基因编辑位点的自动筛选方法、系统、装置及存储介质与流程
本发明涉及基因编辑技术领域,尤其涉及一种基因编辑位点的自动筛选方法、系统、装置及存储介质。
背景技术:
crispr/cas9通过设计特异性向导rna识别靶序列引导cas9核酸内切酶对靶序列的pam上游进行切割,使靶位点dna双链断裂;dna双链断裂后,利用细胞的非同源末端连接(nhej)或同源重组(hdr)的方式对切割位点进行修复,实现dna水平的基因敲除、敲入或点突变。crispr/cas9以其操作简单、成本低且编辑效率等优势,成为生命科学领域最炙手可热的技术之一,已经广泛应用于诸多模式生物的基因编辑相关功能研究中,如哺乳动物(大鼠、小鼠、猪、兔、猴等)、斑马鱼、干细胞、肿瘤细胞系及细菌真菌等。
crispr/cas9敲除方案的设计看似不难,但如果要达到高效切割且后续蛋白水平敲除效果好,需要考虑众多影响因素。对于基因编辑初学者来说,需要几天时间,效果不一定理想;对于有基因编辑经验的研究者,也可能因为信息来源不全面,而没有选到切割效率比较高的grna或者选择的敲除区域dna水平碱基缺失后蛋白表达仍没有变化。
技术实现要素:
有鉴于此,本发明实施例的目的是提供一种基因编辑位点的自动筛选方法、系统、装置及存储介质,能够自动高效筛选敲除效果好且特异性强和切割效率高的基因编辑位点。
第一方面,本发明实施例提供了一种基因编辑位点的自动筛选方法,包括以下步骤:
获取基因名称及物种名称,并根据所述基因名称及所述物种名称从基因数据库获取所述基因的序列、转录本信息及在染色体的位置信息;
根据所述位置信息及所述转录本信息确定备选敲击区;
对所述备选敲击区进行gc含量分析及复杂度分析确定敲击区;
根据共有转录本数量及所述位置信息对所述敲击区进行排序;
根据所述排序,通过所述敲击区的位置信息从所述基因的序列中截取所述敲击区序列;
从grna在线设计软件获取所述敲击区序列的所有grna及对应的特异性得分及切割效率分数,并根据所述特异性得分及所述切割效率分数确定并展示grna的编辑位点。
可选地,根据所述位置信息及根据所述转录本信息确定备选敲击区,包括步骤:
根据所述位置信息确定敲击区范围;
对所述敲击区范围的所述转录本进行预处理得到参考转录本;
根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区。
可选地,根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区,包括:选择在所述参考转录本预设位置及预设长度的区域作为备选敲击区。
可选地,根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区,包括:根据grna设在外显子的位置确定所述备选敲击区。
可选地,根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区,包括:合并外显子预设范围内的其它外显子后,根据合并后的外显子在参考转录本的编码区的位置和长度确定所述备选敲击区。
可选地,所述对所述备选敲击区进行gc含量分析及复杂度分析确定敲击区,包括:
在备选敲击区选取预设区域长度内的平均gc含量在预设区间;
选取所述预设区间内没有预设复杂序列的区域作为敲击区。
可选地,所述根据共有转录本数量及所述位置信息对所述敲击区进行排序,包括:
根据共有转录本的数量对所述敲击区进行排序;
若共有转录本的数量相同,根据所述位置信息对所述敲击区进行排序。
可选地,所述根据所述特异性得分及所述切割效率分数确定grna的编辑位点,包括:
选取所述特异性得分及所述切割效率分数在预设范围内的grna;
根据所述特异性得分及所述切割效率分数的排序,在所述预设范围内的grna确定若干条grna的编辑位点。
第二方面,本发明实施例提供了一种基因编辑位点的自动筛选装置,包括:
至少一个处理器;
至少一个存储器,用于存储至少一个程序;
当所述至少一个程序被所述至少一个处理器执行,使得所述至少一个处理器实现第一方面实施例所述的基因编辑位点的自动筛选方法。
第三方面,本发明实施例提供了一种存储介质,其中存储有处理器可执行的程序,所述处理器可执行的程序在由处理器执行时用于执行如第一方面实施例所述的基因编辑位点的自动筛选方法。
第四方面,本发明实施例提供了一种基因编辑位点的自动筛选系统,包括计算机设备以及与所述计算机设备连接的基因数据库和grna在线设计软件;其中,
所述基因数据库,用于获取基因的序列和信息、转录本信息及在染色体的位置信息;
所述grna在线设计软件,用于获取敲击区序列的所有grna及对应的特异性得分及切割效率分数;
所述计算机设备包括:
至少一个处理器;
至少一个存储器,用于存储至少一个程序;
当所述至少一个程序被所述至少一个处理器执行,使得所述至少一个处理器实现如第一方面实施例所述的一种基因编辑位点的自动筛选方法。
实施本发明实施例包括以下有益效果:本发明实施例根据基因在染色体上的位置信息及转录本信息确定备选敲击区,并对备选敲击区进行gc含量分析及复杂度分析确定敲击区,根据共有转录本数量及位置信息对敲击区进行排序,从而选择效果好的敲击区;根据排序通过敲击区的位置信息从基因的序列中截取敲击区序列,从grna在线设计软件获取敲击区序列的所有grna及对应的特异性得分及切割效率分数,并根据特异性得分及切割效率分数确定并展示grna的编辑位点,从而确定特异性强且切割效率高的基因编辑位点;另外,整个基因筛选过程实现自动化,高效快捷。
附图说明
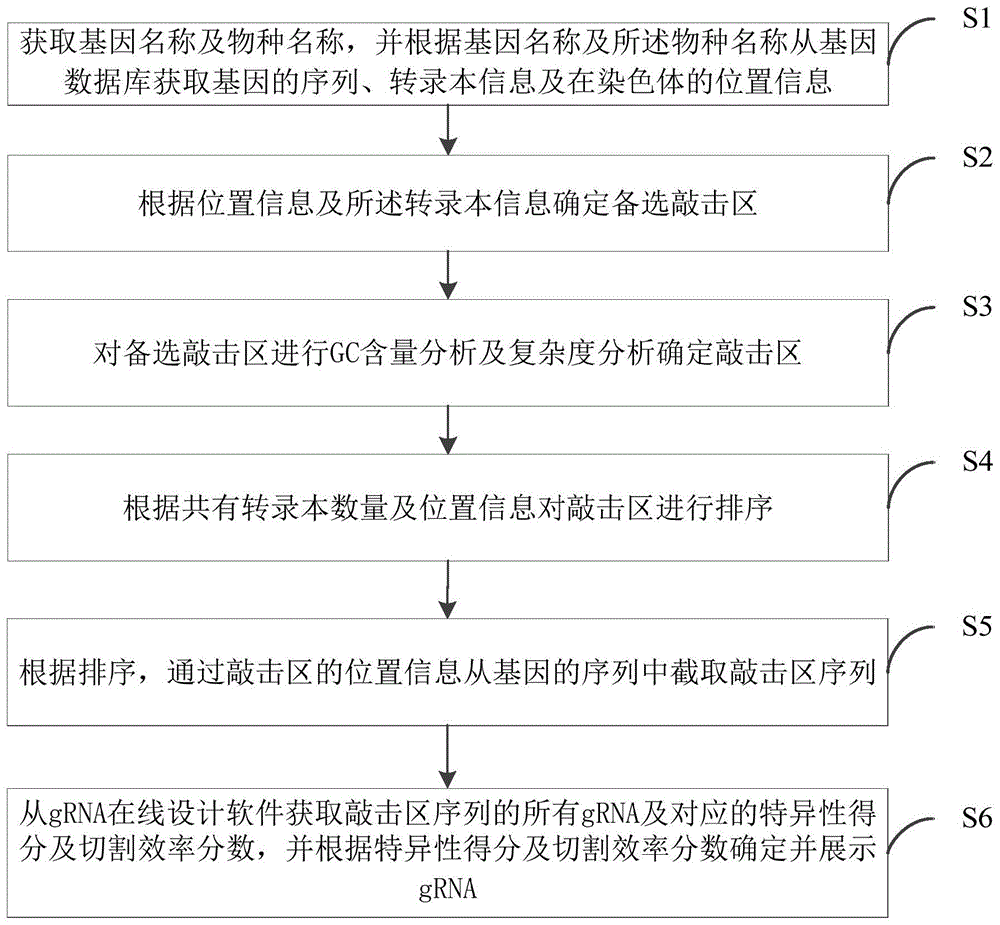
图1是本发明实施例提供的一种基因编辑位点的自动筛选方法的步骤流程示意图;
图2是本发明实施例提供的一种基因编辑位点的自动筛选装置的结构框图;
图3是本发明实施例提供的一种基因编辑位点的自动筛选系统的结构框图。
具体实施方式
下面结合附图和具体实施例对本发明做进一步的详细说明。对于以下实施例中的步骤编号,其仅为了便于阐述说明而设置,对步骤之间的顺序不做任何限定,实施例中的各步骤的执行顺序均可根据本领域技术人员的理解来进行适应性调整。
如图1所示,本发明实施例提供了一种基因编辑位点的自动筛选方法,其包括的步骤如下所示。
s1、获取基因名称及物种名称,并根据所述基因名称及所述物种名称从基因数据库获取所述基因的序列、转录本信息及在染色体的位置信息;
s2、根据所述位置信息及所述转录本信息确定备选敲击区;
s3、对所述备选敲击区进行gc含量分析及复杂度分析确定敲击区;
s4、根据共有转录本数量及所述位置信息对所述敲击区进行排序;
s5、根据所述排序,通过所述敲击区的位置信息从所述基因的序列中截取所述敲击区序列;
s6、从grna在线设计软件获取所述敲击区序列的所有grna及对应的特异性得分及切割效率分数,并根据所述特异性得分及所述切割效率分数确定并展示grna的编辑位点。
需要说明的是,基因数据库可以是ensembl数据库,也可以是其它的数据库,本发明实施例不做具体限制。
需要说明的是,grna在线设计软件的网址为http://crispor.tefor.net/,也可以是其它的软件,本发明实施例不做具体限制。
实施本发明实施例包括以下有益效果:本发明实施例根据基因在染色体上的位置信息及转录本信息确定备选敲击区,并对备选敲击区进行gc含量分析及复杂度分析确定敲击区,根据共有转录本数量及位置信息对敲击区进行排序,从而选择效果好的敲击区;根据排序通过敲击区的位置信息从基因的序列中截取敲击区序列,从grna在线设计软件获取敲击区序列的所有grna及对应的特异性得分及切割效率分数,并根据特异性得分及切割效率分数确定并展示grna的编辑位点,从而确定特异性强且切割效率高的基因编辑位点;另外,整个基因编辑位点的筛选过程实现自动化,高效快捷。
可选地,根据所述位置信息及根据所述转录本信息确定备选敲击区,包括步骤:
根据所述位置信息确定敲击区范围;
对所述敲击区范围的所述转录本进行预处理得到参考转录本;
根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区。
具体地,位置信息是指目标基因在染色体上的位置信息和外显子在转录本上的位置信息,敲击区范围指适合做敲除方案的范围,预处理指排除非编码蛋白的转录本和不完整的转录本,蛋白编码区的位置指蛋白编码区在参考转录本的预设位置,长度根据起始位置及终止位置计算。
可选地,根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区,包括:选择在所述参考转录本预设位置及预设长度的区域作为备选敲击区。
可选地,根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区,包括:根据grna设在外显子的位置确定所述备选敲击区。
需要说明的是,grna设在外显子的位置包括grna设在外显子上和grna设在外显子两端的内含子上,grna设在外显子的不同位置,备选敲击区的长度的计算方法不同。
可选地,根据所述参考转录本的蛋白编码区的位置及长度和/或外显子的位置及长度确定所述备选敲击区,包括:合并外显子预设范围内的其它外显子后,根据合并后的外显子在参考转录本的编码区的位置确定所述备选敲击区。
需要说明的是,合并外显子预设范围内的其它外显子后,需要删除重复的区域。
可选地,所述对所述备选敲击区进行gc含量分析及复杂度分析确定敲击区,包括:
在备选敲击区选取预设区域长度内的平均gc含量在预设区间;
选取所述预设区间内没有预设复杂序列的区域作为敲击区。
需要说明的是,预设区域长度及预设长度范围根据具体情况设置,本发明实施例不做具体限制;另外,复杂的区域包括多种情况,可根据具体情况设置,本发明实施例不做具体限制。
可选地,所述根据共有转录本数量及所述位置信息对所述敲击区进行排序,包括:
根据共有转录本的数量对所述敲击区进行排序;
若共有转录本的数量相同,根据所述位置信息对所述敲击区进行排序。
需要说明的是,排序时优先考虑共有转录本的数量,再考虑前后位置信息。
可选地,所述根据所述特异性得分及所述切割效率分数确定grna的编辑位点,包括:
选取所述特异性得分及所述切割效率分数在预设范围内的grna;
根据所述特异性得分及所述切割效率分数的排序,在所述预设范围内的grna确定若干条grna的编辑位点。
需要说明的是,确定的grna数量根据具体情况确定,如两条或一条grna,本发明实施例不做具体限制。
如图2所示,本发明实施例提供了一种基因编辑位点的自动筛选装置,包括:
至少一个处理器;
至少一个存储器,用于存储至少一个程序;
当所述至少一个程序被所述至少一个处理器执行,使得所述至少一个处理器实现上述方法实施例所述的基因编辑位点的自动筛选方法步骤。
可见,上述方法实施例中的内容均适用于本装置实施例中,本装置实施例所具体实现的功能与上述方法实施例相同,并且达到的有益效果与上述方法实施例所达到的有益效果也相同。
此外,本申请实施例还公开了一种计算机程序产品或计算机程序,计算机程序产品或计算机程序存储在计算机可读存介质中。计算机设备的处理器可以从计算机可读存储介质读取该计算机程序,处理器执行该计算机程序,使得该计算机设备执行上述的基因编辑位点的自动筛选方法。同样地,上述方法实施例中的内容均适用于本存储介质实施例中,本存储介质实施例所具体实现的功能与上述方法实施例相同,并且达到的有益效果与上述方法实施例所达到的有益效果也相同。
如图3所示,本发明实施例提供了一种基因编辑位点的自动筛选系统,包括计算机设备以及与所述计算机设备连接的基因数据库和grna在线设计软件;其中,
所述基因数据库,用于获取基因的序列和信息、转录本信息及在染色体的位置信息;
所述grna在线设计软件,用于获取敲击区序列的所有grna及对应的特异性得分及切割效率分数;
所述计算机设备包括:
至少一个处理器;
至少一个存储器,用于存储至少一个程序;
当所述至少一个程序被所述至少一个处理器执行,使得所述至少一个处理器实现上述的基因编辑位点的自动筛选方法。
具体地,对于所述计算机设备,其可为不同类型的电子设备,包含但不限于有台式电脑、手提电脑等终端。
可见,上述方法实施例中的内容均适用于本系统实施例中,本系统实施例所具体实现的功能与上述方法实施例相同,并且达到的有益效果与上述方法实施例所达到的有益效果也相同。
下面以具体实施例对上述基因编辑位点的自动筛选方法进行说明,基因编辑位点的自动筛选方法首先是确定敲除区域,然后根据敲除区域确定grna,最后将确定的敲除区域及grna画图展示。其中,根据敲击方案的不同,确定敲除区域及grna的方法不同,本实施例提供三种敲除方案及其对应的grna确定方案,三种敲除方案包括移码敲除方案、小片段敲除方案及大片段敲除方案,具体实施方式如下。
需要说明的是,自动编辑基因之前,需要获取用户输入的目标基因名称及物种名称,并且可以利用ensembl数据库提供的接口从ensembl上获取该基因的相关信息。
实施方式一:移码敲除方案
(1)确定敲除区域
s111、判断目标基因是否适合设计敲除方案。具体方法如下:从ensembl上获取目标基因在染色体上的位置,根据这个位置在染色体上检索目标基因所在的区段内是否有其他的基因;如果有其它基因,则继续获取这些与目标基因重叠的其它基因的类型,如果其它基因是编码蛋白的基因,则该目标基因不适合设计敲除方案。
s112、对该目标基因的转录本进行预处理。具体方法为:从ensembl上获取该目标基因的转录本信息,将非编码蛋白的转录本和不完整的转录本排除。
s113、找出各个转录本的蛋白编码区,即cds。具体方法为:通过每条转录本的起始位置和终止位置,编码区的起始位置和终止位置,以及这条转录本的外显子的位置,找出该转录本的cds,其中,外显子与编码区的重叠部分即为cds。
s114、计算cds区域在所有转录本上的共有情况,即有多少个转录本是有这个cds区域的。具体方法为:采用将所有转录本的cds区域放入一个表格中,每个cds区域都有起始位置和终止位置,遍历表格,根据不同转录本的cds之间的位置关系进行判断,如果某个转录本的cds与另一个转录本的cds有重叠部分,则这个cds区域为两个转录本共有。
s115、选择备选敲击区。选取最长的转录本为参考转录本,将在参考转录本前50%的cds作为备选的敲除区域,然后通过这个区域的起始位置和终止位置来计算区域的长度,选择长度为100-300bp的区域作为备选的敲除区域。
s116、gc含量分析,其中,gc含量表示某段序列碱基g和c的占比之和。具体要求为敲除区域上下游800bp内的平均gc含量要在40%-60%之间。
复杂度分析:敲除区域上下游不能出现复杂的区域。有以下情况区域需要排除:1、正向或反向重复大于50bp,从整个敲除区域的dna序列中依次截取50bp的序列与完整dna序列比对,如果在dna序列中出现两次则为重复,则排除该区域;2、连续20个相同的碱基,分别将20个a碱基、20个t碱基、20个g碱基及20个c碱基与dna序列比对,如果出现这样的序列则排除该区域;3、两个环部小于25bp的发夹结构,若某一段序列是另一段序列的反向互补序列,则dna单链分子自身回折形成发夹结构,从序列上依次取10bp的序列(记录最后一个碱基的下标)进行反向互补转化(a变t,t变a,g变c,c变g,然后颠倒序列),然后将其作为pattern在整个序列中查找是否有相同的序列,并得到与pattern相同的序列的索引下标,如果该下标与pattern的下标相差在25以内,则为环部小于25bp的发夹结构,如果这样的结构出现两次则终止迭代,排除该区域。
s117、将剩余的备选敲除区域按照共有转录本的数量和在转录本上的位置的前后顺序来排序,共有转录本的数量多的优先,位置靠前的次之,依次进行grna的设计。
(2)确定grna
s211、从ensembl数据库中获取目标基因的序列,通过敲除区域的起点和终点从基因序列中截取这段敲除区域的序列,然后从grna在线设计软件(http://crispor.tefor.net/)里获取这个区域的所有grna和它们的特异性得分及脱靶情况等。
s212、对grna进行初步筛选。选出特异性得分(mitspecificityscore)在80以上且doench’16在50分以上的grna;然后将每条grna与目标基因的序列进行比对,获取grna在基因上的位置;再进行一次gc含量分析,grna邻近区域的gc含量不能高于80%或低于20%,如以50bp为窗口大小,即每50bp计算一次gc含量(1-50、2-51...),计算该敲除区域及其上下游800bp的区域的局部gc含量,找到所有gc含量大于80%和小于20%的区域,然后将重叠的区域合并,如1-50及21-70合并为1-70,如果grna落在这些区域则排除。
s213、通过在线软件预测grna的切割效率分数,切割分数要求在0.5以上,然后按照特异性得分的高低顺序依次判断靠后的grna是否与靠前的grna重叠,即通过上一步获取到的grna在基因上的位置判断是否重叠,如果有重叠则删除靠后的grna;然后将grna按照特异性得分和切割效率得分综合排序,取最优的两条grna完成设计。如果经过筛选后该敲除区域不能找到两条grna,则换下一个区域继续设计。
实施方式二:小片段敲除方案
(1)确定敲除区域
s121、判断目标基因是否适合设计敲除方案。具体方法如下:从ensembl上获取目标基因在染色体上的位置,根据这个位置在染色体上检索目标基因所在的区段内是否有其他的基因;如果有其它基因,则继续获取这些与目标基因重叠的其它基因的类型,如果其它基因是编码蛋白的基因,则该目标基因不适合设计敲除方案。
s122、对该目标基因的转录本进行预处理。具体方法为:从ensembl上获取该目标基因的转录本信息,将非编码蛋白的转录本和不完整的转录本排除。
s123、找出各个转录本的蛋白编码区,即cds。具体方法为:通过每条转录本的起始位置和终止位置,编码区的起始位置和终止位置,以及这条转录本的外显子的位置,找出该转录本的cds,其中,外显子与编码区的重叠部分即为cds。
s124、计算cds区域在所有转录本上的共有情况,即有多少个转录本是有这个cds区域的。具体方法为:采用将所有转录本的cds区域放入一个表格中,每个cds区域都有起始位置和终止位置,遍历表格,根据不同转录本的cds之间的位置关系进行判断,如果某个转录本的cds与另一个转录本的cds有重叠部分,则这个cds区域为两个转录本共有。
s125、选取位于参考转录本前50%的所有cds作为敲除区域的备选。小片段敲除方案的敲除区域的选择比较复杂,可分成两大类区域。
s125a、将grna设在外显子。通过外显子在转录本上的位置找出在转录本前50%,将转录本的终止位置减去起始位置为转录本的长度,起始位置加上长度的一半即为转录本50%的位置,外显子的终止位置小于转录本50%的位置则符合,且长度为300-500bp的外显子,外显子的终止位置减去起始位置即为该外显子的长度。
s125b、将grna设在外显子两端的内含子上。依次判断每个外显子前后200bp是否有其他外显子,有则合并一起敲除,并计算合并后的区域的长度和这个区域里的外显子长度总合。由于每个外显子都执行这样的操作,所以会出现重复合并,所以根据区域的起始位置和终止位置可以排除重复的区域。
s126、gc含量分析,其中,gc含量表示某段序列碱基g和c的占比之和。具体要求为敲除区域上下游800bp内的平均gc含量要在40%-60%之间。
复杂度分析:敲除区域上下游不能出现复杂的区域。有以下情况区域需要排除:1、正向或反向重复大于50bp,从整个敲除区域的dna序列中依次截取50bp的序列与完整dna序列比对,如果在dna序列中出现两次则为重复,则排除该区域;2、连续20个相同的碱基,分别将20个a碱基、20个t碱基、20个g碱基及20个c碱基与dna序列比对,如果出现这样的序列则排除该区域;3、两个环部小于25bp的发夹结构,若某一段序列是另一段序列的反向互补序列,则dna单链分子自身回折形成发夹结构,从序列上依次取10bp的序列(记录最后一个碱基的下标)进行反向互补转化(a变t,t变a,g变c,c变g,然后颠倒序列),然后将其作为pattern在整个序列中查找是否有相同的序列,并得到与pattern相同的序列的索引下标,如果该下标与pattern的下标相差在25以内,则为环部小于25bp的发夹结构,如果这样的结构出现两次则终止迭代,排除该区域。
s127、将剩余的备选敲除区域按照共有转录本的数量和在转录本上的位置的前后顺序来排序,共有转录本的数量多的优先,位置靠前的次之,依次进行grna的设计。
(2)确定grna
按照敲除区域的分类,不同类的敲除区域设计grna的方法不相同,将grna设在外显子的设计步骤如下。
s221a、用该外显子的序列从grna在线设计软件里获取这个区域的所有grna和它们的特异性得分及脱靶情况等。
s222a、对grna进行初步筛选。选出特异性得分(mitspecificityscore)80以上,doench’16在50分以上的grna,然后将每条grna与目标基因的序列进行比对,获取grna在基因上的位置。再进行一次gc含量分析,grna邻近区域的gc含量不能高于80%或低于20%。
s223a、预测grna切割效率得分,将切割分数低于0.5分的grna排除掉,然后按照特异性得分的高低排序。然后按顺序找到满足以下条件的一对grna:敲除大小≥200bp及特异性得分相差不超过10分,且其中一条位于编码区的前1/2,如果没有符合要求的grna则换下一个敲除区域。
将grna设在外显子两端的内含子上的设计步骤如下。
s221b、用该外显子的上游序列和下游序列从grna在线设计软件里分别获取它们的grna和它们的特异性得分及脱靶情况等。
s222b、对grna进行初步筛选:选出特异性得分(mitspecificityscore)在80以上且doench’16在50分以上的grna,然后将每条grna与目标基因的序列比对,获取grna在基因上的位置;再进行一次gc含量分析,grna邻近区域的gc含量不能高于70%或低于30%,如以30bp为窗口大小,即每30bp计算一次gc含量(1-30,2-31...),计算该敲除区域及其上下游800bp的区域的局部gc含量,找到所有gc含量大于70%和小于30%的区域,然后将重叠的区域合并,如1-30和21-50合并为1-50,如果grna落在这些区域则排除。
s223b、通过在线软件预测grna的切割效率分数,将切割分数低于0.5分的grna排除掉,然后将grna按特异性得分的高低进行排序,选择上游特异性得分最高的grna和下游得分最高的grna,并且特异性得分相差10分以内。先从上游的grna中选择一条特异性得分最高的,然后在下游的grna中按特异性得分的排序选到一条相差10分以内的grna;若下游中没有符合的,则按特异性得分的顺序换一条上游的grna,再找符合要求的下游的grna。
实施方式三:大片段敲除方案
(1)确定敲除区域
s131、判断目标基因是否适合设计敲除方案。具体方法如下:从ensembl上获取目标基因在染色体上的位置,根据这个位置在染色体上检索目标基因所在的区段内是否有其他的基因;如果有其它基因,则继续获取这些与目标基因重叠的其它基因的类型,如果其它基因是编码蛋白的基因,则该目标基因不适合设计敲除方案。
s132、对该目标基因的转录本进行预处理。具体方法为:从ensembl上获取该目标基因的转录本信息,将非编码蛋白的转录本和不完整的转录本排除。
s133、找出各个转录本的蛋白编码区,即cds。具体方法为:通过每条转录本的起始位置和终止位置,编码区的起始位置和终止位置,以及这条转录本的外显子的位置,找出该转录本的cds,其中,外显子与编码区的重叠部分即为cds。
s134、计算cds区域在所有转录本上的共有情况,即有多少个转录本是有这个cds区域的。具体方法为:采用将所有转录本的cds区域放入一个表格中,每个cds区域都有起始位置和终止位置,遍历表格,根据不同转录本的cds之间的位置关系进行判断,如果某个转录本的cds与另一个转录本的cds有重叠部分,则这个cds区域为两个转录本共有。
s135、选出参考转录本的所有cds,判断每个外显子前后200bp是否有其他外显子,有则合并,并计算整个区域的长度和这个区域里的外显子长度总合,然后根据区域的起始位置和终止位置删除重复的区域。
s136、算出参考转录本的编码区的1/2位置,计算公式:(start+(end-start)/2),其中,start表示开始位置,end表示结束位置。若外显子的前端位于前1/2内,则这些外显子的前端可作为敲除区域的起点,所有外显子的后端作为终点,然后进行两两配对,要求满足以下条件:整个区域的大小要在5kb-15kb之间。将得到的所有敲除区域按位置前后顺序排序。
s137、gc含量分析,其中,gc含量表示某段序列碱基g和c的占比之和。具体要求为敲除区域上下游800bp内的平均gc含量要在40%-60%之间。
复杂度分析:敲除区域上下游不能出现复杂的区域。有以下情况区域需要排除:1、正向或反向重复大于50bp,从整个敲除区域的dna序列中依次截取50bp的序列与完整dna序列比对,如果在dna序列中出现两次则为重复,则排除该区域;2、连续20个相同的碱基,分别将20个a碱基、20个t碱基、20个g碱基及20个c碱基与dna序列比对,如果出现这样的序列则排除该区域;3、两个环部小于25bp的发夹结构,若某一段序列是另一段序列的反向互补序列,则dna单链分子自身回折形成发夹结构,从序列上依次取10bp的序列(记录最后一个碱基的下标)进行反向互补转化(a变t,t变a,g变c,c变g,然后颠倒序列),然后将其作为pattern在整个序列中查找是否有相同的序列,并得到与pattern相同的序列的索引下标,如果该下标与pattern的下标相差在25以内,则为环部小于25bp的发夹结构,如果这样的结构出现两次则终止迭代,排除该区域。
(2)确定grna
s231、用该外显子的上游序列和下游序列从grna在线设计软件里分别获取它们的grna和它们的特异性得分。
s232、对grna进行初步筛选:选出特异性得分(mitspecificityscore)80以上且doench’16在50分以上的grna;然后将每条grna与目标基因的序列比对,获取grna在基因上的位置;再进行一次gc含量分析,grna邻近区域的gc含量不能高于70%或低于30%,如以30bp为窗口大小即每30bp计算一次gc含量(1-30、2-31...),计算该敲除区域及其上下游800bp的区域的局部gc含量,找到所有gc含量大于70%和小于30%的区域,然后将重叠的区域合并(如1-30和21-50合并为1-50),如果grna落在这些区域则排除。
s233、通过在线软件预测grna的切割效率分数,将切割分数低于0.5分的grna排除掉,然后将grna按特异性得分的高低进行排序,选择上游特异性得分最高的grna和下游得分最高的grna,并且特异性得分相差10分以内。先从上游的grna中选择一条特异性得分最高的,然后在下游的grna中按特异性得分的排序选到一条相差10分以内的grna;若下游中没有符合的,则按特异性得分的顺序换一条上游的grna,再找符合要求的下游的grna。
(3)画图并展示
根据以上三种实施方式的敲击方案确定敲击区域及grna后,通过以下方法进行画图展示,可以选择其中任一个展示图进行展示,或选择多个展示图进行展示。
s301、展示所有转录本的示意图并框出敲除区域。
展示图主要利用从ensembl上获取的数据信息并加以处理,画出所有转录本的展示图,然后根据设计出的grna的坐标,将其框出。
首先,确定展示图的坐标,具体可以采用如下方法:根据基因的长度确定x轴范围,每条转录本在x轴上的范围都是转录本各自的长度,每条转录本都赋予一个y值,其中,基因的长度即表示碱基数量。另外,可根据ensembl上提供的信息确定颜色:如转录本的来源是“ensembl_havana”,则确定颜色为橙色;转录本是非编码蛋白类型的,则确定为蓝色;其余转录本则是红色。
然后,在每条转录本上画出它们的外显子。综合考虑外显子的位置和编码区的位置,可以将外显子用不同标记表示,如不在编码区里的外显子用空心框表示,在编码区里的外显子用实心框表示;另外,可以在每条转录本下方标上转录本的名字以及方向,颜色均与该转录本的颜色相同。
最后,根据确定的grna的位置,框出要敲除的区域。
s302、展示参考转录本的示意图。
参考转录本的示意图是在所有转录本的示意图中筛选,只画出所述参考转录本的示意图,并在图中框出敲除区域。
s303、展示敲除区域放大图。
根据参考转录本上的外显子的位置和确定的grna位置,只画出在grna敲除区域范围内的外显子。
s304、展示点阵分析图和gc含量图。
对选定的敲除区域的上下游的序列自我比较,将窗口大小设置为预设数量,如20,即每20bp作为一个单位去比对分析,也就是将一条序列的某段20bp的序列和它的反向互补序列与另一条序列进行比对,并将比对结果标注在坐标轴上。另外,正向序列和反向互补序列可以用颜色区分,如红色为正向,绿色为反向互补。
gc图是将窗口大小设置为预设数量,如30,即每30bp碱基计算一次gc含量,然后以碱基位置为x轴,gc含量为y轴作图。
以上是对本发明的较佳实施进行了具体说明,但本发明创造并不限于所述实施例,熟悉本领域的技术人员在不违背本发明精神的前提下还可做作出种种的等同变形或替换,这些等同的变形或替换均包含在本申请权利要求所限定的范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- Egfr基因检测探针及其制备方法和试剂盒的制作方法
- 循环癌细胞的kras致癌基因检测方法
- 焦磷酸测序法检测cyp2c9基因分型的引物对及试剂盒的制作方法
- 一种基于下一代测序技术检测egfr/kras/braf基因突变位点的多重pcr引物和方法及应用
- 一种检测男性不育症Fkbp6基因突变位点的试剂盒及其PCR扩增方法
- 一种检测男性不育症Katnal1基因突变位点的试剂盒及其PCR扩增方法
- 一种华法林药物基因突变位点检测的试剂盒及检测方法
- 用于检测药物性耳聋易感基因突变位点的试剂盒及引物对组的制作方法
- 一种利用pcr扩增后直接dna测序筛查肝豆状核变性致病基因突变位点的方法
- 检测egfr基因突变的双脱氧修饰引物、试剂盒及应用