一种卡利拉嗪长效缓释微球及其制备方法与流程
本发明涉及医药技术领域,具体公开了一种卡利拉嗪长效缓释微球及其制备方法。
背景技术:
一、精神分裂症及双相情感障碍的发病特征精神分裂症是(schizophrenia,sch)是最常见、最复杂且最难做出完整定义的重度精神病,发病率低但患病率高。根据国际精神分裂症试点调查(ipss)资料:一般人群中精神分裂症发病率在0.2
‰
~0.6
‰
,平均为0.3
‰
1.;tusang等在2011年最新的研究结果显示:全球范围内患病率约为5
‰
,整体社会负担在各种疾病中的排名为第四位
2.。根据国家卫生部提供的数字显示中国各类精神疾病患者约一千六百万人,其中精神分裂症患者有七百八十万人,因该病预后不良,约67%精神分裂症患者长期存在明显症状,如阴性症状和认知障碍等
3.。此外,在很长时间里,由于国内重视程度不足、诊断水平落后等,双相障碍(dipolar disorder)长期被误诊为单相抑郁或精神分裂症。双相障碍又称躁郁症,在我国被划为重型精神病之一,双相障碍按发作类型可以分为抑郁、躁狂或混合发作,i型是典型的重躁狂重抑郁。who 1993年所发表的全球疾病负担研究报告指出,双相障碍位列daly(国际上推行以有效生命年的减少来评价疾病负担)减少最多的前10位疾病;中国各种疾病daly减少超过1%的前25位中,双相障碍为第十三位。美国流行病学资料显示,该病的终生患病率为1.2%;中国流行病学数据显示国内双相障碍躁狂症状的发病率为0.042%,而青少年与中年人是高发人群,15岁到30岁是躁郁症的主要发病年龄。双相障碍不只是一种精神类疾病,更可能是器质性病变造成的,本质上是一种生理性疾病,这也意味着靠调节自己的心理或改变想法,并不能缓解、消除病情。双相障碍患者的大脑受损比单相抑郁更严重,复发率很高,很难完全治愈,对健康危害及其极大。二、卡利拉嗪盐酸盐的临床应用2015年9月17日,美国食品药品监督管理局(fda)全球率先批准由匈牙利吉瑞大药厂(gedeon richter)开发,商品名为的盐酸卡利拉嗪胶囊,用于治疗成人躁狂发作或躁狂与抑郁混合发作的双相情感障碍ⅰ型以及精神分裂症。卡利拉嗪(cariprazine)化学名为:氮-[反式-4-[2-[4-(2,3-二氯苯基)-l-哌嗪]乙基]环己基]-氮,氮-二甲基脲,cas号为:839712-12-8,专利cn 108586389 a公开了其合成路径。卡利拉嗪是d2和d3受体的部分激动剂,能发挥认知改善效应,并可以减少阴性症状。作为d2受体部分激动剂,卡利拉嗪与阿立哌唑、依匹哌唑在理论上较其他二代抗精神药物有着先天优势,被认为已超越第二代抗精神病药(非典型抗精神病药)。三、卡利拉嗪及其盐酸盐的口服制剂盐酸卡利拉嗪在水中的溶解度较卡利拉嗪高,其盐酸盐有更高的生物利用度,因此目前市售制剂或专利多采用盐酸盐。
原研制剂,盐酸卡利拉嗪胶囊(商品名为),每日一次。cn 107970217 a发明了一种卡利拉嗪口崩片的制备方法。通过活性成分盐酸卡利拉嗪、粘合剂及崩解剂等处方成分压制成片,使得该卡利拉嗪口崩片能够在口腔中迅速崩解、快速溶出;该制剂为片剂,仍需每天按时一次或者多次服用,无法满足当前临床需求。cn 110769815 a发明了一种口服的卡利拉嗪固体制剂(盐酸卡利拉嗪),用于以低于每日给药剂量调释递送卡利拉嗪,该药物组合物包含有效剂量的卡利拉嗪和至少一种释放调节剂,该释放调节剂适合于降低c
max
并且将auc维持在有效和可耐受的治疗每日剂量的范围内,该发明能够实现14天给药一次,但是该药物组合物的高卡利拉嗪含量在体内释放时易引起局部浓度过高从而可能引起不必要的副反应及毒副作用;而口服制剂的形式无法避免精神分裂患者或者情感障碍患者不配合服药的情况发生。cn 109589315 a公开了一种卡利拉嗪-亲水性材料共混物固体制剂的制备方法,该方法主要是利用卡利拉嗪药学上可接受的辅料提高卡利拉嗪在固体制剂中的溶出速度,提高药品的生物利用度;包括的剂型有片剂、胶囊剂、分散片、口崩片及咀嚼片中的一种,均需一天内多次服药,无法避免精神分裂患者或者情感障碍患者不配合服药的情况发生。四、卡利拉嗪及其盐酸盐的长效制剂和靶向纳米制剂精神分裂症一般治疗周期长,患者经常存在拒绝服药,不按节律服药情况,患者顺应性差,使得病人得不到有效的治疗致使病情恶化或者重新住院。常用的解决方法是监督,这样势必增加患者家属和社会医疗机构的看护负担。因此开发一种能够满足精神疾病患者用药需求、延长给药间隔、减少给药次数、长时间维持有效的血药浓度的缓释制剂是极其必要的,缓释制剂不仅能够够延长患者给药时间,且与普通制剂相比释药平缓,能够有效避免血药浓度的明显波动,减少血药“峰谷”现象,从而减少或避免因血药浓度过高引发的不良反应或毒副作用。长效注射制剂可有效降低精神分裂患者的复发率。有临床研究发现:口服帕利哌酮组的中位复发时间为58天(42-114天),1个月长效注射为172天(134
–
222天),而3个月长效注射组为395天(大于274天),两两比较具有统计学意义(p《0.0001)。相对于口服帕利哌酮的患者而言,pp1m组的复发风险降低56%,而pp3m的复发风险降低了79%((p《0.001))。相对于pp1m组的患者而言,pp3m组的复发风险也显著降低,约为52%(p《0.001)。(peter j.weiden,edward kim,md,jason bermak,et al.does half-life matter after antipsychotic discontinuation?a relapse comparison in schizophrenia with 3different formulations of paliperidone.j clin psychiatry 2017;78(7):e813
–
e820)cn 107049932 a发明了一种小分子药物原位相变凝胶缓释系统及其制备方法,该发明主要通过一种简单的方法将磷脂、司盘、药物活性成分及不同浓度的乙醇溶液制备成磷脂司盘缓释制剂,其制剂生物相容性良好、能够延长释放时间。但是凝胶剂在贮存过程中由于遇水即发生固化易对溶液的稳定性产生影响;原位相变凝胶在注射部位易引起局部浓度过高;且由于乙醇的存在,导致该产品安全性较差,放大生产的可能性不高;另外,专利中表明该制剂虽然可以抑制突释,但半衰期最长也达不到一周,仍然无法满足精神疾病患者需要减少给药次数、增加患者依从性等方面的需求。cn 108261394 a发明了一种盐酸卡利拉嗪注射制剂,该制剂为盐酸卡利拉嗪混悬
水溶液,盐酸卡利拉嗪的浓度较高,通过调整粒径分布实现1-6周长效作用。但是该混悬性注射剂需通过加入稳定剂、助悬剂等维持稳定性,长时间的放置易造成该混悬制剂的不稳定、粒径变大及可注射性变差等问题。us20180344849a1公开了一种相变纳米缀合物的制备及使用方法。该相变纳米缀合物由两部分组成,内部包括一种气相前体,外部为一种或者多种纳米粒,两部分通过某种连接物接合在一起。其中外部的纳米粒可包括治疗、预防及诊断型纳米粒。该专利的发明特点在于通过超声辐射等手段将相变液体纳米粒转变为气体从而实现靶向递送,包括细胞、组织或器官等,而外部的纳米粒可以涵盖多种功能性,因此不同活性成分能够靶向递送至不同区域,实现治疗、预防或者诊断功能。但是,该制剂主要实现的是定点释放,并没有缓释功能。四、微球微球为药物溶解或分散于高分子材料中(生物可降解)形成的微小球体或类球体。长效缓释微球能显著延长药物制剂的释放和作用时间,减少用药次数;长时间提供平稳血药浓度,避免过高达峰药物浓度引发的副作用。与口服相比,精神类药品长效注射制剂可减少给药次数,一周到几个月注射一次,提高患者用药依从性,减轻监护人的负担。目前主要的长效制剂主要有纳米晶体、凝胶剂(含原位凝胶)和植入剂,相比纳米晶体,微球成品为冻干粉末,稳定性更好;与原位凝胶相比,缓释效果更长;与植入剂相比,无需手术,患者服药方便。目前,市场上还没有卡利拉嗪及其盐酸盐的微球产品,也没有相关的文献报道。针对卡利拉嗪的适用症而言,精神疾病患者服用药物的次数越少越好,因此卡利拉嗪长效制剂的开发很有必要。利用盐酸卡利拉嗪能够提高该药在体内的生物利用度,但是盐酸卡利拉嗪的长效制剂目前还很难满足治疗需求。本发明通过精细、创新的制剂学设计和研究,包括原料药的选择(卡利拉嗪与卡利拉嗪盐酸盐),制剂和工艺研究,以期达到卡利拉嗪微球产品长效缓释的目的。
技术实现要素:
针对现有技术中缺少可长效缓释的卡利拉嗪微球长效注射剂,本发明的目的在于提供一种卡利拉嗪长效缓释微球及其制备方法。本发明的卡利拉嗪微球长效注射剂具有长效缓释的作用,在动物体内可以实现一周到三个月的缓释,能够减少患者给药次数,提高患者的依从性,降低患者家属和国家医疗机构的看护负担。本发明旨在制备可长效缓释的卡利拉嗪制剂,发明人前期实验考察了卡利拉嗪及其盐酸盐的凝胶,发现缓释的效果不佳;也尝试了纳米晶体,但稳定性不理想;继而,发明人以微球制剂作为主要研发方向,考察了卡利拉嗪和其盐酸盐在制备微球时的可行性,也探索了不同溶剂对微球制剂的影响,通过精细、创新的制剂学设计才得出以下可行的方案。本发明的卡利拉嗪长效缓释微球包含卡利拉嗪和聚乳酸-羟基乙酸共聚物(plga);卡利拉嗪的含量为20~80%。卡利拉嗪与聚乳酸-羟基乙酸共聚物有较好亲和力,使得该缓释微球有高载药量。所述的卡利拉嗪为原料本身,非卡利拉嗪的药用盐(包括卡利拉嗪的盐酸盐)。优选的,所述卡利拉嗪和聚乳酸-羟基乙酸共聚物在制备微球时溶解所用的有机
溶剂为二氯甲烷或如下组合:苯甲醇和二氯甲烷、苯甲醇和乙酸乙酯、二氯甲烷和乙醇、二氯甲烷和乙酸乙酯,或乙醇和乙酸乙酯;且两种溶剂的体积比为20%-80%∶80%-20%。进一步优选的,所述有机溶剂为苯甲醇和二氯甲烷,且体积比为苯甲醇∶二氯甲烷为30%~60%∶70%~40%,更优选30%~40%∶70%~60%。进一步优选的,所述有机溶剂为乙醇和二氯甲烷,且体积比为乙醇∶二氯甲烷为20%~50%∶80%~50%,更优选20%~30%∶80%~70%。进一步优选的,所述有机溶剂为苯甲醇和乙酸乙酯,且体积比为苯甲醇∶乙酸乙酯为30%~70%∶70%~30%,更优选40%~60%∶60%~40%,更更优选50%∶50%。优选的,所述卡利拉嗪长效缓释微球的d10粒径为0.1~100μm,d90粒径与d10粒径之差为1~500μm;进一步优选的,d10粒径为1~20μm;更进一步优选的,d90粒径与d10粒径之差为5~300μm更优选50~100μm。其中,d10粒径、d90粒径是指颗粒累积分布为10%、90%的粒径。优选的,卡利拉嗪的含量为30~70%。优选的,聚乳酸-羟基乙酸共聚物的数均分子量为5000~200000,进一步优选10000~100000,更优选20000~70000。本发明的所述卡利拉嗪长效缓释微球可通过以下步骤制备:(a)将卡利拉嗪和聚乳酸-羟基乙酸共聚物溶解于有机溶剂中,得油相;以0.5~5wt%优选0.5~3wt%更优选1~2wt%的聚乙烯醇(pva)的水溶液作为水相;搅拌条件下将油相加入水相中,制成微球乳液;(b)继续搅拌,调整温度至10~45℃,减压固化搅拌1~24小时,过滤收集微粒,以纯化水洗涤,冷冻干燥。优选的,步骤(a)中,卡利拉嗪和聚乳酸-羟基乙酸共聚物质量比为1~3∶1,优选1~2∶1。优选的,步骤(a)中,卡利拉嗪和聚乳酸-羟基乙酸共聚物质量之和与有机溶剂体积的比为1g∶5~30ml,优选1g∶10~20ml。优选的,步骤(a)中,油相与水相之比为1∶100~500,优选1∶100~300。优选的,步骤(a)中,所述有机溶剂为二氯甲烷。或者,步骤(a)中,所述有机溶剂选自苯甲醇、乙醇、二氯甲烷、乙酸乙酯中的两种及两种以上。一些优选的实施例中,所述有机溶剂为:苯甲醇和二氯甲烷、苯甲醇和乙酸乙酯、二氯甲烷和乙醇、二氯甲烷和乙酸乙酯,或
乙醇和乙酸乙酯;且两种溶剂的体积比为20%-80%∶80%-20%。进一步优选的,所述有机溶剂为苯甲醇和二氯甲烷,且体积比为苯甲醇∶二氯甲烷为30%~60%∶70%~40%,更优选30%~40%∶70%~60%。进一步优选的,所述有机溶剂为乙醇和二氯甲烷,且体积比为乙醇∶二氯甲烷为20%~50%∶80%~50%,更优选20%~30%∶80%~70%。进一步优选的,所述有机溶剂为苯甲醇和乙酸乙酯,且体积比为苯甲醇∶乙酸乙酯为30%~70%∶70%~30%,更优选40%~60%∶60%~40%,更进一步优选50%∶50%。另一些较佳实施中,所述有机溶剂为:苯甲醇、二氯甲烷和乙酸乙酯,且三种溶剂的体积比为20-60%∶20%-60%∶20%-60%,优选30-40%∶30%-40%∶30%-40%。优选的,步骤(b)中,调整温度至35~45℃,减压固化搅拌8~16小时。现有卡利拉嗪制剂绝大部分采用的是盐酸卡利拉嗪(盐酸盐),本发明在前期实验中也选择盐酸卡利拉嗪,但是盐酸盐的脂溶性以及与微球辅料的相容性并不是很理想。进一步研究发现:相比其他制剂所用的活性成份
‑‑
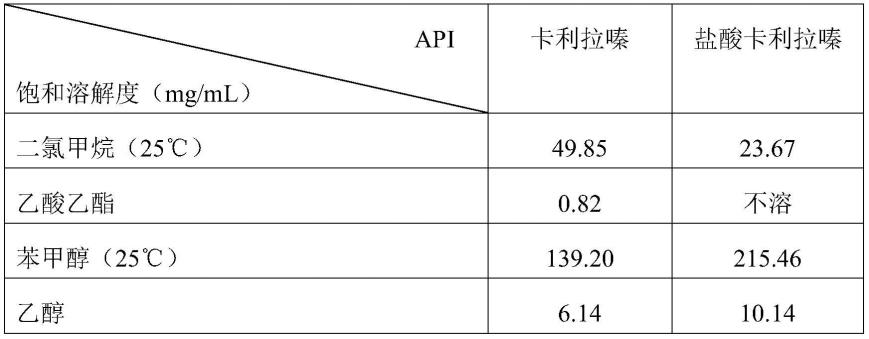
盐酸卡利拉嗪(现有技术认为其溶解度高、生物利用度大),本发明采用卡利拉嗪制备成微球制剂在多种有机溶剂中饱和溶解度更大、适用的溶剂系统更多、与plga的亲和力更高、制备出的微球载药量更高且能够满足体内长期释药的需求。卡利拉嗪和盐酸卡利拉嗪在微球制剂制备中的性能比较实验,具体如下:(1)在溶解度方面卡利拉嗪及盐酸卡利拉嗪在苯甲醇、二氯甲烷中的溶解度差异较大,因此在溶剂系统的选择上,相对于盐酸卡利拉嗪,卡利拉嗪可选的溶剂系统更多,这有利于更大范围选择微球制备工艺。卡利拉嗪及盐酸卡利拉嗪的溶解度见下表。表1两种api在不同溶剂中的饱和溶解度测定值通过上表数据可见,在亲水性有机溶剂苯甲醇和乙醇中盐酸卡利拉嗪的饱和溶解度高于卡利拉嗪;但是,在微球制备主要疏水性溶剂二氯甲烷和乙酸乙酯溶解度均低于卡利拉嗪,而二氯甲烷和乙酸乙酯是微球辅料溶解的必需溶剂,因此,无论在二氯甲烷或乙酸乙酯的单一或与其他溶剂的混合溶剂中,盐酸卡利拉嗪制备的微球均比卡利拉嗪微球产量低或载药量低。相对而言,卡利拉嗪制备微球可选的溶剂系统更多,有利于通过多个溶剂系统筛选出适宜的处方和工艺,并实现预期的体内缓释有效血药浓度和维持时间。
(2)与plga的相似相容性结构分析api与plga的相似相容性对于微球的成球性影响较大,plga的亲脂性较强,而盐酸卡利拉嗪与卡利拉嗪在结构上相比多了一个盐酸基团,该基团增强了盐酸卡利拉嗪的亲水性,减弱了与plga的亲和力,因此盐酸卡利拉嗪微球的成球性及致密度不如卡利拉嗪微球。plga、卡利拉嗪及盐酸卡利拉嗪的结构如下。(3)在理化性质方面相比于盐酸卡利拉嗪,卡利拉嗪在理化性质上与plga的相容性更好,这使得制备的微球载药量也更高;在同一剂量时,载药量高的微球体用量少,可以减少患者的注射疼痛感,并降低生产成本。本发明分别对卡利拉嗪和盐酸卡利拉嗪采用不同的溶剂体系进行了一系列的微球制备实验,同粒径载药量及产率对比见下表。表2卡利拉嗪和盐酸卡利拉嗪采用不同溶剂制备微球的载药量和产率
从上表可见,在相同处方及工艺条件下,二氯甲烷系统及乙醇-二氯甲烷系统无法成功制备盐酸卡利拉嗪微球,而在苯甲醇-二氯甲烷及苯甲醇-乙酸乙酯系统中,卡利拉嗪微球的载药量要显著高于盐酸卡利拉嗪微球,甚至在苯甲醇-乙酸乙酯系统中卡利拉嗪的载药量能够达到盐酸卡利拉嗪的8倍以上;另外,卡利拉嗪的产率同样也高于盐酸卡利拉嗪,在苯甲醇乙酸乙酯系统中,卡利拉嗪的产率能够达到盐酸卡利拉嗪产率的5倍以上,因此从载药量及产率方面来讲,卡利拉嗪相较盐酸卡利拉嗪有更加明显的优势。(4)在缓释效果方面由于精神疾病的特殊性,作为缓释制剂的微球能够减少服药次数,一周一次、一月一次甚至三月一次,给药次数越少越能够增加患者顺应性。相同工艺制备的盐酸卡利拉嗪和卡利拉嗪体外释放结果表明盐酸卡利拉嗪释放快于卡利拉嗪,而卡利拉嗪微球的释放更加缓慢,更易达到延长释放周期、减少给药间隔的制剂设计目标。本发明的积极进步效果在于:本发明的卡利拉嗪长效缓释微球作为卡利拉嗪微球长效注射剂具有长效缓释的作用,在动物体内可以实现一周到三个月的缓释,能够减少患者给药次数,提高患者的依从性,降低患者家属和国家医疗机构的看护负担。
附图说明
图1-图2为本发明实施例8的微球电镜图片;图3为实施例1-4的卡利拉嗪长效缓释微球的体外缓释结果示意图;图4为实施例3、4的卡利拉嗪长效缓释微球的体内缓释结果示意图;图5为实施例5-8的卡利拉嗪长效缓释微球的体外缓释结果示意图;图6为实施例5、8的卡利拉嗪长效缓释微球的体内缓释结果示意图;
图7为实施例9-13的卡利拉嗪长效缓释微球的体外缓释结果示意图;图8为实施例9、13的卡利拉嗪长效缓释微球的体内缓释结果示意图;图9为实施例14-16的卡利拉嗪长效缓释微球的体外缓释结果示意图;图10为实施例16的卡利拉嗪长效缓释微球的体内缓释结果示意图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。实施例1-4(1)以二氯甲烷溶剂系统制备卡利拉嗪长效缓释微球制备步骤如下:将卡利拉嗪与plga溶解于二氯甲烷中,得油相;将聚乙烯醇溶解于纯化水中,制备浓度为0.5~3wt%的溶液,然后溶液经滤膜过滤,得水相;在25℃下,搅拌转速350转/分钟,将油相加入水相中,制成微球乳液;继续搅拌10min,调整转速200转/分钟,调整温度至40℃,控制真空度为-0.095mpa,减压固化搅拌12小时,过滤收集微粒,以纯化水洗涤,冷冻干燥。实施例1~4中卡利拉嗪的用量、plga的用量以及数均分子量、二氯甲烷的用量,水相pva的含量,以及油水比如下表3所示。表3实施例1~4的原料配比(2)载药量的测定采用高效液相色谱法进行微球中载药量(含药量)的测定,色谱条件为:流动相:0.01mol/l磷酸二氢钾水溶液ph值7.0-乙腈(30∶70)(v/v);色谱柱:waters xbridge c18 4.6*150mm*3.5um或相类似的色谱柱;柱温:25℃;流速:1.0ml/min;检测波长:220nm;进样量:20μl。测得实施例1-4的微球载药量如下表4所示。表4实施例1-4的微球载药量 实施例1实施例2实施例3实施例4载药量(%)52.853.636.036.9(3)体外释放实验
采用摇瓶法实施释放度实验,并用高效液相色谱法进行测定。计算方法如下:含药量mg=w样*载药量累积释放量%=(cn*vn+vs*(cn-1+
…
+c1))/含药量*100式中,w样:每份微球的称样量,mg;载药量:每批微球样品测得的含量,%;cn:当前点的浓度mg/ml;vn:当前点的介质体积ml,(本释放实验为200ml);cn-1:前一个时间点的浓度mg/ml;vs:每一个时间点的取样体积ml(本释放实验取样体积为170ml)。色谱测试条件:流动相:0.01mol/l磷酸二氢钾水溶液ph值7.0(称取1.36g磷酸二氢钾加水稀释至1000ml,用氢氧化钾溶液调节ph值为7.0)-乙腈(30∶70);色谱柱:waters xbridge c18 4.6*150mm*3.5um或相类似的色谱柱;柱温:30℃;流速:1.0ml/min;检测波长:220nm;进样量:100μl;运行时间:8分钟。实施例1-4的微球的体外缓释结果如图3所示。体外释放结果表明,以二氯甲烷溶剂系统制备微球时,聚乳酸-羟基乙酸共聚物的数均分子量会在一定程度上影响微球体外释放速度,实施例3中聚乳酸羟基乙酸共聚物的数均分子量最高,其释放速度最慢,实施例1和实施例2的释放速度相对较快,因为二者所使用plga其分子量差别较小,因此对于释放速度可能并未起到决定性影响,实施例2的油水比较小,说明在固化过程中耗费时间更长,微球结构更为致密,即释放相对偏慢。(4)体内释放实验动物:雄性sd大鼠,体重250-300g样品配制:采用20ml注射针头、2ml注射器吸取1ml溶媒,将溶媒缓慢注入样品瓶中,将混悬液轻轻左右振摇使混悬均匀给药方式:皮下注射取血:设置多个取样点,检测不同时间大鼠体内血药浓度分析方法:lc-ms/ms实施例3-4的体内释放曲线如图4所示。体内释放结果表明实施例3在90天时血药浓度仍在有效范围内,能够实现体内90天缓释,实施例4的体内血药浓度在90天时,基本释放完全,说明我们在制备时所掺用的小分子量plga对于其体内释放有一定的促进作用导致其体内释放速度比实施例3快。因此采用二氯甲烷溶剂系统制备微球时,采用高分子量plga(13w)能够实现体内缓释90天的目标,低分子量plga能够促进体内外释放速度。实施例5-8以二氯甲烷-乙醇溶剂系统制备卡利拉嗪长效缓释微球,制备步骤如下:将卡利拉嗪与plga溶解于二氯甲烷和乙醇中,得油相;将聚乙烯醇溶解于纯化水中,制备浓度为0.5~3wt%的溶液,然后溶液经滤膜过滤,得水相;在25℃下,搅拌转速350转/分钟,将油相加入水相中,制成微球乳液;继续搅拌10min,调整转速200转/分钟,调整温度至40℃,控制真空度为-0.095mpa,减压固化搅拌12小时,过滤收集微粒,以纯化水洗涤,冷冻干燥。
实施例5~8中卡利拉嗪的用量、plga的用量以及数均分子量、二氯甲烷和乙醇的用量,水相pva的含量,以及油水比如下表5所示。表5实施例5~8的原料配比表5实施例5~8的原料配比参照实施例1-4的测试方法,测试实施例5-8的微球载药量、体外释放和体内释放效果。图1-2示出了实施例8的微球电镜图片,图片显示微球外观圆整、光滑、均一且无粘连,平均粒径为50~200μm,表6示出了实施例5-8的微球载药量,其中实施例7-1和7-2制备油相时无法完全溶解,图5示出了实施例5-8的微球的体外缓释结果,图6示出了实施例5和实施例8的微球的体内缓释结果。表6实施例5-8的微球载药量 实施例5实施例6实施例7实施例7-1实施例7-2实施例8载药量(%)55.256.555.2不溶不溶55.8结果显示以二氯甲烷-乙醇溶剂系统制备的微球释放趋势总体一致,从体外释放结果来看,均能够在30天完成释药近50%。而通过体内缓释结果来看,实施例5及实施例8在20天左右血药浓度开始下降,能够实现体内30天的缓释,具有良好的体内缓释效果。实施例9-13以二氯甲烷-苯甲醇溶剂系统制备卡利拉嗪长效缓释微球,步骤如下:将卡利拉嗪与plga溶解于二氯甲烷和苯甲醇中,得油相;将聚乙烯醇溶解于纯化水中,制备浓度为0.5~3wt%的溶液,然后溶液经滤膜过滤,得水相;在25℃下,搅拌转速350转/分钟,将油相加入水相中,制成微球乳液;继续搅拌10min,调整转速200转/分钟,调整温度至40℃,控制真空度为-0.095mpa,减压固化搅拌12小时,过滤收集微粒,以纯化水洗涤,冷冻干燥。实施例9-13中卡利拉嗪的用量、plga的用量以及数均分子量、二氯甲烷和苯甲醇的用量,水相pva的含量,以及油水比如下表7所示。表7实施例9~13的原料配比
参照实施例1-4的测试方法,测试实施例9-13的微球载药量、体外释放和体内释放效果。表8示出了实施例9-13的微球载药量,图7示出了实施例9-13的微球的体外缓释结果,图8示出了实施例9和实施例13的微球的体内缓释结果。表8实施例9-13的微球载药量 实施例9实施例10实施例11实施例12实施例13载药量(%)56.658.952.443.851.8体外释放结果表明苯甲醇含量最高的实施例11体外释放速度最快,在12天左右即可释放35%,采用同一型号plga的实施例9、实施例10及实施例12随着plga的占比增大,释放也逐渐加快,因此在此系统内,苯甲醇的含量对于释放速度有一定的影响。体内释放结果表明实施例9在25天左右开始出现明显的下降趋势,而实施例13在20天左右即出现明显的下降趋势,说明实施例9基本能够实现30天的缓释,但是实施例13可实现3周缓释。实施例14-16以乙酸乙酯-苯甲醇溶剂系统制备卡利拉嗪长效缓释微球,步骤如下:将卡利拉嗪与plga溶解于乙酸乙酯和苯甲醇中,得油相;将聚乙烯醇溶解于纯化水中,制备浓度为2wt%的溶液,然后溶液经滤膜过滤,得水相;在25℃下,搅拌转速350转/分钟,将油相加入水相中,制成微球乳液;继续搅拌10min,调整转速200转/分钟,调整温度至40℃,控制真空度为-0.095mpa,减压固化搅拌12小时,过滤收集微粒,以纯化水洗涤,冷冻干燥。实施例14~16中卡利拉嗪的用量、plga的用量以及数均分子量、乙酸乙酯和苯甲醇的用量,水相pva的含量,以及油水比如下表9所示。表9实施例14~16的原料配比
参照实施例1-4的测试方法,测试实施例14-16的微球载药量、体外释放和体内释放效果。表10示出了实施例14-16的微球载药量,其中,实施例14-1制备油相时未能完全溶解,实施例14-2制得的产品不成球形,图9示出了实施例9-13的微球的体外缓释结果,图10示出了实施例16的微球的体内缓释结果。表10实施例14-16的微球载药量 实施例14实施例14-1实施例14-2实施例15实施例16载药量(%)45.75不溶解不成球形43.3742.5在苯甲醇乙酸乙酯系统中,实施例16的体内血药浓度在30天内保持平稳释放趋势,达到一个月缓释目标。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1