5n的稀盐酸洗涤后,有大量固体析出,抽滤得到固体化合物用二氯甲烷和水洗涤后得到4.4g目标化合物11。
[0050]
步骤9:化合物12的合成
[0051][0052]
将化合物11(2.9g,5mmol)溶解于20ml二氯甲烷中,接着向体系中加入二氧六环/浓盐酸的混合液(1/1,50ml),常温反应20小时,tlc监测反应完全后,抽滤得到固体化合物,滤饼用二氯甲烷洗涤、除杂、烘干后得到2.2g白色固体12。
[0053]
步骤10:化合物117的合成
[0054][0055]
将化合物12(2.6g,5mmol)溶解于20ml四氢呋喃中,接着向体系中1n的氢氧化钠溶液40ml,伴随大量白色固体的析出,反应继续常温反应2.5小时,用4n盐酸调节ph=1,加入30ml水,用乙酸乙酯萃取三次后,合并有机相、无水硫酸钠干燥、过滤、旋干后,重新用15ml二氯甲烷溶解得到的固体化合物,抽滤,滤饼用二氯甲烷洗涤后,晾干得到1.8g目标化合物117。
[0056]
实施例二药理试验
[0057]
本发明的化合物的动物体内活性实验
[0058]
选用雄性c57/bl6小鼠,18-22g。实验过程简述如下:取生长良好的小鼠黑色素瘤b16的荷瘤小鼠,颈椎脱臼处死,无菌条件下剥取长势良好的瘤块,匀浆,用生理盐水1:4稀释,给每只小鼠腋下背部接种0.2ml瘤液(约2
×
106细胞),次日将动物随机分组并开始给药,分别是对照组、紫杉醇组(20mg/kg)、117组(200mg/kg)、紫杉醇(20mg/kg)联合117组(200mg/kg),每组含动物9只,紫杉醇和117均于接种24h后给药,其中紫杉醇为腹腔注射,117为灌胃给药。连续给药10天后,颈椎脱臼处死,分别称量体重、瘤重。计算肿瘤生长抑制率(%),并将结果进行统计学处理。
[0059][0060]
表1本发明化合物对小鼠黑色素瘤b16移植瘤瘤重和动物体重的影响
[0061]
[0062][0063]
注:*:p<0.05,与对照组相比;#:p<0.05,与紫杉醇组相比
[0064]
上述实验结果表明,具有本发明通式的化合物117具有增加紫杉醇体内抑制小鼠黑色素瘤b16移植瘤生长的药理学活性。
[0065]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种等同变换,这些等同变换均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1