用乌帕替尼治疗脊柱关节炎和银屑病的制作方法
用乌帕替尼治疗脊柱关节炎和银屑病
1.相关申请的交叉引用
2.本技术要求于2020年5月29日提交的美国临时专利申请第63/032,042号;2020年1月 31日提交的美国临时专利申请第62/968,849号;2019年10月29日提交的美国临时专利申请第62/927,548号;以及2019年9月30日提交的美国临时专利申请第62/908,163号的优先权,所有这些都通过引用整体并入本文。
背景技术:
3.中轴型脊柱关节炎(axspa)包括一系列炎症性累及中轴骨骼。根据国际脊柱关节炎评估协会(asas)的axspa标准,该疾病可通过放射学表现进一步分为2类:强直性脊柱炎 (as)和“早期”形式的中轴型spa,称为非放射学中轴型脊柱关节炎(nr-axspa)。nr-axspa 和as患者具有共同的流行病学、遗传和临床疾病特征,包括疾病活动性和对治疗的相似应答。参见例如,poddubnyy和sieper,《风湿病学时论(curr opinrheumatol.)》(2014)26:377-383。
4.根据国际治疗建议,非甾体抗炎药(nsaid)是axspa的一线治疗。参见,例如,van derheijde d等人,《风湿病学年鉴(ann rheum dis.)》(2017)76:978-991;ward等人,《关节炎和风湿病(arthritis rheumatol.)》(2016)68:282-298。在最多四个星期内服用两种非甾体抗炎药失败后,生物疾病改善抗风湿药(bdmard)是下一个推荐的治疗选择。在axspa,传统的合成的改善疾病的抗风湿药(csdmard)和长期使用皮质类固醇无效,因此不推荐用于治疗中轴症状。参见,例如,van der heijde d等人,《风湿病学年鉴(ann rheum dis.)》(2017) 76:978-991。此外,只有大约45%至50%的患者表现出国际脊柱关节炎评估协会40(asas40) 应答,并且只有大约15%至20%在未接受过生物治疗的患者中达到缓解状态,而对bdmard 应答不足的axspa患者的应答率甚至更低。参见,例如,sieper和poddubnyy,《柳叶刀杂志 (lancet)(2017)390:73-84;sieper等人,《风湿病学年鉴》(2017)76:571-592;rudwaleit等人,《关节炎研究与治疗(arthritis res ther.)》(2010)12:r117;deodhar等人,《关节炎风湿病》(2019)71:599-611。迄今为止,除nsaid外,尚无口服靶向疗法获批用于治疗强直性脊柱炎(as)或非放射学axspa。
5.银屑病关节炎(psa)是一种慢性全身性炎症性疾病,属于脊柱关节炎(spa)的一种亚型,其特征是关节炎和银屑病相关。psa的病程通常以发作和缓解为特征。如果不加以治疗, psa患者可能会出现持续性炎症、进行性关节损伤、残疾和预期寿命缩短。肌肉骨骼症状的初始治疗由非甾体抗炎药(nsaid)和局部皮质类固醇注射组成,而外用疗法则用于银屑病的初始治疗。对于这些措施缺乏疗效或毒性的受试者,建议对应答不足的受试者使用非生物疾病改善抗风湿药(非生物dmard)(例如,甲氨蝶呤[mtx]、来氟米特[lef]、柳氮磺胺吡啶[ssz])和环孢素a进行系统治疗,然后进行抗肿瘤坏死因子(tnf)治疗。其它生物疗法(例如,il-12/23或il-17抑制剂)也被推荐作为选定psa受试者的抗tnf抑制剂的替代品。参见,例如,gossec等人,《风湿病学年鉴》(2016)75:499-510;coates等人,《关节炎风湿病》(2016)68:1060-71。然而,尽管目前可用的生物制剂取得了有益的结果,但大约40%的患者
在美国风湿病学会(acr)评分中没有至少20%的改善,并且只有58%至61%的psa患者在接受治疗1年后能够达到临床缓解,只有大约43%的患者达到至少1年的持续缓解。参见,例如,gossec等人,《风湿病学年鉴》(2016)75:499-510;alamanos等人,《风湿病学杂志(jrheumatol.)》(2003)30:2641-2644;savolainen等人,《风湿病学杂志》(2003)30:2460-8;sandborn,《消化疾病(digdis.)》(2010)28:536-42;sabre等人,《关节炎研究与治疗(arthritisrestherapy)》(2010)12:r94;perrotta等人,《风湿病学杂志》(2016)43:350-5。
[0006]
因此,对于治疗非放射学中轴型脊柱关节炎(nr-axspa)、强直性脊柱炎(as)、银屑病关节炎(psa)和银屑病(pso)(包括作为psa皮肤体征的pso)的其它治疗选择,仍然存在明确的医疗需求。
技术实现要素:
[0007]
本公开解决了上述需求并提供了用于治疗中轴型脊柱关节炎(axspa),包括非放射学axspa(nr-axspa)和强直性脊柱炎(as)的方法,以及用于治疗银屑病关节炎(psa)和银屑病(pso),包括作为psa皮肤体征的pso的方法。
[0008]
以下列举的实施例1至77阐述了如本文所述的方法的某些方面。
[0009]
实施例1:在某些方面,提供了一种在有需要的受试者中治疗活动性强直性脊柱炎(as)的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少14周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用第一剂后的14周内达到国际脊柱关节炎评估协会40(asas40)应答。
[0010]
实施例2:根据实施例1所述的方法,其中当所述方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的14周内达到asas40应答。在实施例1所述的方法的某些实施例中,其中当所述方法用于治疗受试者群体时,所述治疗群体中统计学显着的所述受试者群体在施用所述第一剂后的14周内达到asas40应答。
[0011]
实施例3:根据实施例1或2所述的方法,其中所述治疗群体中在基线时患有活动性as的所述受试者或多名受试者在施用所述第一剂后的14周内进一步达到选自由以下组成的组的至少一个结果:
[0012]
a.强直性脊柱炎疾病活动性评分(asdas)较基线有所改善;
[0013]
b.脊柱的磁共振成像(mri)加拿大脊柱关节炎研究协会(sparcc)评分(mri-spinesparcc)较基线有所改善;
[0014]
c.asas部分缓解(pr);
[0015]
d.巴氏强直性脊柱炎疾病活动指数50(bathankylosingspondylitisdiseaseactivityindex50,basdai50)应答;
[0016]
e.巴氏强直性脊柱炎功能指数(bathankylosingspondylitisfunctionalindex,basfi)较基线有所改善;
[0017]
f.asdas低疾病活动性(lda);
[0018]
g.asdas非活动性疾病(id);
[0019]
h.asdas重大改善(mi);和
[0020]
i.asdas临床重要改善(cii)。
[0021]
在实施例1或2的方法的某些实施例中,当所述方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的14周内达到这些结果中的至少一个。在实施例1或2的方法的其它实施例中,当所述方法用于治疗受试者群体时,所述治疗群体中统计学显着的受试者群体在施用第一剂后的14周内达到至少一个结果。
[0022]
实施例4:根据实施例3所述的方法,其中所述治疗群体中在基线时患有活动性as的所述受试者或多名受试者在施用第一剂后的14周内进一步达到每个结果。
[0023]
实施例5:根据实施例1至4中任一项所述的方法,其中所述治疗群体中的所述受试者或多名受试者在基线时满足1984年修订的强直性脊柱炎纽约标准。
[0024]
实施例6:根据实施例1至5中任一项所述的方法,其中所述治疗群体中的所述受试者或多名受试者在基线时满足2009年asas分类标准。
[0025]
实施例7:根据实施例1至6中任一项所述的方法,其中所述治疗群体中的所述一名受试者或多名受试者在基线时满足选自由以下组成的组的至少一个标准:
[0026]
a.巴氏强直性脊柱炎疾病活动指数(basdai)评分≥4;
[0027]
b.强直性脊柱炎疾病活动性评分(asdas)≥2.1;以及
[0028]
c.患者对总背痛的评估(总背痛评分)≥4,基于0至10数字评定量表。
[0029]
实施例8:根据实施例1至7中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时未接受过生物疾病改善抗风湿药(bdmard)。
[0030]
实施例9:根据实施例1至7中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时对生物疾病改善抗风湿药(bdmard)应答不足或不耐受。
[0031]
实施例10:根据实施例9所述的方法,其中在施用所述第一剂之前,所述治疗群体中的所述一名或多名受试者已经施用一种bdmard,并且由于不耐受或缺乏疗效而停止使用 bdmard。
[0032]
实施例11:根据实施例10所述的方法,其中所述bdmard是肿瘤坏死因子(tnf)抑制剂或白细胞介素(il)-17抑制剂。
[0033]
实施例12:根据实施例1至11中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时对至少两种nsaid应答不足或不耐受、对nsaids不耐受和/或对 nsaid有禁忌症。
[0034]
实施例13:在其它方面,提供了一种在有需要的受试者中治疗活动性非放射学中轴型脊柱关节炎的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少14周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用第一剂后的14周内达到asas40应答。在某些实施例中,当所述方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的14周内达到asas40应答。在某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的14周内达到asas40应答。
[0035]
实施例14:在其它方面,提供了一种在有需要的受试者中治疗活动性非放射学中轴型脊柱关节炎的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替
尼游离碱或其药学上可接受的盐,持续至少52周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的52周内达到asas40应答。在某些实施例中,当所述方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的52周内达到asas40应答。在某些实施例中,所述治疗群体中统计学显着的受试者群体在施用第一剂后的52周内达到asas40应答。
[0036]
实施例15:根据实施例13或14所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时满足2009年asas中轴型脊柱关节炎分类标准,但不满足1984年修订的强直性脊柱炎纽约标准的放射学标准。
[0037]
实施例16:根据实施例13或14所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时满足选自由以下组成的组的至少一个标准:
[0038]
a.巴氏强直性脊柱炎疾病活动指数(basdai)评分≥4;
[0039]
b.强直性脊柱炎疾病活动性评分(asdas)≥2.1;
[0040]
c.患者对总背痛的评估(总背痛评分)≥4,基于0至10数字评定量表;以及
[0041]
d.炎症活动的客观征像,选自由以下组成的组:
[0042]
i.骶髂(si)关节mri上活动性炎症的客观征象,和
[0043]
ii高敏c反应蛋白>正常上限(uln)。
[0044]
实施例17:根据实施例13至16中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时未接受过bdmard。
[0045]
实施例18:根据实施例13至16中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时对bdmard应答不足或不耐受。
[0046]
实施例19:根据实施例18所述的方法,其中在施用所述第一剂之前,所述治疗群体中的所述一名或多名受试者已经施用一种bdmard,并且由于不耐受或缺乏疗效而停用所述 bdmard。
[0047]
实施例20:根据实施例19所述的方法,其中所述bdmard是肿瘤坏死因子(tnf)抑制剂或白细胞介素(il)-17抑制剂。
[0048]
实施例21:根据实施例13至20中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时对至少2种nsaid应答不足或不耐受、对nsaids具有不耐受和/或对nsaid有禁忌症。
[0049]
实施例22:根据实施例13至21中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的14周内达到选自由以下组成的组的至少一个额外结果:
[0050]
a.强直性脊柱炎疾病活动性评分(asdas)较基线有所改善;
[0051]
b.si关节的磁共振成像(mri)加拿大脊柱关节炎研究协会(sparcc)评分(mri-si 关节sparcc)较基线有所改善;
[0052]
c.asas部分缓解(pr);
[0053]
d.巴氏强直性脊柱炎疾病活动指数(basdai)50应答;
[0054]
e.巴氏强直性脊柱炎功能指数(bath ankylosing spondylitis functional index,basfi) 较基线有所改善;
[0055]
f.强直性脊柱炎生活质量(asqol)较基线有所改善;
[0056]
g.asas健康指数(hi)较基线有所改善;
[0057]
h.马斯特里赫特强直性脊柱炎附着点炎评分(maastrichtankylosingspondylitisenthesitisscore,mases)较基线有所改善;以及
[0058]
i.线性巴氏强直性脊柱炎计量指数(basmi
lin
)较基线有所改善。
[0059]
在实施例13至21中任一项所述的方法的某些实施例中,其中当所述方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的14周内达到至少一个结果。在实施例13至21中任一项所述的方法的某些实施例中,其中当所述方法用于治疗受试者群体时,所述治疗群体中统计学显着的受试者群体在施用第一剂后的14周内达到至少一个结果。
[0060]
实施例23:在其它方面,提供了一种在有需要的受试者中治疗活动性银屑病关节炎(psa)的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少12周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的12周内达到美国风湿病学会20%(acr20)应答。
[0061]
实施例24:在又其它方面,提供了一种在有需要的受试者中治疗活动性银屑病关节炎(psa)的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少12周,其量足以递送30mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的12周内达到美国风湿病学会20%(acr20)应答。
[0062]
实施例25:根据实施例23或24所述的方法,其中当所述方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的12周内达到acr20应答。在实施例23或24所述的方法的其它实施例中,当所述方法用于治疗受试者群体时,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的12周内达到acr20应答。
[0063]
实施例26:根据实施例23至25中任一项所述的方法,其中所述治疗群体中在基线时患有活动性psa的所述一名或多名受试者进一步达到选自由以下组成的组的至少一个结果:
[0064]
a.在施用所述第一剂后的12周内,健康评估问卷残疾指数(haq-di)较基线有所改善;
[0065]
b.在施用所述第一剂后的16周内,银屑病静态研究者总体评估(staticinvestigatorglobalassessment,siga)达到0或1,且较基线改善至少2分(对于基线siga≥2的受试者);
[0066]
c.在施用所述第一剂后的16周内达到银屑病面积严重程度指数(psoriasisareaseverityindex,pasi)75应答(对于基线时bsa≥3%的银屑病受试者);
[0067]
d.在施用所述第一剂后的24周内sharp/vanderheijde评分(shs)较基线有所改善;
[0068]
e.在施用所述第一剂后的24周内达到最小疾病活动性(mda);
[0069]
f.在施用所述第一剂后的24周内,利兹附着点炎指数(leedsenthesitisindex,lei)较基线有所改善,优选地其中所述改善是在施用所述第一剂后的24周内附着点炎消
退(lei= 0)(对于基线存在附着点炎(lei>0)的受试者);
[0070]
g.在施用所述第一剂后的12周内达到acr 20应答(乌帕替尼与阿达木单抗 (adalimumab)相比的非劣效性);
[0071]
h.在施用所述第一剂后的12周内,36项简表健康调查(sf-36)较基线有所改善;以及
[0072]
i.在施用所述第一剂后的12周内,慢性病治疗疲劳功能评估(facit-f)问卷较基线有所改善。
[0073]
在某些实施例中,其中当根据实施例23至25所述的方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少 45%的所述受试者在施用所述第一剂后的14周内达到至少一个结果。在其它实施例中,其中当根据实施例23至25所述的方法用于治疗受试者群体时,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的14周内达到至少一个结果。
[0074]
实施例27:根据实施例26所述的方法,其中所述治疗群体中在基线时患有活动性psa 的所述一名或多名受试者进一步达到每个结果。
[0075]
实施例28:根据实施例23至27中任一项所述的方法,其中所述治疗群体中在基线时患有活动性psa的所述一名或多名受试者进一步达到选自由以下组成的组的至少一个结果:
[0076]
a.在施用所述第一剂后的12周内,acr 20应答并优于阿达木单抗(每隔一周40mg);以及
[0077]
b.在施用所述第一剂后的24周内,利兹指趾炎指数(leeds dactylitis index,ldi)较基线有所改善,优选地其中所述改善是在施用所述第一剂后的24周内指趾炎消退(ldi=0) (对于基线存在指趾炎(ldi>0)的受试者)。
[0078]
在实施例28的某些实施例中,其中当所述方法用于治疗受试者群体时,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、或至少45%的所述受试者在施用所述第一剂后的14周内达到至少一个结果。在实施例28的其它实施例中,其中当所述方法用于治疗受试者群体时,所述治疗群体中统计学显着的受试者群体在施用第一剂后的14周内达到至少一个结果。
[0079]
实施例29:根据实施例28所述的方法,其中所述治疗群体中患有活动性psa的所述一名或多名受试者在施用所述第一剂后的12周内进一步达到acr 20应答并优于阿达木单抗 (每隔一周40mg)。在实施例29的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用第一剂后的12周内达到acr20应答并优于阿达木单抗。在实施例29的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的12周内达到acr20应答并优于阿达木单抗。
[0080]
实施例30:根据实施例29所述的方法,其中所述治疗群体中患有活动性psa的所述一名或多名受试者每天一次口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少12周,其量足以递送30mg的乌帕替尼游离碱当量。
[0081]
实施例31:根据实施例23至30中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的12周内达到acr 50%应答(acr50)。在实施例31
的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的12周内达到 acr50应答。在实施例31的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者在施用所述第一剂后的12周内达到acr50应答。
[0082]
实施例32:根据实施例23至30中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的12周内达到acr 70%应答(acr70)。在实施例32的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的12周内达到acr70应答。在实施例32的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的12 周内达到acr70应答。
[0083]
实施例33:根据实施例23至32中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时满足银屑病关节炎分类标准(caspar)的标准。
[0084]
实施例34:根据实施例23至33中任一项的权利要求所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时具有选自由≥3个压痛关节(基于68个关节计数)和≥3个肿胀关节(基于66个关节计数)组成的组的至少一个标准。
[0085]
实施例35:根据实施例34所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时具有≥5个压痛关节(基于68个关节计数)和≥5个肿胀关节(基于66个关节计数)。
[0086]
实施例36:根据实施例23至35中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时具有选自由以下组成的组的至少一个标准:如通过中央成像检查确定的≥1个x光片侵蚀,和hs-crp>实验室定义的正常上限(uln)。
[0087]
实施例37:根据实施例23至36中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者具有活动性斑块状银屑病的诊断,或所述受试者在基线时具有斑块状银屑病的记录病史。
[0088]
实施例38:根据实施例23至36中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时对至少一种生物疾病改善抗风湿药(bdmard)应答不足或不耐受。
[0089]
实施例39:根据实施例38所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂之前已经停止了所有bdmard。
[0090]
实施例40:根据实施例23至36中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时对先前或同时使用至少一种非生物dmard的治疗应答不足或不耐受,或对非生物dmard不耐受或有禁忌症。
[0091]
实施例41:根据实施例23至40中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时具有中度至重度活动性银屑病关节炎。
[0092]
实施例42:在其它方面,提供了一种在有需要的受试者中治疗活动性银屑病的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)75应答。
[0093]
实施例43:在其它方面,提供了一种在有需要的受试者中治疗活动性银屑病的方
法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16周,其量足以递送30mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)75应答。
[0094]
实施例44:根据实施例42或43所述的方法,其中当所述方法用于治疗受试者群体时,所述治疗群体中的一部分受试者在施用第一剂后的16周内达到pasi 75应答。在实施例44 的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的16周内达到pasi 75应答。在实施例44的其它实施例中,所述治疗群体中统计学显着的所述受试者群体在施用第一剂后的16周内达到pasi 75应答。
[0095]
实施例45:根据实施例1至44或46至77中任一项所述的方法,其中所述受试者是成年受试者,或所述治疗群体中的所述受试者是成年受试者。
[0096]
实施例46:根据实施例1至12中任一项所述的方法,其中在第14周后通过继续施用每日剂量来维持或改善所述asas40应答。在一个方面,所述asas40应答被维持或改善直至第64周(包括第64周)。
[0097]
实施例47:根据实施例1至12中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的2周内进一步达到asas40。
[0098]
实施例48:根据实施例1至12中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的2周内进一步达到asas40,并且其中在第14周后通过继续施用每日剂量来维持或改善所述asas40。
[0099]
实施例49:在另一方面,提供了一种在有需要的受试者中治疗活动性强直性脊柱炎的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少14周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的14周内达到asas部分缓解(pr)、asdas低疾病活动性(lda)、asdas 非活动性疾病(id)、asdas重大改善(mi)和/或asdas临床重要改善(cii)。
[0100]
实施例50:根据实施例49所述的方法,其中当所述方法用于治疗受试者群体时,所述治疗群体中的一部分受试者在施用所述第一剂后的14周内达到asas部分缓解(pr)、asdas 低疾病活动性(lda)、asdas非活动性疾病(id)、asdas重大改善(mi)和/或asdas 临床重要改善(cii)。在实施例50的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的14周内达到asas部分缓解(pr)、asdas低疾病活动性(lda)、asdas非活动性疾病(id)、asdas重大改善(mi)和/或asdas临床重要改善(cii)。在实施例50的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的14周内达到 asas部分缓解(pr)、asdas低疾病活动性(lda)、asdas非活动性疾病(id)、asdas 重大改善(mi)和/或asdas临床重要改善(cii)。
[0101]
实施例51:根据实施例49或50所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的14周内进一步达到每个结果。
[0102]
实施例52:根据实施例49至51中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时满足1984年修订的强直性脊柱炎纽约标准。
[0103]
实施例53:根据实施例49至51中任一项所述的方法,其中所述治疗群体中的所述
一名或多名受试者在基线时满足2009年asas分类标准。
[0104]
实施例54:根据实施例49至53中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时满足选自由以下组成的组的至少一个标准:
[0105]
a.巴氏强直性脊柱炎疾病活动指数(basdai)评分≥4;
[0106]
b.强直性脊柱炎疾病活动性评分(asdas)≥2.1;以及
[0107]
c.患者对总背痛的评估(总背痛评分)≥4,基于0至10数字评定量表。
[0108]
实施例55:根据实施例49至54中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时未接受过生物疾病改善抗风湿药(bdmard)。
[0109]
实施例56:根据实施例49至55中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在基线时对生物疾病改善抗风湿药(bdmard)应答不足或不耐受。
[0110]
实施例57:根据实施例56所述的方法,其中在施用所述第一剂之前,所述群体中的所述一名或多名受试者已经施用一剂bdmard,并且由于不耐受或缺乏疗效而停用所述 bdmard。
[0111]
实施例58:根据实施例57所述的方法,其中所述bdmard是肿瘤坏死因子(tnf)抑制剂或白细胞介素(il)-17抑制剂。
[0112]
实施例59:根据实施例49至58中任一项所述的方法,其中所述群体中的所述一名或多名受试者在基线时对至少两种nsaid应答不足或不耐受、对nsaids不耐受和/或对nsaid 有禁忌症。
[0113]
实施例60:根据实施例49至59中任一项所述的方法,其中所述asas部分缓解(pr)、 asdas低疾病活动性(lda)、asdas非活动性疾病(id)、asdas重大改善(mi)和/或 asdas临床重要改善(cii)在第14周后通过继续施用每日剂量得以维持或改善。
[0114]
实施例61:根据实施例49至60中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的2周内进一步达到asas部分缓解(pr)、asdas低疾病活动性(lda)、asdas非活动性疾病(id)、asdas重大改善(mi)和/或asdas临床重要改善(cii)。在实施例61的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的2周内达到asas部分缓解(pr)、asdas低疾病活动性(lda)、asdas 非活动性疾病(id)、asdas重大改善(mi)和/或asdas临床重要改善(cii)。在实施例 61的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的2周内达到asas部分缓解(pr)、asdas低疾病活动性(lda)、asdas非活动性疾病(id)、asdas重大改善(mi) 和/或asdas临床重要改善(cii)。
[0115]
实施例62:根据实施例23至41中任一项所述的方法,其中在第12周后通过继续施用每日剂量来维持或改善所述acr评分。
[0116]
实施例63:根据实施例23至41或62中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的2周内进一步达到acr20。
[0117]
实施例64:根据实施例42至44中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)90应答。
[0118]
实施例65:根据实施例42至44或64中任一项所述的方法,其中在第16周后通过继
续施用每日剂量来维持或改善所述pasi应答。
[0119]
实施例66:在另一方面,提供了一种在有需要的受试者中治疗活动性银屑病关节炎的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上的接受剂,持续至少24周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的24周内达到最小疾病活动性(mda)。
[0120]
实施例67:在另一方面,提供了一种在有需要的受试者中治疗活动性银屑病关节炎的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少24周,其量足以递送30mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的24周内达到最小疾病活动性(mda)。
[0121]
实施例68:根据实施例66或67的方法,其中当所述方法用于治疗受试者群体时,所述治疗群体中的一部分受试者在施用所述第一剂后的24周内达到最小疾病活动性(mda)。在实施例68的某些实施例中,所述治疗群体中统计学显着的所述受试者群体在施用所述第一剂后的24周内达到最小疾病活动性(mda)。在实施例68的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的24周内达到最小疾病活动性(mda)。
[0122]
实施例69:根据实施例66至68中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的16周内进一步达到选自pasi 75应答、pasi 90应答和 pasi 100应答的银屑病面积严重程度指数(pasi)应答,并且在第16周后通过继续施用每日剂量来维持或改善所述pasi应答。在实施例69的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的16周内达到pasi 75应答、pasi 90应答和pasi 100 应答。在实施例69的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少 25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的16周内达到pasi 75应答、pasi 90应答和pasi 100应答。
[0123]
实施例70:根据实施例23至42中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)75应答。在实施例70的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的16周内达到pasi 75应答。在实施例70的某些实施例中,所述治疗群体中至少10%、至少 15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的16周内达到pasi 75应答。
[0124]
实施例71:根据实施例23至42或70中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)90 应答。
[0125]
实施例72:根据实施例23至42或70至71中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数 (pasi)100应答。
[0126]
实施例73:在另一方面,提供了一种在有需要的受试者中治疗活动性银屑病关节炎的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16周,其量足以递送15mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)75应答。
[0127]
实施例74:在另一方面,提供了一种在有需要的受试者中治疗活动性银屑病关节炎的方法,所述方法包含每天一次向所述受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16周,其量足以递送30mg的乌帕替尼游离碱当量,其中所述受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)75应答。
[0128]
实施例75:根据实施例73或74所述的方法,其中当所述方法用于治疗受试者群体时,所述治疗群体中的一部分受试者在施用所述第一剂后的16周内达到pasi 75应答。在实施例 75的某些实施例中,所述治疗群体中统计学显着的受试者群体在施用所述第一剂后的16周内达到pasi 75应答。在实施例75的某些实施例中,所述治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的所述受试者在施用所述第一剂后的16周内达到pasi 75应答。
[0129]
实施例76:根据实施例73至75中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)90应答。
[0130]
实施例77:根据实施例73至76中任一项所述的方法,其中所述治疗群体中的所述一名或多名受试者在施用所述第一剂后的16周内达到银屑病面积严重程度指数(pasi)100应答。
附图说明
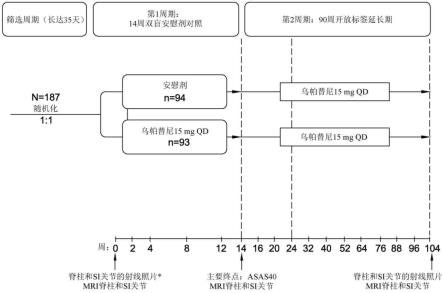
[0131]
图1描绘了2/3期强直性脊柱炎(select-axis 1)临床研究计划。星号(*)表示在筛选期间进行了射线照相。asas40=国际脊柱关节炎评估协会40%应答。mri=磁共振成像。qd=每天一次。si=骶髂关节。
[0132]
图2描绘了2/3期强直性脊柱炎(select-axis 1)临床试验中使用hochberg程序的多重性对照分析。星号(*)表示结果在多重性对照分析中具有统计学显着性,否则示出标称p值。多重性对照的终点以顺序方式进行测试,最初分配的α=0.05。只有当序列中的前一个终点满足统计显着性要求时,才能为排序较低的终点声明统计显着性(p<0.05)。只有当通过hochberg 程序测试的终点组都具有统计显着性时,才能评估asashi。在hochberg程序中,basdai50、 basfi和asas pr达到了所需的统计显着性;但wpai、mases、basmi和asqol不符合统计显着性要求,因此未测试asas hi。根据hochberg程序,根据标称p值的大小,所有终点都使用分配的α进行测试,从最大值开始。如果某个终点被拒绝,则p值较小的所有终点都将被拒绝。如果一个终点发生故障,则程序前进到下一个终点。asas=国际脊柱关节炎评估协会。asas40=asas 40%应答。asas hi=asas健康指数。asas pr=asas部分缓解。 asdas=强直性脊柱炎疾病活动性评分。asqol=强直性脊柱炎生活质量。basdai50=巴氏强直性脊柱炎疾病活动指数较基线改善50%。basfi=巴氏强直性脊柱炎功能指数。basmi=巴氏强直性脊柱炎计量指数。mases=马斯特里赫特强直性脊柱炎附着点炎评分。mri=磁共振成像。qd=每天一次。sparcc=加拿大脊柱关节炎研究协会。wpai=工作效率和活动障碍。
[0133]
图3a至3c和图3d至3n分别描绘了2/3期强直性脊柱炎(select-axis 1)关键临床疗效终点的临床试验的第14周结果,以及直到第64周(包括第64周)的关键终点的时间进程。图3a描绘了在第14周的asas20、asas40、asas pr和basdai50应答;图3b描绘了第14周时
sparcc mri脊柱和si关节评分相对于基线的变化;以及图3c描绘了第14周的其它多重性对照的关键次要疗效终点。结果表明,该研究达到了其主要终点,接受乌帕替尼游离碱治疗的患者与安慰剂相比,在统计学上显着更多患者在低14周达到asas40应答 (48/93[51.6%]与24/94[25.5%]相比;p=0.0003),治疗差异(95%ci)为26.1%(12.6至39.5%)。除asas20和sparcc mri si关节外,所有终点均受多重性对照。多重性对照的次要终点以顺序方式进行测试:asdas、sparcc mri脊柱、hochberg程序测试的一组终点(basdai50、 asqol、asas pr、basfi、basmi、mases和wpai)和asas hi。对于图3l至3n,星号(***)表示p<0.001,星号(**)表示p<0.01;和星号(*)表示p<0.05。对于其他附图,星号(*)表示在多重性对照分析中具有统计学显着性,否则示出标称p值。考虑多重性调整,在第14周的asdas(图3c)、sparcc mri脊柱(图3b)和basfi(图3c)相对于基线的变化以及达到basdai50(图3a)和asas pr(图3a)的患者比例对于乌帕替尼游离碱与安慰剂相比具有统计学显着性。图3d至3k描绘了asas40(图3d)、asas20(图 3e)、asas部分缓解(pr)(图3f)、basdai50应答(图3g)、asdas非活动性疾病(id) (图3h)、asdas低疾病活动性(lda)(图3i)、asdas重大改善(mi)(图3j)和asdas 临床重要改善(ch)(图3k)直至第64周(包括第64周)的时间进程。在第14周,安慰剂组获救并施用15mg的乌帕替尼游离碱(安慰剂
→
乌帕替尼15mg qd)。与从第0天开始持续接受乌帕替尼游离碱的患者相比,在第14周从安慰剂转为乌帕替尼的患者表现出相似的疗效应答。数据表明,15mg qd乌帕替尼,在第14周表现出疗效,并在直至且包括第64周维持甚至改善该疗效,这将有助于解决as患者(包括非放射学中轴型脊柱关节炎(nr-axspa) 患者)未得到满足的需求,尤其是在那些患有活动性疾病且对非甾体抗炎药应答不足的患者中。与安慰剂相比,有显着更高比例的接受乌帕替尼的患者最早在第2周达到患者总体疼痛评估(ptpain)减轻≥30%(图3l)和减轻≥50%(图3m),以及最早在第4周减轻≥70%(图3n),并且此后所达到的疗效得以维持。在第14周从安慰剂转为开放标签乌帕替尼的患者在第14 周后通常达到与最初随机接受乌帕替尼相同的疼痛减轻水平。mases评估包括基线附着点炎患者;wpai评估包括目前受聘的患者;统计分析计划中预先指定的sparcc mri评估群体 (基线包括研究药物首次给药后≤3天的mri数据,第14周包括直至第2期研究药物首次给药的mri数据)。asas20=国际脊柱关节炎评估协会20应答。asas40=国际脊柱关节炎评估协会40应答。asqol=强直性脊柱炎生活质量评分。asdas=强直性脊柱炎疾病活动性评分。 basdai50=巴氏强直性脊柱炎疾病活动指数较基线改善50%。basfi=巴氏强直性脊柱炎功能指数。basmi=巴氏强直性脊柱炎计量指数。hi=健康指数。mases=马斯特里赫特强直性脊柱炎附着点炎评分。mri=磁共振成像。mmrm=重复测量的混合模型。nri=无应答者插补。 ao=如观察到的。pr=部分缓解。qd=每天一次。si,骶髂关节。sparcc=加拿大脊柱关节炎研究协会。wpai=工作效率和活动障碍。
[0134]
图4a至4e描绘了在2/3期强直性脊柱炎(select-axis 1)临床试验的第2、4、8、12 和14周测量的asas域(asas40、ptga、背痛、basfi和炎症)的数据。早在第一次基线检查后(第2周)就观察到乌帕替尼游离碱与安慰剂在asas40中的显着差异(图4a)及其四个单独域中的每一个的平均变化(图4b至4e),并且这种差异一直持续到第14周,第14 周在多重性对照分析中实现了统计学上的显着差异。背痛定义为基于以下问题的数字评定量表(0至10),“在上周的任何时间,您经历的背痛程度是多少?炎症定义为basdai问题5 和6的平均值。basdai=巴氏强直性脊柱炎疾病活动指数。basfi=巴氏强直性脊柱炎功能指数。bl=基
线。lsm=最小二乘均值。mmrm=重复测量的混合模型。nri=无应答者插补。 ptga=疾病活动性的患者总体评估。qd=每天一次。
[0135]
图5描绘了2/3期强直性脊柱炎(select-axis 1)临床试验的预先指定和补充的sparccmri分析。在统计分析计划中预先指定了sparcc mri评估群体(基线包括研究药物首次给药后≤3天的mri数据,并且第14周包括直至第2期研究药物首次给药的mri数据)。补充 sparcc mri分析包括在基线和第14周的标称检查时收集的所有mri数据,并证实了脊柱和si关节的主要sparcc mri分析的结果。mmrm=重复测量的混合模型。mri=磁共振成像。qd=每天一次。si=骶髂关节。sparcc=加拿大脊柱关节炎研究协会。
[0136]
图6a至6d描绘了如图5所述的2/3期强直性脊柱炎(select-axis 1)的sparcc评分变化的累积概率图,表明与安慰剂相比,接受乌帕替尼的患者的sparcc mri脊柱和si关节评分从基线到第14周的改善程度更大。初级mri分析(图6a至6b)和补充mri分析(图 6b至6c)的结果是一致的。
[0137]
图7a至7c描绘了在2/3期强直性脊柱炎(select-axis 1)临床试验中,在第14周达到 asdas lda、asdas id、asdas cii和asdas mi的患者百分比(图7a)、平均asdas 较基线随时间的变化(图7b)和asdas mi随时间的编号(图7c)。在第14周,与安慰剂相比,乌帕替尼游离碱达到asdas lda、asdas id、asdas cii和asdas mi的患者比例更高(标称p<0.0001)(图7a)。asdas=强直性脊柱炎疾病活动性评分。bl=基线。cii=临床重要改善(较基线下降≥1.1分)。id=非活动性疾病(评分<1.3)。lda=低疾病活动性 (<2.1)。mi=重大改善(较基线下降≥2分)。mmrm=重复测量的混合模型。nri=无应答者插补。qd=每天一次。
[0138]
图8a至8d描绘了在2/3期强直性脊柱炎(select-axis 1)临床试验中各个asdas组分较基线的最小二乘均值(lsm)随时间的变化。早在第2周就观察到平均asdas(图7b) 和各个asdas组分(图8a至8d)的改善,使用乌帕替尼游离碱后持续改善直至第14周。脊柱疼痛=basdai问题2。外周疼痛/肿胀=basdai问题3。晨僵持续时间=basdai问题6。 asdas=强直性脊柱炎疾病活动性评分。basdai=巴氏强直性脊柱炎疾病活动指数。bl=基线。hscrp=高敏c反应蛋白。mmrm=重复测量的混合模型。ptga=疾病活动性的患者总体评估。qd=每天一次。
[0139]
图9描绘了用于治疗as和nr-axspa受试者的3期研究设计。as=强直性脊柱炎;asas=国际脊柱关节炎评估协会;bdmard-ir=生物疾病改善抗风湿药应答不足;ema=欧洲药品管理局;fda=美国食品和药物管理局;mri=磁共振成像;nr-axspa=非放射学中轴型脊柱关节炎;qd=每天一次;si=骶髂关节;upa=乌帕替尼游离碱。
[0140]
图10描绘了select-psa1在患有活动性psa且先前对至少一种非生物dmard(psadmard-ir)应答不足的受试者中的3期研究设计。a.所有受试者将在筛选、第24周、第56 周、第104周和第152周接受手脚x光检查。b.在第16周,将向分类为无应答者(定义为在第12周和第16周的压痛关节计数(tjc)和肿胀关节计数(sjc)之一或两者均未达到至少 20%的改善)的受试者提供救援治疗。c.在第24周,无论应答如何,所有安慰剂受试者都将转为乌帕替尼游离碱15mg qd或30mg qd(1∶1比例)。
[0141]
图11a至11b描绘了3期select-psa1(psa dmard-ir)中用于主要终点(第12周的 acr20应答)和排序次要终点的图形测试程序。总体i型错误率控制在双侧0.05水平(包括用于中期无效分析的0.0001α)。图11a的第1部分描述了psa症状和放射学进展中具有足够动
力的排序次要终点的测试顺序。图11b的第2部分描述了其它排序次要终点的测试顺序,包括与阿达木单抗的优效性测试。仅在达到所有第1部分端点的统计显着性后,才测试第2 部分端点。
[0142]
图12a至12ee提供了select-psa1(psa dmard-ir)3期临床试验的主要和关键次要疗效结果的总结。图12a描绘了直至第24周(包括第24周)的acr20应答的时间进程和相关联的95%置信区间(ci)。通过检查呈现具有95%ci的应答率(%)。对于所有检查,upa 剂量与安慰剂相比的标称p值<0.05。upa=乌帕替尼游离碱;ada=阿达木单抗;qd=每天一次;eow=每隔一周。符号:***,乌帕替尼与安慰剂相比,p<0.001;###,乌帕替尼与阿达木单抗相比,p<0.001;ζζζ,非劣效性乌帕替尼与阿达木单抗相比,p<0.001。其它关键的次要疗效结果包括:患者达到acr50应答(图12b);患者达到acr70应答(图12c);患者达到最小疾病活动性(mda)(图12d);患者在第12周在acr20中达到乌帕替尼与阿达木单抗的非劣效性(图12e);患者在24周内在acr标准的核心分量达到相对于基线的ls平均变化:压痛关节计数(tjc68)(图12f)、肿胀关节计数(sjc66)(图12g)、医生对疾病活动性的总体评估(phga)(图12h)、患者对疾病活动性的总体评估(ptga)(图12i)、疼痛(ptpain)(0分表示“无疼痛”,10分表示“可能最严重的疼痛”)(图12j)和高敏c反应蛋白(hs-crp)(图12k);在24周内达到银屑病面积严重程度指数pasi 75(图12l)、pasi 90(图12m)和pasi 100(图12n)的患者比例,其中在第16周之后进行评估并且患者能够根据研究者的判断使用专门针对银屑病的伴随治疗;在24周内达到静态研究者总体评估 (siga)的患者比例(图12o);在24周内银屑病症状的自我评估(saps)相对于基线的变化(图12p);患者在24周内达到健康评估问卷残疾指数(haq-di)(图12q)和sf-36身体分量综合评分(pcs是从八个sf-36域计算的两个综合评分之一)相对于基线的变化。采用线性算法计算pcs,其标准平均值为50,分数越高表示结果越好)(图12r);患者在24 周内达到慢性病治疗疲劳功能评估(facit-f)(facit-f分数范围为0至52,分数越高表示疲劳程度越低)相对于基线的变化(图12s);患者在24周内达到晨僵(basdai问题5和6 的平均值)(图12t)、晨僵严重程度(basdai问题5)(图12u)和晨僵持续时间(basdai 问题6)(图12v)相对于基线的变化;根据利兹附着点炎指数(lei)(图12w)、加拿大脊柱关节炎研究协会(sparcc)(图12x)得出的24周内附着点消退的患者比例,以及根据利兹指趾炎指数(ldi)得出的指趾炎缓解的患者比例(图12y);患者在第24周达到放射学终点(mtss=修改过的总sharp/van der heijde评分,jsn=关节间隙变窄评分)相对于基线的变化(图12z);以及在第24周无放射学进展mtss<0.0(图12aa)和mtss<0.5(图12bb) 的患者比例。与安慰剂相比,有显着更高比例的接受乌帕替尼游离碱(15mg qd或30mg qd) 的患者早在第2周达到患者总体疼痛评估(ptpain)减轻≥30%和≥50%,并且此后所达到的疗效得以维持(参见图12cc和12dd)。与安慰剂相比,有显着更高比例的接受乌帕替尼游离碱的患者早在第2周(30mg qd乌帕替尼)或早在第4周(15mg qd乌帕替尼)达到ptpain 减轻≥70%,兵器此后所达到的疗效得以维持(参见图12ee)。符号:*,在0.05水平上具有统计学显着性(upa 15mg qd);ζ,在0.05水平上具有统计学显着性(upa 30mg qd);#,在0.05水平有统计学显着性(ada 40mg eow);***,乌帕替尼与安慰剂相比,p<0.001; ###,乌帕替尼与阿达木单抗相比,p<0.001;ζζζ,非劣效性乌帕替尼与阿达木单抗相比, p<0.001;pbo=安慰剂;ada=阿达木单抗;upa=乌帕替尼游离碱;eow=每隔一周;qd=每天一次;ci=置信区间。
[0143]
图13描绘了select-psa2在患有活动性psa且先前对至少一种bdmard(psabdmard-ir)应答不足的受试者中的3期研究设计。a.在第16周,将向分类为无应答者(定义为在第12周和第16周的压痛关节计数(tjc)和肿胀关节计数(sjc)之一或两者均未达到至少20%改善)的受试者提供救援治疗。b.在第24周,无论应答如何,所有安慰剂受试者都将转为乌帕替尼游离碱15mg qd或30mg qd(1∶1比例)。
[0144]
图14a至14dd提供了select-psa2(psabdmard-ir)3期临床试验的主要和关键次要疗效结果的总结。图14a描绘了主要终点acr20直至第24周(包括第24周)的时间进程。通过检查呈现具有95%ci的应答率(%)。对于所有检查,upa剂量与安慰剂相比的标称p值<0.05。upa=乌帕替尼游离碱。符号:*=乌帕替尼15mg qd与安慰剂相比,p≤0.05;#=乌帕替尼30mg qd与安慰剂相比,p≤0.05;=在多重性对照分析中显着。其它关键的次要疗效结果包括:患者达到acr50应答(图14b);患者达到acr70应答(图14c);在24周内达到最小疾病活动性(mda)的患者比例(图14d);患者达到利兹附着点炎指数(lei)基线变化(图14e);患者达到附着点炎消退(lei=0)(图14f);根据加拿大脊柱关节炎研究协会 (sparcc)得出的24周内附着点炎消退的患者比例(图14g);患者达到利兹指趾炎指数(ldi) 基线改变(图14h);患者达到指趾炎消退(ldi=0)(图14i);在24周内达到银屑病面积严重程度指数pasi 75(图14j)、pasi 90(图14k)和pasi 100应答(图14l)的患者比例;患者在银屑病关节炎(dapsa)评分中达到相对于基线的疾病活动性变化(图14m);患者在24周内达到acr标准核心分量相对于基线的变化:压痛关节计数(tjc68)(图14n)、肿胀关节计数(sjc66)(图14o)、医生对疾病活动性的总体评估(phga)(图14p)、患者对疾病活动性的总体评估(ptga)(图14q)、疼痛(ptpain)(0分表示“无疼痛”,10分表示“可能最严重的疼痛”)(图14r)、高敏c反应蛋白(hs-crp)(图14s);在24周内达到静态研究者总体评估(siga)0/1的患者比例(图14t);患者在24周内达到银屑病症状的自我评估(saps)相对于基线的变化(图14u);患者在24周内达到健康评估问卷残疾指数(haq-di)(图14v)相对于基线的变化;患者在24周内达到sf-36身体分量综合(图14w) 相对于基线的变化;患者在24周内达到慢性病治疗疲劳功能评估(facit-f)(facit-f分数范围为0至52,分数越高表示疲劳程度越低)相对于基线的变化(图14x);患者在24周内达到晨僵(basdai问题5和6的平均值)相对于基线的变化(图14y);患者在24周内达到晨僵严重程度(basdai问题5)相对于基线的变化(图14z);以及患者在24周内达到晨僵持续时间(basdai问题6)相对于基线的变化(图14aa)。与安慰剂相比,有显着更高比例的接受乌帕替尼游离碱(15mg qd或30mg qd)的患者早在第2周达到患者对总体疼痛评估(ptpain)减轻≥30%、≥50%和≥70%,并且此后所达到的疗效得以维持(参见图14bb至 14dd)。upa=乌帕替尼;pbo=安慰剂;qd=每天一次;nri=无应答者插补。
[0145]
定义
[0146]
此章节和整个公开中使用的章节标题不旨在是限制性的。
[0147]
1.临床终点定义
[0148]
美国风湿病学会(acr)标准
[0149]
acr标准是基于对一组核心测量的改进计算得出的复合测量。acr20定义为压痛和肿胀关节计数(tjc和sjc)至少改善20%(与基线值相比),其余5项核心测量(受试者对疼痛的评估、受试者对疾病活动性的总体评估、医生对疾病活动性的总体评估、受试者对身体机能的评估和急性期反应物hscrp)中的3项至少改善20%。acr50和acr70的定义类似,分别具
有至少50%和70%的改善。如果满足以下条件,受试者将被归类为acr20(acr50或acr70)应答者:
[0150]
1.压痛关节计数(tjc68)较基线改善≥20%(50%或70%),并且
[0151]
2.肿胀关节计数(sjc66)较基线改善≥20%(50%或70%),并且
[0152]
3.以下5项中的至少3项较基线改善≥20%(50%或70%):
[0153]
a.患者对疼痛的总体评估(ptpain)
[0154]
b.患者对疾病活动性的总体评估(ptga)
[0155]
c.医生对疾病活动性的总体评估(pga)
[0156]
d.患者对身体机能的自我评估(即,通过健康评估问卷haq-di评分来衡量)
[0157]
e.急性期反应物值高敏crp(hscrp)
[0158]
国际脊柱关节炎评估协会(asas)、asas20、asas40、asas-pr和asas5/6应答。
[0159]
用于asas应答的域如下:
[0160]
a.患者的整体评估-以ptga疾病活动性表示(nrs评分为0至10)
[0161]
b.疼痛-以患者对总背痛的评估表示(总背痛,nrs评分为0至10)
[0162]
c.功能-以basfi表示(nrs评分为0至10)
[0163]
d.炎症-以2次晨僵相关basdai的平均值表示(basdainrs评分为0至10的问题5和6的平均值)
[0164]
asas20应答:在上述4个域中的≥3个域中,与基线相比,改善≥20%和绝对改善≥1个单位(评分范围为0至10;0=无疼痛且10=可能最严重的疼痛),在潜在的剩余域中没有恶化(定义为≥20%的恶化和≥1个单位的净恶化)。
[0165]
asas40应答:在上述4个域中的≥3个域中,与基线相比,改善≥40%和绝对改善≥2个单位(评分范围为0至10;0=无疼痛且10=可能最严重的疼痛),在潜在的剩余域中没有恶化(定义为净恶化>0个单位)。
[0166]
asas部分缓解(pr):对于上述4个域中的每一个,与基线相比,绝对评分≤2个单位(评分范围为0至10;0=无疼痛且10=可能最严重的疼痛)。
[0167]
asas5/6应答:在以下6个域中的5个域中,与基线相比,改善≥20%:basfi、患者对总背痛的评估、ptga疾病活动性、炎症(basdai的问题5和6的平均值])、basmi的腰椎侧屈,以及hs-crp。
[0168]
asas健康指数(hi)
[0169]
asashi是一个线性综合测量,具有两种回答选项:“我同意”和“我不同意”,列出17个问题。asashi上的每条陈述都给出“1”=“我同意”或“0”=“我不同意”的评分。asashi的总和范围为0至17,评分越低表明健康状况越好。问题7和8并不适用于所有患者。对于那些勾选回答“不适用”的患者,总分分别基于n=16或n=15进行分析。如果不超过20%的数据(即,3个问题)缺失,则可以分析总分。对于最多缺失三个答案的受访者,总分计算如下:sum.score=x/(17-m)*17,其中x是问题总分,m是缺失问题的数量,m≤3。缺失超过三个答案(m>3)的案例不能分配总分,并且总分将被设置为缺失。17个asas健康指数问题如下:
[0170]
1.疼痛有时会扰乱我的正常活动。
[0171]
2.我发现长时间站立很难。
[0172]
3.我跑步有问题。
[0173]
4.我在使用厕所设施时遇到问题。
[0174]
5.我经常筋疲力尽。
[0175]
6.我不太愿意做任何需要体力的事情。
[0176]
7.我对性失去了兴趣。
[0177]
8.我很难操作汽车的踏板。
[0178]
9.我发现很难与人接触。
[0179]
10.我无法在平坦的地面上户外行走。
[0180]
11.我觉得很难集中注意力。
[0181]
12.由于行动不便,我的旅行受到限制。
[0182]
13.我经常感到沮丧。
[0183]
14.我觉得洗头很困难。
[0184]
15.由于我的风湿病,我经历了财务变化。
[0185]
16.我晚上睡得不好。
[0186]
17.我无法克服我的困难。
[0187]
强直性脊柱炎疾病活动性评分(asdas)
[0188]
用于计算asdas的参数:
[0189]
1.患者对总背痛的评估(basdai问题2,nrs评分为0至10),
[0190]
2.晨僵持续时间(basdai问题6,nrs评分为0至10),
[0191]
3.疾病活动性的患者整体评估(ptga,nrs评分为0至10),
[0192]
4.外周疼痛/肿胀(basdai问题3,nrs评分为0至10),以及
[0193]
5.高敏c反应蛋白(hscrp)(以mg/ml为单位)或红细胞沉降率(esr)。
[0194]
asdas的计算:
[0195]
asdas
hs-c
rp
=0.121
×
总背痛+0.110
×
ptga+0.073
×
外周疼痛/肿胀+0.058
×
晨僵持续时间 +0.579
×
ln(hs-crp+1)。
[0196]
asdas
esr
=0.113
×
患者整体+0.293
×
√esr+0.086
×
外周疼痛/肿胀+0.069
×
晨僵持续时间 +0.079
×
总背痛。
[0197]
为了计算观察到的asdas评分,将首先计算观察到的分量值。然后,这些分量将根据 asdas公式包括在计算中。如果窗口中缺少任何观察到的分量,则观察到的asdas评分将缺失。
[0198]
当常规crp低于检测限或高灵敏度crp<2mg/l时,应采用2mg/l的恒定值计算asdas-crp。
[0199]
asdas评分按以下asdas疾病活动状态分类:
[0200]
·
asdas非活动性疾病(id):asdas<1.3
[0201]
·
asdas中度疾病:1.3≤asdas<2.1
[0202]
·
asdas低疾病活动性(lda):asdas<2.1
[0203]
·
asdas高疾病:2.1≤asdas≤3.5
[0204]
·
asdas极高疾病:asdas>3.5
[0205]
asdas应答类别定义如下:
[0206]
·
asdas重大改善(mi)(相对于基线的变化≤-2.0;相对于基线降低≥2分)
[0207]
·
asdas临床重要改善(cii)(相对于基线的变化≤-1.1;(相对于基线降低≥1.1分)
[0208]
强直性脊柱炎生活质量问卷(asqol)
[0209]
asqol的18项陈述(见下文)中的每一项都给出“1”(是)或“0”(否)的评分。测量的概念包括日常生活活动、情绪功能、疼痛、疲劳和睡眠问题。如果问题得到肯定(回答“是”),则得“1”分,表示不良的qol。所有问题评分相加得出总分或指数。得分范围从0 (良好的qol)至18(较差的qol),得分越高表示功能越差。缺失超过三个答案(即,超过 20%)的案例不能分配总分。对于有1至3个缺失答案的案例,总分计算如下:t=18x/18-m 其中:t是总分,x是肯定问题的总分,m是缺失问题的数量。
[0210]
1.我的病情限制了我可以去的地方
[0211]
2.我有时想哭
[0212]
3.我穿衣有困难
[0213]
4.我做家务有困难
[0214]
5.无法入睡
[0215]
6.我无法与我的朋友/家人一起参加活动
[0216]
7.我总是感到很累
[0217]
8.我必须停止我正在做的事情来休息
[0218]
9.我有难以忍受的痛苦
[0219]
10.早上起来需要很长时间
[0220]
11.我无法做家务
[0221]
12.我很容易疲倦
[0222]
13.我经常感到沮丧
[0223]
14.痛苦一直都在
[0224]
15.我觉得我错过了很多
[0225]
16.我觉得洗头很困难
[0226]
17.我的病情让我失望
[0227]
18.我担心让人们失望
[0228]
巴氏强直性脊柱炎疾病活动指数(basdai)、basdai 50应答和晨僵评分
[0229]
basdai由1至10的量表组成(1表示没有问题,10表示最严重的问题),用于回答与 5种症状有关的6个问题:疲劳、脊柱疼痛、关节疼痛/肿胀、局部压痛区域(也称为附着点炎或肌腱和韧带炎症)、晨僵持续时间和晨僵严重程度。评分越低表明疾病活动性越低。
[0230]
六个basdai问题(分量)如下:
[0231]
q1.您如何描述您所经历的疲劳/疲倦的总体程度?
[0232]
q2.您如何描述您所经历的as颈部、背部或臀部疼痛的总体程度?
[0233]
q3.除了颈部、背部或臀部之外,您如何描述关节疼痛/肿胀的总体程度?
[0234]
q4.您如何描述您对任何压痛或触痛部位所产生的总体不适程度?
[0235]
q5.您如何描述从您醒来时起的晨僵的总体程度?
[0236]
q6.从醒来的那一刻起,您的晨僵会持续多久?
[0237]
问题1至5的回答范围从0(无)到10(非常严重);问题6的回答范围从0(0小时) 到
10(2小时或更多),其中5表示1小时。
[0238]
basdai的评分:basdai将报告为0至10。评分的最大值为10,病情计算如下:
[0239]
basdai评分=0.2(q1+q2+q3+q4+q5/2+q6/2)
[0240]
如果5个问题(问题1至问题4,炎症)中有一个缺失,则评分是4个非缺失问题的平均值(4个非缺失问题的总和除以4)。如果5个问题中有1个以上缺失,则basdai评分缺失。问题5和问题6共同构成问题5(炎症)。如果问题5和6都缺失,并且问题1至4没有缺失,则只有一个问题将被视为缺失。basdai评分仍然可以计算为问题1至4的平均值。但是,例如,如果问题6和问题1都缺失,则2个问题将被视为缺失,因为炎症计算将不完整。在这种情况下,basdai评分将被视为缺失。
[0241]
basdai 50应答是基于basdai的分类应答,表示basdai较基线至少改善了50%。
[0242]
晨僵评分是basdai问题5和6的平均值,范围为0至10。
[0243]“basdai和basdai问题(分量)相对于基线的变化,包括basdai问题5和6的平均值相对于基线的变化”是指(1)basdai评分与基线相比的变化,(2)所有basdai问题与基线相比的变化,以及(3)问题5和6(代表炎症)的平均值与基线相比的变化。
[0244]
巴氏强直性脊柱炎功能指数(basfi)
[0245]
basfi由以下10个问题组成,评估完成穿衣、弯腰、伸手、转身和爬梯等活动的能力,每个问题的答案范围从0(容易)到10(不可能):
[0246]
1.在没有帮助或辅助(例如,穿袜辅助器)的情况下穿上您的袜子或紧身衣。
[0247]
2.从腰部向前弯曲,在没有帮助的情况下从地板上捡起一支笔。
[0248]
3.在没有帮助或辅助(例如,援助之手)的情况下伸向高架子。
[0249]
4.不用手或任何其它帮助从无扶手的餐厅椅子上站起来。
[0250]
5.在没有帮助的情况下仰卧起身。
[0251]
6.无支撑站立10分钟无不适。
[0252]
7.在不使用扶手或助行器的情况下爬12到15级台阶。每一步踩一脚。
[0253]
8.不转身就从肩膀上看过去。
[0254]
9.进行体力要求较高的活动(例如,理疗、锻炼、园艺或运动)。
[0255]
10.无论是在家还是在工作中,都可以进行一整天的活动。
[0256]
参见,例如,sieper等人,《风湿病学年鉴》(2009)68(suppl ii):iil-ii44。 doi:10.1136/ard.2008.104018。
[0257]
basfi的计分。basfi评分将根据问题1到10的平均值得出。如果最多缺失2个问题,则相应的评分将替换为其余非缺失问题的平均值。如果缺失3个或更多问题,basfi将被视为缺失。
[0258]
巴氏强直性脊柱炎计量线性指数(basmi
lin
)
[0259]
线性basmi(basmi
lin
)综合评分将使用basmi分量进行计算。下表列出了basmi
lin
的分量和评分的评估范围。
[0260][0261]
basmi
lin
=耳屏对壁、颈椎旋转和腰椎侧屈的评估测量是左右测量的平均值;a=评估测量
[0262]
每个评估的评分范围从0到10,且basmi
lin
总分将是5个评估评分的平均值。如果缺失 1个问题,则basmi
lin
将计算为剩余4个问题的平均值。因此,basmi
lin
总分的范围应该在 0到10之间。如果缺失2个或更多问题,则basmi
lin评分
将被视为缺失。参见例如,van der heijde 等人,《关节炎护理和研究(arth.care&res.)》(2012)64:1919-1922和van der heijde等人,《风湿病学年鉴》(2008)67:489-93。
[0263]
体表面积-银屑病(bsa-ps)
[0264]
应选择受试者的右手或左手作为测量装置。出于临床估计的目的,假定手掌加上五个手指的总表面约为相当于1%。通过想象分散的斑块是否移动以使它们彼此相邻,然后估计所累及的总面积,有助于测量总受累面积。参见,例如,参见,例如,bozek和reich,《高级临床实验医学(adv.clin.exp.med.)》(2017)26:851-856。
[0265]
银屑病关节炎疾病活动性(dapsa)评分
[0266]
dapsa是衡量银屑病关节炎疾病活动性的连续终点。dapsa由五个分量组成:压痛关节计数68、肿胀关节计数66、患者对疼痛的总体评估(pt pain)(0至10nrs)、疾病活动性 ptga(0至10nrs)和hscrp(以mg/dl为单位)。dapsa评分的计算如下:
[0267]
dapsa=sjc66+tjc68+pt pain(0-10nrs)+ptga(0-10nrs)+hscrp(以mg/dl为单位)
[0268]
为了计算观察到的dapsa评分,将首先计算观察到的分量值。然后,这些分量将根据 dapsa公式包括在计算中。如果窗口中缺失任何观察到的分量,则观察到的dapsa评分将缺失。
[0269]
疾病活动性评分(das28)
[0270]
das28(crp)和das28(esr)是分别使用实验室值(hscrp)或红细胞沉降率(esr) 测量高敏c反应蛋白来评估psa疾病活动性的综合指标。das28提供介于0到10之间的评分,表明测量时的关节炎疾病活动性。das28(crp)和das28(esr)的计算基于压痛关节计数、肿胀关节计数、疾病活动性ptga(0-100)和hscrp(以mg/l为单位)或esr(mm/hr)。由于疾病活动性ptga是以0至10nrs的量表收集的,因此在使用das28公式之前,该变量需要乘以10。为了计算观察到的das28评分,将首先计算观察到的分量值。然后,这些分量将根据所选的das公式包括在计算中。如果窗口中缺失任何观察到的分量,则观察到的 das28评分将缺失。das28(crp)和das28(esr)的计算由以下等式提供:
[0271]
das28(crp)=0.56
×
√(tjc28)+0.28
×
√(sjc28)+0.36
×
ln(hscrp+1)+0.014
×
ptga +0.96
[0272]
das28(esr)=0.56
×
√(tjc28)+0.28
×
√(sjc28)+0.70
×
ln(esr)+0.014
×
ptga
[0273]
其中√是平方根,且ln是自然对数;tjc28是指受试者在提供的28个评估关节中的总压痛关节计数;sjc28是指受试者在提供的28个评估关节中的总肿胀关节计数;das28(crp) 等式中的hscrp单位表示为mg/l;das28(esr)等式中的esr单位表示为mm/hr;ptga 是指患者对疾病活动性的总体评估。
[0274][0275]
利兹指趾炎指数(ldi)、指趾炎计数和压痛指趾炎计数
[0276]
指趾炎评分将是在基线时存在指趾炎、指趾炎计数(在基线存在指趾炎的受试者中)(“总指趾炎计数”或“压痛指趾炎计数”)和指趾炎的消退(在基线存在指趾炎的受试者中)。基线时存在指趾炎的定义如下:至少一个受累且压痛的指,其周长超过参考指≥10%。指趾炎计数将计算为存在指趾炎的指数(手和脚),范围从0到20。
[0277]
利兹指趾炎指数(ldi)是基于指周长和压痛的评分,对所有指趾炎的指进行评估和汇总。评估应从手和脚的目视检查开始。对于其中一个或两个手指出现指趾炎的每一对指,使用指长仪评估受累指(右侧和左侧)的周长。另外,通过在掌指骨和近侧指间关节之间挤压指轴来评估受累的指对的压痛,并记录为是或否压痛。压痛不应通过挤压关节线来评估。对于受试者的20个指中的每一个,需要先计算一个指的最终评分。对于未受累的指,指最终评分设置为0。对于受累的指,如果a/b≥1.1,则指最终评分计算为(a/b-1)*100*c,如果a/b <1.1,则指最终评分=0,其中a表示指圆周,b表示参考周长,且c表示压痛评分。参考周长可以是未受累的对侧指的周长(如果有),或者来自参考表(如果没有)。对于没有可用的指长仪测量的任何指,标准参考值将用于计算ldi。ldi是所有20个指的最终评分之和。指趾炎消退的受试者比例定义为ldi=0的受试者比例。注射皮质类固醇的指在注射后90天内将被视为不可评估。如果一个指缺失并且它的对侧指患有指趾炎,则将缺失的指记录为“指不存在”。
[0278]
附着点炎评分:加拿大脊柱关节炎研究协会(sparcc)附着点炎指数、利兹附着点炎指数(lei)、附着点炎总计数和马斯特里赫特强直性脊柱炎附着点炎评分(mases)
[0279]
对于加拿大脊柱关节炎研究协会(sparcc)附着点炎指数,评估了16个部位,如下表第1至8行所示。每个部位的检查压痛记录为存在(编码为1)、不存在(编码为0)或未评估(na)。sparcc附着点炎指数是通过对16个部位的评分求和来计算的。sparcc评分范围从0到16。
[0280]
利兹附着点炎指数评估下表第2、7和9行所示的6个附着点部位的附着点炎。对于6个部位的每一个,检查时的压痛记录为存在(编码为1)、不存在(编码为0)或未评估(na)。 lei是通过对6个部位的评分求和来计算的。lei的范围从0到6。
[0281]
总附着点炎计数是通过对下表中所有18个部位的压痛评分求和来计算的。
[0282]
lei中包括的附着点炎部位消退的受试者比例定义为lei=0的受试者比例;sparcc附着点炎指数和附着点炎总计数的消退的比例类似定义(评分=0)。
[0283][0284]
存在=1;不存在=0;na=未评估
[0285]
将测量马斯特里赫特强直性脊柱炎附着点炎评分(mases),以评估13个不同部位(左 /右第一肋软骨关节、左/右第七肋软骨关节、左/右髂后上棘、左/右髂前上棘、左/右髂嵴、第五腰椎棘突和左/右跟腱近侧插入)是否存在(1)或不存在(0)附着点炎,注意受试者的应答,总分范围为0至13。如果缺少一个或多个位置,将使用可用数据计算评分。如果所有位置都缺失,则mases设置为缺失。
[0286]
euroqol-5d(eq-5d-5l)
[0287]
eq-5d-5l问卷是衡量健康相关生活质量最常用的问卷之一。它由问卷和视觉模拟量表 (vas)组成。自我评估问卷测量健康状况的5个维度(活动能力、自我保健、日常活动、疼痛/不适和焦虑/抑郁)。要求as受试者将他们自己当前在每个维度中的功能等级分为3个残疾程度中的一个(重度、中度或无)。要求psa受试者将自己当前在每个维度中的功能等级分为5个等级(分别对应1级到5级的无问题、轻微问题、中度问题、严重问题和极端问题),并包括eq视觉模拟量表(eq vas)。将健康状况的5个维度转换为单个指标值。使用vas,受试者记录对当前感知健康状况的感知,等级范围从0(可能的最差健康状况)到100(可能的最佳健康状况)。
[0288]
facit-疲劳问卷(facit-f)
[0289]
facit疲劳问卷是一个包含13个问题的工具,用于测量个人在过去一周的日常活动中的疲劳程度。疲劳程度以四分制进行测量(4=完全不疲劳至0=非常疲劳)。疲劳量表范围从0 到52,评分越高表示疲劳程度越低。每个问题的问题评分是通过从4中减去或添加0来计算的,具体取决于它是否是反转问题。facit疲劳量表的计算方法是将所有问题评分相加,乘以13,然后除以回答的问题数。如果回答的问题少于7个,则不计算量表。
[0290]
1.我感到疲劳
[0291]
2.我感到浑身无力
[0292]
3.我感到无精打采(“筋疲力尽”)
[0293]
4.我感到疲倦
[0294]
5.我很难开始做事,因为我感到疲倦
[0295]
6.我很难完成事情,因为我感到疲倦
[0296]
7.我有精力
[0297]
8.我能够进行日常活动
[0298]
9.我白天需要睡觉
[0299]
10.我累得吃不下了
[0300]
11.我需要帮助以进行日常活动
[0301]
12.我因为太累而无法做我想做的事情而感到沮丧
[0302]
13.我必须限制我的社交活动因为我感到疲倦
[0303]
健康评估问卷残疾指数(haq di)
[0304]
haq-di是一种自我报告的患者结果测量方法。它计算为以下8个类别的评分的平均值,范围为0至3:穿衣和打扮、起身、饮食、步行、卫生、伸展、抓握和活动。评分越高,反映出残疾程度越高。每个类别中所有问题的最高评分被视为该类别的评分。haq-di考虑了受试者在残疾类别评分算法中使用的辅助工具或设备或协助。对于每个类别,都有一个aidsor devices伴随变量,用于记录受试者在他/她的日常活动使用的援助类型(如果有的话)。如果检查某个类别的辅助工具或设备和/或其它人的帮助,如果原始分数为0(无困难)或1 (有些困难),则该类别的评分设置为2(非常困难)。然后通过将调整后的类别评分相加并除以回答的类别数来计算haq-di。如果受试者没有至少6个类别的评分,则无法计算haq-di。
[0305]
高敏c反应蛋白(hs-crp)
[0306]
在血浆中测量的c反应蛋白(crp)是一种急性期蛋白,出现在血液循环中以响应炎症,并作为全身炎症的生物标志物。然而,常规的crp检测方法(浊度法、比浊法)在检测crp 浓度低于6-10mg/l时显示较差的灵敏度。参见,例如,poddubnyy等人,《风湿病学年鉴 (ann.rheum.dis.)》(2010)69:1338-1341。
[0307]
高敏c反应蛋白(hs-crp)测定是比常规crp更精确的测量方法。可以使用几种不同的测试来测量待治疗受试者的hs-crp正常范围与异常值;因此,正常上限(uln)将由实验室为所用的hs-crp测试确定,并且可能因实验室而异。
[0308]
psa的卫生资源利用(hru)问卷
[0309]
hru问卷含有以下类别中关于医疗保健利用的三个问题:计划外医疗保健专业人员就诊、急诊室就诊和住院。从hru问卷收集的数据将用于计算在每个变量(即,计划外的psa 相关医疗保健专业人员就诊次数、急诊室就诊次数、入院次数和住院总天数)中观察到的每单位时间(例如,受试者-年)的个人累计使用次数,如下所示:(i)受试者的观察时间将被定义为“最后一次hru未缺失的就诊日期——基线就诊日期。”以及(ii)将汇总每个受试者在基线后的使用次数。
[0310]
失眠严重程度指数(isi)
[0311]
isi是一份7个问题的问卷,用于评估(1)入睡困难,(2)睡眠维持困难(3)早醒问题, (4)对睡眠模式的满意度,(5)由于睡眠问题对日常工作的干扰,(6)睡眠问题对其它人的注意程度,以及(7)睡眠问题造成的痛苦程度。问题1(失眠的严重程度问题)采用李克特量表评分,(0)“无”、1
‑“
轻度”、2
‑“
中度”、3
‑“
严重”和4
‑“
非常严重”。其余的关于满意度、损伤的注意程度、对睡眠和干扰的担忧/苦恼的问题以5分李克特型量表形式进行类似的评分。请参阅下面的isi实例:
[0312]
1.请评价您失眠问题的当前(即,过去2周)严重程度:
[0313]
a.入睡困难
[0314]
b.难以维持睡眠
[0315]
c.早醒的问题
[0316]
2.您对当前的睡眠模式是否满意/不满意?(0=非常满意;1=满意;2=中等满意;3=不满意;5=非常不满意)
[0317]
3.您认为您的睡眠问题在影响您的生活质量方面对其它人来说有多明显?(0=完全不明显;1=有一点;2=有点;3=很多;5=非常明显)
[0318]
4.您对当前的睡眠问题有多担心/苦恼?(0=完全不担心;1=有一点担心;2=有点担心;3=非常担心;4=非常担心)
[0319]
5.您认为您的睡眠问题在多大程度上会干扰您的日常功能(例如,白天的疲劳、情绪、工作/日常家务的能力、注意力、记忆力、情绪等)?(0=完全没有干扰;1=有一点;2=有点;3=很多;4=非常干扰)。
[0320]
评分/解释指南:添加所有七个问题的评分。如果缺失一两个问题,则可以将其值替换为剩余问题的平均评分。如果有两个以上没有回答的问题,则优先考虑总分缺失。总分类别:
[0321]
0-7=无临床显着失眠
[0322]
8-14=阈下失眠
[0323]
15-21=临床失眠症(中度严重)
[0324]
22-28=临床失眠症(严重)
[0325]
关节计数评估:sjc和tjc
[0326]
肿胀关节计数评估(sjc或sjc66):将通过体检对66个关节进行评估。检查肿胀的关节与检查压痛的关节相同,但不包括髋关节。关节肿胀将被分类为存在(“1”)、不存在(“0”)、替换(“9”)或无评估(“na”)。注射皮质类固醇的关节在注射后90天内将被视为不可评估。 sjc66的范围为0到66。
[0327]
压痛关节计数评估(tjc或tjc68):将在体检时通过压力操作对68个关节的压痛进行评估。关节疼痛/压痛将被分类为:存在(“1”)、不存在(“0”)、替换(“9”)或无评估(“na”)。注射皮质类固醇的关节在注射后90天内将被视为不可评估。tjc68的范围为0到68。
[0328]
在每次研究就诊时评估解剖关节的肿胀和压痛。下表中的34个解剖关节在本研究中针对身体的左侧和右侧进行了评估。
[0329][0330]
a.髋关节不进行肿胀评估。
[0331]
改良stoke强直性脊柱炎脊柱评分(msasss)
[0332]
msasss是一种评分方法,用于测量强直性脊柱炎患者脊柱的放射学进展。msasss的范围在0到72之间,是通过对从t12下缘到s1上缘的腰椎前部和从c2下缘到t1上缘的颈椎前部进行评分得出的,评分为0(正常)、1(糜烂、硬化或变方)、2(韧带骨赘)、3(桥接韧带骨
赘)或na椎体不可评估。将分析脊柱胶片的x光片的放射学进展(从基线到随访时间点)。
[0333]
银屑病关节炎的最小疾病活动性(mda-psa)
[0334]
当满足以下7项标准中的5项(应答者)或如果7项标准中的至少3项未满足(无应答者),则将患者分类为psa具有最小疾病活动性(mda):tjc68≤1;sjc66≤1;pasi≤1或 bsa-ps≤3%;患者对疼痛的总体评估(pt pain)_≤1.5(0至10nrs);患者对疾病活动性的总体评估≤2(0至10nrs);haq-di评分≤0.5;利兹附着点炎指数≤1。
[0335]
改良银屑病关节炎应答标准(改良psarc)
[0336]
改良psarc是psa特异性的综合应答者指数。要获得应答,受试者必须完成以下4个问题中的2个,其中一个必须是压痛关节计数68或肿胀关节计数66,并且任何测量均未恶化:tjc68改善≥30%;sjc66改善≥30%;疾病活动性nrs的ptga改善;疾病活动性nrs的 pga改善。
[0337]
nsaid评分
[0338]
为了计算nsaid评分,收集以下信息:(1)nsaid的类型和重量系数;(2)剂量(mg/ 剂);(3)频率(每周施用天数);(4)施用天数(nsaid结束日期-nsaid开始日期+1);(5) 研究天数(最后就诊日期-基线日期+1)。
[0339]
然后可以使用以下等式计算nsaid评分:
[0340]
=(等效nsaid评分)x(施用天数)/(研究天数)
[0341]
其中等效nsaid评分通过以下等式计算:
[0342]
=(重量系数)x(剂量)x(频率)
[0343]
nsaid的权重因子由每种nsaid的最大剂量决定,以达到100分,其中0表示未施用, 100表示每天剂量150mg双氯芬酸,或1000mg萘普生,或200mg醋氯芬酸,或400mg塞来昔布等。例如,150mg双氯芬酸相当于1000mg萘普生,因此双氯芬酸的重量系数为 100/150,且萘普生的重量系数为100/1000。另见dougados等人,《风湿病学年鉴》(2011) 70:249-251。
[0344]
·
双氯芬酸,重量系数:100/150
[0345]
·
萘普生,重量系数:100/1000
[0346]
·
醋氯芬酸,重量系数:100/200
[0347]
·
塞来昔布,重量系数:100/400
[0348]
·
依托度酸,重量系数:100/600
[0349]
·
依托昔布,重量系数:100/90
[0350]
·
氟比洛芬,重量系数:100/200
[0351]
·
布洛芬,重量系数:100/2400
[0352]
·
吲哚美辛,重量系数:100/150
[0353]
·
酮洛芬,重量系数:100/200
[0354]
·
美洛昔康,重量系数:100/15
[0355]
·
尼美舒利,重量系数:100/200
[0356]
·
保泰松,重量系数:100/400
[0357]
·
吡罗昔康,重量系数:100/20
[0358]
·
替诺昔康,重量系数:100/20
[0359]
患者对夜间背部疼痛的评估(夜间背部疼痛)、患者对总背痛的评估(总背痛评分)
和患者对疼痛的总体评估(pt pain或“疼痛”)数值评定量表(nrs)
[0360]
疼痛将使用0至10数值评定量表(nrs)针对夜间背部疼痛nrs(0=无疼痛,10=可能最严重的疼痛)、总背痛(0=无疼痛,10=剧烈疼痛)和疼痛(即,总体疼痛)(0=无疼痛, 10=剧烈疼痛)的量表问题进行测量。
[0361]
asas(例如,asas20、asas40、asas pr)使用上述评分方法评估患者对总背痛的评估(总背痛评分)。对于asdas,患者对总背痛的评估通过回答basdai问题2来评估总背痛。
[0362]
患者对疾病活动性数字评定量表的总体评估(ptga-nrs)
[0363]
受试者将使用患者对疾病nrs的总体评估来评估其疾病活动性。范围为0到10,0表示无活动性,10表示严重活动性。
[0364]
医生对疾病活动性数字评定量表的总体评估(pga或phga-疾病活动性nrs)
[0365]
将进行pga-疾病活动性以评估受试者当前的疾病活动性,同时考虑关节炎和银屑病活动性,独立于受试者的自我评估,使用0至10nrs。范围为0到10,0表示无活动性,10表示严重活动性。
[0366]
体力活动评估
[0367]
体力活动评估测量步数和体力活动(例如,通过数字健康技术可穿戴装置测量)和脊柱运动任务范围(例如,通过倾斜计测量)。
[0368]
银屑病关节炎疾病活动性评分(pasdas)
[0369]
pasdas是联合关节、指趾炎和附着点炎评估、医生和患者对关节炎、sf36-pcs和hscrp 测量的总体评估的连续量表。pasdas根据以下等式计算:
[0370]
=(((0.18√(pga))+0.159√(ptga)-0.253√(sf36-pcs)+0.101ln(sjc66+1)+0.048ln (tjc68+1)+0.23ln(lei+1)+0.37ln(压痛指趾炎计数+1)+0.102ln(hscrp+1)+2)*1.5
[0371]
其中√是平方根,ln是自然对数;ptga在0至100的范围内;pga在0至100的范围内(因为ptga和pga是以0至10nrs的范围收集的,它们的值需要乘以10才能用于pasdas 公式);sf36-pcs是sf36仪器中的物理分量刻度;hscrp的单位是mg/l;lei的范围为0 到6。
[0372]
银屑病面积严重程度指数(pasi)
[0373]
pasi是衡量银屑病严重程度的指标。四个解剖部位-头部、上肢、躯干和下肢-使用5分制评估红斑、硬结和脱屑(0=无症状;1=轻微;2=中度;3=显著;4=非常显著)。根据给定解剖部位的病变程度,受影响的面积被分配一个数值(0=无受累;1=<10%;2=10%至29%; 3=30%至49%;4=50%至69%;5=70%至89%;6=90%至100%)。由于头部、上肢、躯干和下肢分别约占体表面积的10%、20%、30%和40%;pasi评分使用以下公式计算:
[0374]
=0.1(eh+ih+dh)ah+0.2(eu+iu+du)au+0.3(e
t
+i
t
+d
t
)a
t
+0.4(e
l
+i
l
+d
l
)a
l
[0375]
其中e、i、d和a分别表示红斑、硬结、脱屑和面积,h、u、t和1分别表示头部、上肢、躯干和下肢。
[0376]
pasi评分范围从0.0到72.0,最高评分代表最严重程度的完全性红皮病。通常,3分或以下代表轻度疾病,3分以上(包括15分)代表中度疾病,15分以上被认为与重度疾病有关。如果缺失一个问题,则不计分pasi。如果就诊时pasi评分(≥pasi 75/50/90/100应答)相对于基线pasi评分至少降低75%(50%、90%、100%),则达到pasi 75(pasi 50、pasi 90、pasi100)应答。参见,例如,feldman等人,《皮肤病学研究杂志(j invest dermatol.)》
1996;106(1):183-6。
[0377]
银屑病症状自我评估(saps)
[0378]
银屑病症状自我评估(saps)包含11个以症状为中心的问题,如下所示。每个问题从0到10进行评分,0表示最不严重,10表示最严重。总分是通过对11个问题求和生成的。总分数范围为0到110。
[0379]
1.在过去的24小时内,您在受银屑病影响的区域经历的最严重的疼痛是什么?
[0380]
2.在过去24小时内,您在受银屑病影响的区域经历的最严重的瘙痒是什么?
[0381]
3.在过去24小时内,您在受银屑病影响的区域经历的最严重的发红情况是什么?
[0382]
4.在过去24小时内,您在受银屑病影响的区域经历的最严重的脱屑是什么?
[0383]
5.在过去24小时内,您在受银屑病影响的区域经历的最严重的剥落(鳞屑从皮肤上脱落)是什么?
[0384]
6.在过去24小时内,您在受银屑病影响的区域经历的最严重出血是什么?
[0385]
7.在过去24小时内,您在受银屑病影响的区域经历的最严重的烧伤是什么?
[0386]
8.在过去24小时内,您在受银屑病影响的区域经历的最严重的刺痛是什么?
[0387]
9.在过去24小时内,您在受银屑病影响的区域经历的最严重的压痛是什么?
[0388]
10.在过去24小时内,您因受银屑病影响的区域皮肤开裂而经历的最严重的疼痛是什么?
[0389]
11.在过去24小时内,您因银屑病而经历的最严重的关节疼痛是什么?
[0390]
sharp/vanderheiide评分(shs)评分(相当于改良shard总评分,mtss)
[0391]
将根据sharp的方法(针对psa的vanderheijde改良)对放射学结果进行评估和评分。为了获得shs总分,手和脚的侵蚀和jsn评分将相加。评分范围总结如下。无psa放射学进展的发现表明shs与基线相比无变化或改善(三0)。
[0392][0393]
侵蚀评估。将评估每只手(每只手20个位置)和每只脚(每只脚6个位置)的侵蚀。shs方法评估的位置包括:4个远侧指骨间关节(2至5);5个掌指关节(1至5);4个近侧指骨间关节(2至5);拇指的指骨间关节;近侧第一掌骨;桡骨;尺骨;大多角骨和小多角骨(作为一个单元;多角骨);舟骨;月骨;5个跖趾关节(1至5);第一脚趾指骨间关节。
[0394]
关节间隙变窄评估。将在每只手(每只手20个位置)和每只脚(每只脚6个位置)中评估关节间隙变窄(jsn)。shs方法评估的位置包括:4个远侧指骨间关节(2至5);4个近侧指骨间关节(2至5);5个掌指关节(1至5);拇指的指骨间关节(ip);3个腕掌关节(3至5);桡腕关节;多角舟关节;头状-舟状-月状关节;5个跖趾关节;第一脚趾指骨间关节。
[0395]
对于评估的每个关节和骨骼,评分范围如下:侵蚀:0至5(手/手腕)或0至10(脚)表示侵蚀程度(其中0表示没有侵蚀);关节间隙变窄:0至4表示关节间隙变窄(jsn)的程度(其中0表示没有变窄)。
[0396]
加拿大脊柱关节炎研究协会(sparcc)脊柱评估(mrisparcc-脊柱或mri-spine
sparcc)和骶髂(si)关节(mri sparcc-关节或mri-si关节sparcc)
[0397]
脊柱和骶髂(si)关节的sparcc评分是通过分别对脊柱和si关节的骨髓水肿病变的存在、深度和强度的评估得出的二分结果相加来计算的。
[0398]
在脊柱的mri sparcc评分中,使用短-ti反转恢复(stir)图像序列评估整个脊柱的活动性炎症(骨髓水肿)。评估了23个椎间盘单位(dvu),并选择了6个受影响最严重的 dvu并用于计算mri脊柱sparcc评分。对于六个dvu中的每一个,在四个象限中评估3 个连续的矢状切片,以评估所有三个维度的炎症程度:
[0399]
1.针对stir上信号增加的存在对每个象限进行评分(1=信号增加;0=正常信号)
[0400]
2.在每个矢状切片上,在任何椎间盘单位中存在表现出高信号强度(与脑脊液相当)的病变,则额外评分为1分。
[0401]
3.包括病变的切片显示从终板延伸的深度≥1cm的连续增加信号将被评分为每切片+1。
[0402]
任何单个切片的最高可能评分为6,所有6个椎间盘单元的最高评分为108。
[0403]
si关节的mri sparcc评分是在stir图像序列的6个连续切片上进行的。髂骨内和骶骨内直至骶骨孔的所有病变都将被评分。si关节分为4个象限:上髂骨、下髂骨、上骶骨和下骶骨。每个连续切片在所有四个象限中分别为左右关节评分,如下所示:
[0404]
1.针对stir上信号增加的存在对每个象限进行评分(1=信号增加;0=正常信号)
[0405]
2.包括在stir序列上表现出强烈信号的病变的关节被评分为每切片+1。
[0406]
3.包括病变的关节显示出距离关节表面深度≥1cm的连续增加信号将被评分为每切片 +1。
[0407]
任何单个切片的最高可能评分为12,所有6个切片的最高评分为72。
[0408]
银屑病静态研究者总体评估(siga)
[0409]
siga是一个5分制的评分,范围从0到4,基于对所有银屑病病变的平均高度、红斑和鳞屑的评估。评估被认为是“静态的”,它指的是评估时患者的疾病状态,不与患者之前的任何疾病状态进行比较,无论是在基线时还是在之前的就诊时。分数越低,表明银屑病的严重程度越轻(0=清除,1=几乎清除,2=轻度,3=中度和4=重度)。基于siga的psa-1和psa-2 临床研究的二元临床终点是达到0或1的siga评分和至少比基线改善2分的受试者比例。该终点在基线siga得分≥2的受试者中计算。
[0410]
银屑病关节炎(wpai-psa)和中轴型脊柱关节炎(wpai-axial spa)的工作效率和活动障碍问卷
[0411]
工作效率和活动障碍问卷(wpai)旨在衡量整体健康状况和特定症状对工作和工作之外的效率的影响而开发的。它由6个问题组成。较低的wpai评分表明有所改善。4个主要障碍评分(s1到s4)表示为基于6个问题的障碍百分比。
[0412]
s0.就业:定义如下
[0413]
s1.缺勤率:由于psa或中轴型spa而错过的工作时间百分比:
[0414][0415]
s2.出勤率:由于psa或中轴型spa导致的工作障碍百分比:
[0416][0417]
s3.由于psa或中轴型spa造成的总体工作障碍百分比:
[0418][0419]
s4.psa或中轴型spa导致的活动障碍百分比:
[0420][0421]
s5.受试者是否误工(定义如下)。这是得出误工的受试者比例所必需的。
[0422]
在计算wpai评分时,应遵循以下计算说明。
[0423]
ο将就业定义为二元“是”或“否”变量,其中“是”对应于“就业”,“否”对应于“失业”。
[0424]
ο如果q1=“是”或q2>0或q4>0,则受试者将在给定就诊中被视为“就业”。
[0425]
ο如果q1=“否”并且在q2和q4下没有记录到正小时数(即,如果q1=“否”且q2 ≤0且q4≤0,则为失业),则受试者将在给定就诊中被视为“失业”。
[0426]
ο如果q1=缺失并且在q2和q4下没有记录正小时数,则在给定就诊中将认为受试者的就业状况“缺失”。
[0427]
ο如果受试者为“失业”或就业状态为“缺失”,则s1、s2和s3将设置为“缺失”。
[0428]
ο如果q2=0且q4=0或缺失,则q2/(q2+q4)=缺失(即,s1=缺失)。
[0429]
ο如果q2=0且q4=0,则将s3设置为缺失。
[0430]
ο如果q2缺失或q4缺失,则将s1和s3设置为缺失。
[0431]
ο如果q4=缺失,则不要设置q5=缺失。
[0432]
ο如果q5缺失,则应用以下规则:
[0433]
ο如果q2>0,q4=0且q5=缺失,则s3=100%。
[0434]
ο如果q2=0,q4>0且q5=缺失,则s3缺失。
[0435]
ο如果q2>0,q4>0且q5=缺失,则s3缺失。
[0436]
ο确定受试者是否误工(基于q2)以分析误工的受试者比例:
[0437]
ο创建一个二元(是或否)“误工”变量。
[0438]
ο如果q2大于0,则受试者将被视为误工。
[0439]
ο如果q2=缺失,则误工=缺失。
[0440]
ο如果q2>0,则误工=“是”。
[0441]
ο如果q2=0,则误工=“否”。
[0442]
ο因此,将根据误工=“是”的受试者数计算误工的受试者比例。
[0443]
36项简表健康调查(表sf-36)
[0444]
36项简表,第2版(sf-36v2)(质量指标)健康调查包括36个一般健康问题。它有两个分量:身体和精神。对于每个分量,使用8个子域计算转换后的综合评分:身体机能、角色-生理、身体疼痛、一般健康、活力、社会功能、角色-心理和心理健康。范围从0到100,分数越高表示结果越好。sf-36的编码和评分将使用供应商提供的软件。
[0445]
2.附加定义
[0446]
在阐述数值范围的情况下,明确设想的是所述范围内的每个中间数字具有相同的精确度。例如,对于范围6到9,设想了除了6和9之外的数字7和8,并且对于范围6.0到7.0,明确地设想了数字6.0、6.1、6.2、6.3、6.4、6.5、6.6、6.7、6.8、6.9和7.0。以相同的方式,所有所阐述比率还包含落入更宽比率内的所有子比率。
[0447]
除非上下文另外清楚地指明,否则单数形式“一个(a)”、“一种(an)”和“所述(the)”包含复数指示物。
[0448]
术语“约”通常是指本领域技术人员认为等同于所阐述值(即,具有相同的功能或结果) 的数字范围。在许多情况下,术语“约”可以包含四舍五入到最接近的有效数字的数字。
[0449]“受试者”是指人。术语“患者”和“受试者”在本文可互换使用。
[0450]“成年受试者”是指18岁或以上的受试者。
[0451]“幼年”或“小儿”受试者是指1至<18岁的受试者。待治疗的幼年受试者是被诊断患有幼年as(jas)、幼年psa(jpsa)和/或幼年pso(jpso)并经医生确定需要治疗的受试者 (分别为“活动性幼年as”和“活动性幼年psa”、“活动性幼年pso”)。幼年as可根据国际风湿病协会联盟(ilar)进行分类(定义了18岁之前开始的7种不同的关节炎类别:全身性关节炎、少关节炎、多发性关节炎(类风湿因子[rf]-阴性)、多发性关节炎(rf-阳性)、 psa、附着点炎相关jia(或幼年附着点炎相关关节炎[era])和未分化关节炎)。参见,例如, petty等人,《风湿病学杂志(j rheumatol.)》(2004)31:390-2。幼年psa可根据儿科国际风湿病协会联盟(ilar)和/或成人标准[银屑病关节炎分类标准(caspar)]进行分类。参见例如,aviel等人,《小儿风湿病学(pediatric rheumatology)》(2013)11:11;zisman等人,《风湿病学杂志》(2017)44:342-351。
[0452]
用于对患有中轴型脊柱关节炎(中轴型spa或axspa)的受试者进行分类的“2009asas 分类标准”描述于rudwaleit等人《风湿病学年鉴(ann.rheum.dis.)》(2009)68:777-783。该标准要求受试者患有慢性背痛(≥3个月)且发病年龄<45岁,受试者还具有以下病情(1)通过放射学或磁共振成像(mri)存在骶髂关节炎加上至少一项spa特征(“成像组”)或(2) 存在人类白细胞抗原(hla)b27加上至少两个spa特征(“临床组”)。影像学上的骶髂关节炎是指mri上的活动性(急性)炎症,高度提示与spa相关的骶髂关节炎,或明确的放射学骶髂关节炎。spa特征选自由以下组成的组:炎症性背痛、关节炎、附着点炎(足跟)、葡萄膜炎、指趾炎、银屑病、克罗恩病或溃疡性结肠炎、对nsais的良好应答(在背部全剂量服用nsaid后24至48小时,疼痛不再出现或好多了)、spa家族史、hla-b27阳性和c 反应蛋白升高(在存在背痛且排除其它升高原因后高于正常上限)。另见deodhar等人,《关节炎和风湿病(arth.&rheum.)》(2014)66:2649-2656。
[0453]
van der linden等人,《关节炎和风湿病》(1984)27:361-368中描述了用于对患有强直性脊柱炎(as)的受试者进行分类的“1984年修订的纽约标准”,它有两个分量:诊断和分级;诊断分量还具有两个标准:临床和放射学。临床标准要求:(i)腰痛和僵硬超过3个月,运动后症状改善,但休息不能缓解;(ii)腰椎在矢状面和额状面上的运动受限,以及(iii)相对于年龄和性别校正后的正常值而言,胸部扩张受限。放射学标准要求双侧骶髂关节炎分级 》2级或单侧骶髂关节炎分级3至4级。分级分量要求:(i)如果放射学标准与至少一项
临床标准相关,则确诊为强直性脊柱炎;(ii)如果存在3个临床标准,并且存在放射学标准而没有任何符合临床标准的体征或症状,则可能为强直性脊柱炎。另见deodhar等人,《关节炎和风湿病(arth.&rheum.)》(2014)66:2649-2656。
[0454]
术语“中轴型脊柱关节炎”(中轴型spa或axspa)包括“强直性脊柱炎”(as)和“非放射学中轴型脊柱关节炎”(nr-中轴型spa,或nr-axspa)。患有“活动性中轴型脊柱关节炎
”ꢀ
(活动性axspa)的受试者是指临床诊断为活动性as或活动性nr-中轴型spa并且需要由医生确定的治疗的受试者。
[0455]
患有“活动性强直性脊柱炎”(活动性as)的受试者是指临床诊断为as并且需要由医生确定的治疗的受试者。在某些实施例中,被诊断为患有as的受试者被进一步分类为(例如,在美国)满足1984年修订的纽约as标准和/或满足2009年asas分类标准。在某些实施例中,具有高as疾病活动性的受试者具有基于基线时的0至10数字评定量表的巴氏强直性脊柱炎疾病活动性指数评分≥4和/或asdas≥2.1和/或患者对总背痛的评估(总背痛评分) ≥4。参见,例如,van der heijde等人,《风湿病学年鉴》(2017)76:978-991;sieper和poddubnyy,《柳叶刀杂志(lancet)》(2017)73-84。
[0456]
患有“活动性非放射学中轴型脊柱关节炎”(活动性nr-中轴型spa或活动性nr-axspa) 的受试者是指临床诊断为nr-中轴型spa并且需要由医生确定的治疗的受试者。在某些实施例中,被诊断为患有nr-中轴型spa的受试者被进一步分类为(例如,在美国)满足2009年 asas的axspa分类标准但不满足1984年修订的as纽约标准的放射学标准。在某些实施例中,具有高nr-中轴型spa疾病活动性的受试者具有基于基线时的0至10数字评定量表的巴氏强直性脊柱炎疾病活动性指数评分≥4和/或asdas≥2.1和/或患者对总背痛评分的评估(总背痛评分)≥4;和选自由以下组成的组的炎症活动性的客观体征:(i)si关节mri上活动性炎症的客观体征或(ii)基线时的hscrp>正常上限(uln)。参见,例如,van der heijde等人,《风湿病学年鉴》(2017)76:978-991sieper等人,《风湿病学年鉴(ann.rheum.dis.)》(2009) 68增刊2:ii1-44。doi:10.1136/ard.2008.104018;van der heijde等人,《风湿病学年鉴》(2017) 76:978-991;sieper和poddubnyy,《柳叶刀杂志》(2017)73-84。
[0457]“caspar标准”(银屑病关节炎的分类标准(参见,例如,taylor等人,《关节炎和风湿病》(2006)54:2665-2673))要求受试者在基线时患有炎性关节疾病(关节、脊柱或附着点),具有以下5个类别中≥3分:
[0458]
1.目前银屑病的证据、银屑病个人病史或银屑病家族史(以下之一)。
[0459]
a.目前的银屑病被定义为根据风湿病学家或皮肤科医生的判断为目前存在的银屑病皮肤或头皮疾病。目前的银屑病评分为2分;所有其它特征的评分为1。
[0460]
b.银屑病个人病史定义为可以从患者、家庭医生、皮肤科医生、风湿病学家或其它合格的医疗保健提供者处获得的银屑病病史。
[0461]
c.根据患者报告,银屑病家族史定义为一级或二级亲属有银屑病史。
[0462]
2.目前体检中观察到的典型银屑病指甲营养不良,包括甲剥离、凹陷和角化过度。
[0463]
3.根据当地实验室参考范围,通过除乳胶以外的任何方法,但优选得通过酶联免疫吸附测定或比浊法,对类风湿因子的存在进行阴性测试结果。
[0464]
4.当前的指趾炎,定义为整个指肿胀,或风湿病学家记录的指趾炎病史。
[0465]
5.近关节新骨形成的放射学证据,在手或足的平片上表现为关节边缘附近不明确的骨化 (但不包括骨赘形成)。
[0466]
患有“活动性银屑病关节炎”(活动性psa)的受试者是指临床诊断为psa并且需要由医生确定的治疗的受试者。在某些实施例中,被诊断为患有psa的受试者被进一步分类为(例如,在美国)在基线时满足psa分类标准(caspar)标准。在某些实施例中,受试者在基线时可能具有≥3个压痛关节(基于68个关节计数)和≥3个肿胀关节(基于66个关节计数)。在某些实施例中,受试者在基线时可能具有≥5个压痛关节(基于68个关节计数)和≥5个肿胀关节(基于66个关节计数)。在某些实施例中,受试者可能具有≥3%的体表面积患有银屑病 (bsa-ps)。在某些实施例中,排除对psa具有最小疾病活动性的受试者(应答者和非应答者)。“活动性psa”包括由医生确定在患有活动性psa时功能正常的受试者和由医生确定在患有活动性psa时功能不正常的受试者。例如,腹部患有银屑病的受试者可能被认为表现出活动性 psa但能够正常发挥功能,而面部患有银屑病的受试者可能被认为表现出活动性psa但不能正常发挥功能(例如,由于情绪困扰)。“中度至重度活动性psa”是“活动性psa”的一个子集,涉及由医生确定受试者在患有活动性psa时不能正常发挥功能。
[0467]
患有“活动性银屑病”(活动性pso)的受试者是指临床诊断为pso并且需要由医生确定的治疗的受试者。在某些实施例中,活动性银屑病是活动性斑块状银屑病。在某些实施例中,受试者具有斑块状银屑病的记录病史。“活动性pso”包括由医生确定在患有活动性pso时功能正常的受试者和由医生确定在患有活动性pso时功能不正常的受试者。例如,腹部患有银屑病的受试者可能被认为表现出活动性pso但能够正常发挥功能,而面部患有银屑病的受试者可能被认为表现出活动性pso但不能正常发挥功能(例如,由于情绪困扰)。“中度至重度活动性pso”是“活动性pso”的一个子集,涉及由医生确定受试者在患有活动性pso时不能正常发挥功能。在某些实施例中,受试者在基线时可能具有≥3%的体表面积患有银屑病 (bsa-ps)。
[0468]
缩写“as”是指强直性脊柱炎。
[0469]
在施用第一剂jak1抑制剂的“x周内”达到结果,其中x是大于0的整数(例如,1、 2、4、6、8、10、12、14、16、18、20、22、24、52、......64周等)表示结果发生在从施用 jak1抑制剂的第一剂(第0周)开始到给定指定周的最后一天(包括最后一天)结束的时间范围内。可在受试者给定的第x周的任何时间点以及第一天和最后一天(包括第一天和最后一天)进行测量或评分,以确定结果是否在“第x周”(例如,在第1、2、4、6、8、10、12、14、16、18、20、22、24、52周、......64周等)达到。
[0470]
缩写“axspa”是指中轴型脊柱关节炎。
[0471]
缩写“bdmard”和“生物dmard”是指生物疾病改善抗风湿药。bdmard的实例包括但不限于生物肿瘤坏死因子抑制剂(例如,阿达木单抗、依那西普(etanercept))和白细胞介素(il)-17抑制剂(例如,苏金单抗(secukinumab)、依克珠单抗(ixekizumab))。
[0472]
术语“bdmard-ir”是指对bdmard应答不足的受试者。bdmard-ir受试者包括对至少一种bdmard治疗应答不足的受试者,或对bdmard不耐受或有禁忌症的受试者。 bdmard-ir受试者包括由于不耐受或缺乏疗效而停止使用至少一种bdmard治疗的受试者。
[0473]
术语“未接受过bdmard”是指之前未暴露于任何生物疗法(包括任何bdmard)的受试者,这些生物疗法可能对正在治疗的疾病或病症产生潜在的治疗影响。
[0474]
术语“基线”或“bl”是指第一次给药jak1抑制剂之前的时间。在施用第一剂jak1 抑制剂(即,乌帕替尼游离碱或其药学上可接受的盐)之前收集基线测量值(即,“基线就诊”或“筛选就诊”),并且可以包括在第一次给药jak1抑制剂的当天但之前进行的测量。
[0475]
特定评分或测量的术语“相对于基线变化”是指与基线时的评分或测量相比,评分或测量有所改善(例如,在受试者或受试者群体中表现出积极的临床效果)。
[0476]
缩写“cii”表示临床重要改善。
[0477]
当提及除施用jak1抑制剂之外的治疗时,短语“伴随施用”或“伴随治疗”是指在基线时和/或在用jak1抑制剂治疗期间进行额外的治疗。
[0478]
缩写“dmard”和“非生物dmard”是指非生物疾病改善抗风湿药。非生物dmard 包括但不限于甲氨蝶呤(mtx)、柳氮磺胺吡啶(ssz)、来氟米特(lef)、阿普斯特、羟氯喹(hcq)、布西拉明和艾拉莫德。“非生物dmard”和“常规合成疾病改善抗风湿药
”ꢀ
(csdmard)在本文中可互换使用。
[0479]
术语“dmard-ir”或“非生物dmard-ir”是指对非生物dmard应答不足的受试者。 dmard-ir受试者包括对至少一种非生物dmard治疗应答不足,或对非生物dmard不耐受或有禁忌症的受试者。
[0480]
缩写“ema”是指欧洲药品管理局。
[0481]
缩写“fda”是指美国食品和药物管理局。
[0482]
缩写“hscrp”是指高敏c反应蛋白。
[0483]
缩写“id”表示非活动性疾病。
[0484]
在具有活动性psa的受试者中,短语“改善身体机能”是指与基线相比,活动或任务有所改善。
[0485]“需要治疗”或“由医生确定需要治疗......”是指医生认为,在基线时,病情没有得到足够好的控制,例如通过其它药物治疗(例如,通过其它治疗或先前用于治疗该病症的疗法)。
[0486]
在患有活动性psa的受试者中,短语“抑制结构损伤的进展”或“防止结构进展”是指与基线相比,在x光片上证明预防了骨变化。
[0487]“jak1抑制剂”是指化合物乌帕替尼((3s,4r)-3-乙基-4-(3h-咪唑并[1,2-a]吡咯并[2,3-e] 吡嗪-8-基)-n-(2,2,2-三氟乙基)吡咯烷-1-甲酰胺)游离碱或其药学上可接受的盐。本文进一步描述了jak1抑制剂的固态形式。
[0488]
缩写“lda”是指低疾病活动性。
[0489]
缩写“mi”是指重大改善。
[0490]
缩写“mri”是指磁共振成像。
[0491]
如果施用jak1抑制剂保留了至少50%的减去安慰剂的阿达木单抗效果,则认为结果与阿达木单抗相比具有“非劣效性”(ni)。
[0492]
缩写“nr-axspa”是指非放射学中轴型脊柱关节炎。
[0493]
缩写“nri”是指无应答者插补。
[0494]
缩写“nsaid”是指非甾体抗炎药。nsaid的实例包括但不限于传统的nsaid(例如,布洛芬)和水杨酸盐(例如,阿司匹林)。
[0495]
术语“药学上可接受的”(如在“药学上可接受的盐”或“药学上可接受的稀释剂”中
的叙述中)是指与向人类受试者施用相容的材料,例如,该材料不会引起不良的生物效应。stahl 和wermuth在“药用盐手册:性质、选择与使用(handbook of pharmaceutical salts:properties, selection,and use)”(德国,魏因海姆,wiley-vch,2002)中描述了药学上可接受的盐的实例。在“《药物赋形剂手册(handbook ofpharmaceutical excipients)》”中描述了药学上可接受的赋形剂的实例,编辑rowe等人,(医药出版社(pharmaceutical press),第7版,2012)。
[0496]“药学上可接受的盐”是指那些保留游离碱的生物有效性和性质并且通过与例如盐酸、氢溴酸、硫酸、硝酸和磷酸等无机酸或者如磺酸、羧酸、有机磷酸、甲磺酸、乙磺酸、对甲苯磺酸、柠檬酸、富马酸、马来酸、琥珀酸、苯甲酸、水杨酸、乳酸、单苹果酸、单草酸、如单酒石酸等酒石酸(例如,(+)或(-)-酒石酸或其混合物)、氨基酸(例如,(+)或(-)-氨基酸或其混合物)等有机酸反应获得的盐。可以通过本领域技术人员已知的方法来制备这些盐。
[0497]“受试者群体”是指参与临床试验的一组受试者,所有受试者都患有相同的疾病或要治疗的症状,其中临床试验包含治疗组(用jak1抑制剂治疗的受试者亚组)和安慰剂组(未接受jak1抑制剂治疗的受试者亚组)。当与受试者群体的治疗结合使用时,短语“治疗群体中至少x%的受试者达到”特定应答是指安慰剂校正的x%应答(治疗的受试者-未治疗的受试者)。
[0498]
缩写“pr”是指部分缓解。
[0499]
缩写“psa”是指银屑病关节炎。
[0500]
缩写“pso”是指银屑病。银屑病包括作为psa皮肤表现的银屑病。
[0501]
缩写“qd”意指每日一次。
[0502]
短语“减少体征和症状”是指与基线相比,疾病活动性、功能和/或生活质量有所改善。
[0503]
缩写“si”表示骶髂。
[0504]“统计学显着”表示对于给定的假设检验,当观察到的p值<α时。预先指定的显着性水平,α,是在假设为真的情况下拒绝原假设的概率。α也称为i型错误或假阳性率。它通常设置为等于或小于0.05。观察到的p值是在零假设下,观察到相同大小或更极端效应的概率。当观察到的p值<α时,拒绝零假设,并声称具有统计学显着性。在多重性对照分析中,当调整后的p值<α时,结果具有统计学显着性。在多重性对照分析的固定序列中,只有当序列中的前一个终点满足统计显着性要求时,才能为排序较低的终点声明统计显着性。
[0505]
如果jak1抑制剂和阿达木单抗之间治疗差异的零假设检验的多重性调整p值小于预先指定的显着性水平,则认为结果与阿达木单抗相比“优越”。
[0506]“全脊柱强直”是指c2-t1或t12-s1脊柱总和≥5节段中的桥接韧带骨赘(融合)(例如,将考虑在颈椎融合2节段和在腰椎融合3节段被视为全脊柱强直阳性)。
[0507]
如本文所用,术语“治疗(treating)”、“治疗(treatment)”和“疗法”等意在包括但不限于减轻或缓解受试者正遭受的病症的一种或多种症状(即,中轴型脊柱关节炎(axspa)(例如,非放射学axspa(nr-axspa)、强直性脊柱炎(as)、银屑病关节炎(psa)、银屑病(pso)),包括减缓或停止病情进展,如减缓或停止与病症相关的结构损伤的进展、病症的结构进展和/ 或改善患有病症的受试者的身体机能。
[0508]
术语“乌帕替尼游离碱”是指乌帕替尼的游离碱(非盐、中性)形式。乌帕替尼游离
碱固态形式的实例包括无定形乌帕替尼游离碱和乌帕替尼的结晶游离碱,如乌帕替尼的结晶游离碱溶剂化物、结晶游离碱水合物、结晶游离碱半水合物和结晶游离碱无水物。乌帕替尼游离碱固态形式的具体实例包括但不限于无定形乌帕替尼游离碱、乌帕替尼游离碱溶剂化物形式a、乌帕替尼游离碱水合物形式b、乌帕替尼游离碱水合物形式c(其为半水合物)和乌帕替尼游离碱无水物形式d,各自如wo 2017/066775和wo 2018/165581中所述。
[0509]
术语“乌帕替尼游离碱当量”是指施用的中性乌帕替尼游离碱(活性成分)的量,并且不包括溶剂化物或水合物(包括半水合物)的任何共形成物(例如,溶剂或水分子),并且不包括药学上可接受的盐的任何药学上可接受的盐抗衡阴离子。例如,15.4mg的结晶乌帕替尼游离碱半水合物(每个乌帕替尼游离碱分子的1/2水构象分子)递送15mg的乌帕替尼游离碱当量,而30.7mg的结晶乌帕替尼游离碱半水合物(每个乌帕替尼游离碱分子包括1/2的水构象分子)递送30mg的乌帕替尼游离碱当量。
具体实施方式
[0510]
强直性脊柱炎(as)是一种主要影响中轴骨骼的慢性炎症性风湿性疾病,其特征是慢性背痛(包括夜间背痛)、晨僵、附着点炎、外周关节炎和关节外表现。这种疾病的“早期”形式(非放射学中轴型脊柱关节炎(nr-axspa))具有许多as疾病特征。
[0511]
由于as的长期衰弱性质,经常发生不可逆的结构损伤,对患者的生活产生负面影响。目前尚无治愈as的方法,因此治疗的主要目标是通过控制疾病的体征和症状、预防结构性损伤和维持身体机能,最理想的是通过实现持续的临床缓解或至少降低疾病活动性,来最大限度地提高患者的生活质量。非甾体抗炎药(nsaid)是as的一线治疗药物,其次是生物疾病改善抗风湿药(bdmard),如肿瘤坏死因子(tnf)抑制剂或白细胞介素-17(il-17) 抑制剂,用于对nsaid应答不足的患者。tnf抑制剂和il-17抑制剂对一些as患者有效,但仍有一些患者,这些已获批准的疗法均不能解决个体治疗目标。as是一种难以治疗的疾病,基于il-6抑制剂托珠单抗(tocilizumab)和sarilumab以及il-12/23抑制剂优特克单抗 (ustekinumab)和t细胞阻断抑制剂阿巴西普(abatacept)的低疗效显示。参见,例如,sieper 等人,《风湿病学年鉴(ann.rheum.dis.)》201473:95-100,sieper等人,《风湿病学年鉴(ann. rheum.dis.)》201574:1051-1057;deodhar等人,《关节炎和风湿病》201971:258-270,以及 song等人,《风湿病学年鉴(ann.rheum.dis.)》201170:1108-1110。
[0512]
jak家族由4个成员组成:jak1、2、3和酪氨酸激酶2(tyk2)。这些细胞质酪氨酸激酶协同作用以激活信号转导子和转录激活子(stat),后者转导细胞因子介导的信号,并与多种膜细胞因子受体相关,如常见的γ链(cgc)受体和糖蛋白130跨膜蛋白质。jak3和 jak1是cgc细胞因子受体复合物的组分,负责炎症细胞因子il-2、-4、-7、-9、-15和-21 的信号转导;而il-12和il-23通过jak2和tyk2发出信号。参见ghoreschi等人,《免疫学综述(immunolrev.)》(2009),228:273-87。这些信号的传播对于中轴型脊柱关节炎(axspa) 中炎症反应的放大很重要。乌帕替尼是一种jak抑制剂,旨在提高jak1对jak2、jak3和酪氨酸激酶2的选择性,已经研究用于在随机、安慰剂对照的2/3期select-axis 1研究中治疗对非甾体抗炎药(nsaid)应答不足的未接受过bdmard的as患者。参见本文的实例 2。第二项研究(select-axis-2)正在进行中,将招募范围扩大到非放射学中轴型脊柱关节炎(nr-axspa)患者和as bdmard-ir患者。参见实例3。
[0513]
select-axis 1在第14周达到了其主要终点,即国际脊柱关节炎评估协会(asas40) 应答的显着改善,以及多项疾病活动性测量(asdas、basdai、asas及其分量)、炎症(基于脊柱和骶髂关节的mri以及hscrp)、身体机能(basfi)、生活质量(asqol、asas hi) 和疾病的其它方面(basmi、mases)的显着改善,反映了乌帕替尼与安慰剂相比在结果上的显着改善。此外,对第14周乌帕替尼、第16周生物制剂依克珠单抗和阿达木单抗,以及第12周jak小分子抑制剂托法替尼(tofacitinib)和非戈替尼(filgotinib)的关键主要和次要终点的安慰剂校正数据进行了审查,虽然不是直接比较,但表明乌帕替尼15mg qd与其它两种jak小分子抑制剂相比,在更难达到的终点asas pr、asdas id和asdas lda方面显示出明确的前景,其显著疗效仅与生物制剂所显示的疗效相当。参见实例2,和van derheijde等人,《柳叶刀杂志》(2018)392:2441-2451,van der heijde d等人,《风湿病学年鉴》 ann.rheum.dis.)》(2017)1-8;van der heijde等人,《柳叶刀杂志》(2018)2378-2387。此外,这种疗效一旦在第14周达到,就会随着时间的推移而持续或改善,在这些难以达到的终点(包括asdas重大改善(mi)和asdas临床重要改善(cii))中具有长期疗效,持续或改善直至并包括第64周。在第14周从安慰剂转为乌帕替尼的患者中,与从第0周开始持续接受乌帕替尼的患者相比,直至并包括第64周观察到相似的起效速度和疗效应答程度。基于2/3期研究select-axis 1的结果以及来自其它乌帕替尼临床试验的一致安全性数据,乌帕替尼在 as中的获益-风险概况(尤其是与其它小分子jak抑制剂的风险状况相比),并从tnf抑制剂和il-17抑制剂的获益风险背景来看,为as患者提供了一种有前途的口服靶向治疗选择,特别是对于那些患有活动性疾病且对nsaid应答不足的as患者(以及nr-axspa患者)。
[0514]
银屑病关节炎(psa)是一种全身性炎症性疾病,具有多种临床表现,如斑块状银屑病、关节炎、指趾炎和附着点炎,并且与as相关,因为它具有共同的遗传和临床特征(例如,轴性受累并伴有背痛、外周关节炎或附着点炎、与hla-b27的遗传关联以及存在关节外表现)。多种细胞因子,如il-1、-6、-12、-17、-20和-23,被认为与银屑病病变中表皮角质形成细胞的活化和增殖有关。参见例如,nestle等人,《新英格兰医学杂志(n.engl.j.med.)》 (2009)361:496-509。il-17/il-23细胞因子轴也被认为在psa发病机制中很重要。参见例如, mease,《风湿病学新见(curr.opin.rheumatol.)》(2015)27:127-33。因此,jak1的阻断可以抑制被认为在psa发病机制中重要的中枢细胞因子信号的应答。目前针对psa的治疗指南各不相同,建议将如甲氨蝶呤等传统合成疾病改善抗风湿药(dmard)作为初始治疗,然后是生物dmard或靶向合成dmard,如托法替尼或最初为tnfi,然后是其它批准的疗法。虽然有多种治疗可供选择,但需要额外的选择才能达到更多难以达到的终点,如达到最小疾病活动性(mda),以及作为psa皮肤表现的银屑病治疗,达到pasi 75或pasi 90。jak1 抑制剂乌帕替尼已经研究用于治疗患有活动性银屑病关节炎和先前对至少一种非生物疾病改善抗风湿药(非生物dmard)(select psa1)的应答不足或先前对至少一种生物疾病改善抗风湿药(bdmard)应答不足(select psa2)的患者。参见实例4和5。在这两项试验中,与安慰剂相比,每天一次15mg和30mg的乌帕替尼对包括肌肉骨骼症状(外周关节炎、附着点炎、指趾炎和脊柱炎)的银屑病关节炎的临床表现、银屑病、身体机能、疼痛、疲劳和生活质量具有更好的疗效。早在第2周就观察到疗效。此外,对乌帕替尼和jak小分子抑制剂托法替尼(批准用于治疗psa的剂量较低(5mg bid))的关键主要和次要终点的安慰剂校正数据进行了审查,虽然不是直接比较,但表明乌帕替尼15mg qd和30mg qd对更难达到的最小疾病活动性
(mda)的终点,以及银屑病终点pasi 75或pasi 90,以及某些acr分量(例如,acr20/50/70)显示出明确的前景。参见实例4和5,以及gladman等人,《新英格兰医学杂志》(2017)377:1525-1536和mease p等人,《新英格兰医学杂志》(2017) 377:1537-1550。此外,观察到这种疗效一旦达到,在日常治疗过程中会随着时间的推移而持续或改善。
[0515]
因此,在一个方面,提供了一种用于通过向有需要的受试者施用jak1抑制剂乌帕替尼游离碱或其药学上可接受的盐来治疗各种脊柱关节炎和银屑病病症的方法,包括中轴型脊柱关节炎(axspa)、银屑病关节炎(psa)和银屑病(pso)的类型。在各个方面,提供了一种用于治疗活动性非放射学axspa(nr-axspa)的方法、用于治疗活动性强直性脊柱炎(as) 的方法以及用于治疗活动性银屑病关节炎(psa)和活动性银屑病(pso)(包括作为psa皮肤表现的pso)的方法。
[0516]
在一个实施例中,可用于本文公开的方法的jak1抑制剂是乌帕替尼游离碱。乌帕替尼游离碱固态形式包括无定形乌帕替尼游离碱和乌帕替尼的结晶游离碱。乌帕替尼的结晶游离碱包括选自由乌帕替尼的结晶游离碱溶剂化物、u乌帕替尼的结晶游离碱水合物(例如,乌帕替尼的结晶游离碱半水合物)和乌帕替尼的结晶游离碱无水物组成的组的那些。在一个实施例中,乌帕替尼的结晶游离碱是乌帕替尼的结晶游离碱半水合物。在一个实施例中,乌帕替尼的结晶游离碱是如wo 2018/165581和wo 2017/066775中所述的乌帕替尼游离碱水合物形式c(其为半水合物)。适用于本文公开的方法的jak1抑制剂的固态形式的其它具体实例包括选自由无定形乌帕替尼游离碱、乌帕替尼游离碱溶剂化物形式a、乌帕替尼游离碱水合物形式b、乌帕替尼游离碱无水物形式d和乌帕替尼酒石酸盐水合物组成的组的那些,各自如wo 2018/165581和wo 2017/066775中所述。
[0517]
治疗中轴型脊柱关节炎的方法
[0518]
本文提供了治疗中轴型脊柱关节炎(axspa)的方法。在一个特定方面,提供了治疗活动性axspa的方法,其涵盖治疗患有活动性强直性脊柱炎(as)和活动性非放射学中轴型脊柱关节炎(nr-axspa)的受试者,包含以一定量和/或以一定间隔向有需要的受试者每天一次口服施用一定剂量的jak1抑制剂乌帕替尼游离碱或其药学上可接受的盐。在一个方面,jak1 抑制剂是乌帕替尼游离碱。在一个方面,每天一次口服施用jak1抑制剂,其量足以递送15mg 的乌帕替尼游离碱当量。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52周。
[0519]
axspa的疾病活动性/严重程度可以使用多种指标来衡量,包括国际脊柱关节炎评估协会 (asas)应答(例如,asas20、asas40、asas部分缓解(pr)、asass/6);强直性脊柱炎疾病活动性评分(asdas)、asdas低疾病活动性(lda)、asdas非活动性疾病(id)、 asdas重大改善(mi)、asdas临床重要改善(cii)、脊柱的磁共振成像(mri)加拿大脊柱关节炎研究协会(sparcc)评分(mri-spine sparcc);骶髂(si)关节的mri sparcc 评分(mri-si关节sparcc);巴氏强直性脊柱炎疾病活动指数(basdai);basdai 50 (basdai50)应答;巴氏强直性脊柱炎功能指数(basfi);强直性脊柱炎生活质量问卷 (asqol);asas健康指数(hi);马斯特里赫特强直性脊柱炎附着点炎评分(mases)(附着点炎);线性巴氏强直性脊柱炎计量指数(basmi
lin
)(活动性);工作效率和活动障碍问卷
ꢀ‑
中轴型脊柱关节炎(wpai-axial spa);高敏c反应蛋白水平(hscrp);慢性病治疗疲劳功能评估(facit-f)问卷;失眠严重程度指数(isi);改良stroke强直性脊柱炎脊柱评分 (msasss);患者对总背痛的评估(总背痛
评分);患者对夜间背痛的评估(夜间背痛);患者对疼痛的总体评估(pt pain);医生对疾病活动性的总体评估(pga-疾病活动性);炎症 (basdai的问题5和6的平均值);患者对总背痛的评估(basdai的问题2);外周疼痛/ 肿胀(basdai的问题3);晨僵持续时间(basdai的问题6);患者对疾病活动性的总体评估(ptga);压痛关节计数(tjc68)和肿胀关节计数(sjc66);指趾炎的消退;总指趾炎计数;euroqol-5d-5l(eq-5d-5l)问卷;36项简表健康调查(sf-36);体力活动评估和nsaid 评分。这些指标在临床终点定义和实例中有详细描述。
[0520]
在一个方面,提供了一种治疗axspa,包括活动性axspa的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂,其中受试者在施用 jak1抑制剂后达到了国际脊柱关节炎评估协会40(asas40)应答。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52周。
[0521]
在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14 周)达到asas40应答,并且在第14周后通过继续施用每日剂量维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas40应答,并且在第2周后通过继续施用每日剂量维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂的52周内(包括在第52周)实现asas40应答,并且在第52周后通过继续施用每日剂量维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内、4周内、8周内、12周内、14周内、18周内、24周内、32周内、40周内和/或 52周内(包括在第2周、第4周、第8周、第12周、第14周、第18周、第24周、第32 周、第40周和/或第52周)达到asas40应答。在一个实施例中,受试者在施用第一剂后的 2周内(包括在第2周)达到asas40应答,并维持asas40应答直至施用第一剂后的至少 14周(例如,直至至少第14周)。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jakl抑制剂,持续至少2周、持续至少4周、持续至少8周、持续至少12周、持续至少14周、持续至少18周、持续至少24周、持续至少32周、持续至少40周和/或持续至少52周。在一个实施例中,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52周。
[0522]
在另一方面,提供了一种治疗axspa,包括活动性axspa的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用jak1抑制剂,其中受试者在本文所述的一定间隔达到asas40应答,并且在施用jak1抑制剂后,另外达到下文所述的用于治疗强直性脊柱炎(as)和/或非放射学中轴型脊柱关节炎(nr-axspa)的至少一个结果。在一个方面, jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52周。
[0523]
在另一方面,提供了一种在有需要的受试者群体中治疗axspa,包括活动性axspa的方法,该方法包含如本文所述以一定量和/或以一定间隔向受试者施用一定剂量的jak1
抑制剂,其中治疗群体中的一部分受试者在施用jak1抑制剂后达到asas40应答(例如,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到应答)。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas40应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到 asas40应答,并且在第14周后通过继续向受试者施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到asas40应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到asas40应答,并且在第52周后通过继续向受试者施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas40应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas40应答,并且在第2周后通过继续向受试者施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,治疗群体中的受试者在施用jak1抑制剂后达到下文所述的用于治疗强直性脊柱炎(as)和/ 或非放射学中轴型脊柱关节炎(nr-axspa)的至少一个结果(例如,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少 35%、至少40%或至少45%的受试者达到至少一个结果)。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,以足以递送15mg的乌帕替尼游离碱当量的量向群体施用一定剂量的jak1抑制剂。在一个方面,向受试者每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,向受试者每天一次口服施用jak1抑制剂,持续至少52周。
[0524]
进一步提供了减轻axspa的体征和症状的方法。在一个方面,提供了一种减轻axspa(包括活动性axspa)的体征和症状的方法,其包括以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂。在一个方面,jak1抑制剂是乌帕替尼游离碱或其药学上可接受的盐。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂每天施用一次,持续14周。在一个方面,jak1抑制剂每天施用一次,持续52周。
[0525]
在一个方面,当受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到选自由以下组成的组的至少一个结果:asas40应答;asdas相对于基线的变化(改善);脊柱mri sparcc评分(mri-spine sparcc)相对于基线的变化(改善);asas部分缓解 (pr);basdai50应答;basfi相对于基线的变化(改善);asqol相对于基线的变化(改善);asas健康指数(hi)相对于基线的变化(改善);mases(附着点炎)相对于基线的变化(改善);basmi
lin
(活动度)相对于基线的变化(改善);wpai-axial spa相对于基线的变化(改善);si关节的mri sparcc评分(mri-si关节sparcc)相对于基线的变化(改善);或当受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到asas40应答,施用jak1抑制剂后,axspa(包括活动性axspa)的体征和症状减轻。在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到应答,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在另一方面,受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到应答,并且在第52周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到应答,并且在第2周后
通过继续施用每日剂量的jak1抑制剂来维持或改善应答。
[0526]
对于本文所述的治疗axspa的方法和/或用于减轻axspa的体征和症状的方法中的任一种,治疗群体中的一名受试者和/或多名受试者i)可能未接受过生物疾病改善抗风湿药 (bdmard)或ii)在基线时可能对bdmard应答不足或不耐受(bdmard-ir)。在某些实施例中,受试者(或治疗群体中的多名受试者)在基线时可能先前对nsaid应答不足、不耐受或有禁忌症。
[0527]
治疗强直性脊柱炎(as)的方法
[0528]
进一步提供了治疗强直性脊柱炎(as)的方法。例如,在一个方面,提供了一种治疗包括活动性as在内的as的方法,其包含每天一次以一定量和/或以一定间隔向有需要的受试者口服施用一定剂量的jak1抑制剂。在一个方面,jakl抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,受试者未接受过bdmard。在一个方面,受试者是bdmard-ir。
[0529]
as的疾病活动性/严重程度可以使用多种指标来衡量,包括上述针对axspa治疗的指标。在一个特定方面,提供了一种治疗包括活动性as在内的as的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂,其中受试者在施用 jak1抑制剂后达到国际脊柱关节炎评估协会40(asas40)应答。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,受试者未接受过bdmard。在一个方面,受试者是bdmard-ir。在一个方面,受试者是成年人。
[0530]
在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到 asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14 周)达到asas40应答,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到 asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周) 达到asas40应答,并且在第2周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内、4周内、8周内、12周内、 14周内、16周内、18周内、20周内、24周内、32周内、40周内、52周内、64周内、76周内、88周内、96周内和/或104周内(包括在第2周、第4周、第8周、第12周、第14周、第16周、第18周、第20周、第24周、第32周、第40周、第52周、第64周、第76周、第88周、第96周和/或第104周)达到asas40应答。在一个方面,受试者在施用第一剂jak1 抑制剂后的14周内(包括在第14周)达到asas 40应答,并维持或改善asas40应答直至施用第一剂后的至少64周(例如,直至第64周(包括第64周))。在一个实施例中,受试者在施用第一剂后的2周内(包括在第2周)达到asas 40应答,并维持或改善asas40应答直至施用第一剂后的至少14周(例如,直至至少第14周)。在一个实施例中,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas 40应答,并维持或改善asas40 应答直至施用第一剂后的至少64周(例如,直至第64周(包括第64周))。在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)替代地或额外地达到选自由以下组成的组的至少一个额外结果:asas部分缓解(pr);basdai50应答;脊柱mri sparcc评分(mri-spine sparcc)相对于基线的变化(改善);asdas相对于基线的变化 (改善);
basfi相对于基线的变化(改善);asdas低疾病活动性(lda);asdas非活动性疾病(id);asdas重大改善(mi);以及asdas临床重要改善(cii)。在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到结果,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少2周、持续至少4周、持续至少8 周、持续至少12周、持续至少14周、持续至少16周、持续至少18周、持续至少20周、持续至少24周、持续至少32周、持续至少40周、持续至少52周、持续至少64周、持续至少 76周、持续至少88周、持续至少96周和/或持续至少104周。在一个实施例中,每天一次口服施用jak1抑制剂,持续至少14周。
[0531]
进一步提供了治疗包括活动性as在内的as的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者每天一次口服施用一定剂量的jak1抑制剂,其中受试者在施用jak1抑制剂后达到asas部分缓解(pr)、asdas低疾病活动性(lda)、asdas非活动性疾病(id)、asdas重大改善(mi)和/或asdas临床重要改善(cii)。在一个实施例中,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas pr、asdas lda、asdas id、asdas mi和/或asdas cii。在一个实施例中,受试者在施用第一剂jak1 抑制剂后的14周内(包括在第14周)达到每个结果(例如,asas pr、asdas lda、asdasid、asdas mi和asdas cii)。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内 (包括在第2周)达到asas pr、asdas lda、asdas id、asdas mi和/或asdas cii。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内、4周内、8周内、12周内、14 周内、16周内、18周内、20周内、24周内、32周内、40周内、52周内、64周内、76周内、 88周内、96周内和/或104周内(包括在第2周、第4周、第8周、第12周、第14周、第 16周、第18周、第20周、第24周、第32周、第40周、第52周、第64周、第76周、第 88周、第96周和/或第104周)达到asas pr、asdas lda、asdas id、asdas mi和/ 或asdas ch。在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14 周)达到asas pr、asdas lda、asdas id、asdas mi和/或asdas cii,并且在第14 周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas pr、asdas lda、asdas id、 asdas mi和/或asdas cii,并且在第2周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少2周、持续至少4周、持续至少8周、持续至少12周、持续至少14周、持续至少16周、持续至少18周、持续至少20周、持续至少24周、持续至少32周、持续至少 40周、持续至少52周、持续至少64周、持续至少76周、持续至少88周、持续至少96周和/或持续至少104周。在一个实施例中,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,受试者未接受过bdmard。在一个方面,受试者是bdmard-ir。在一个方面,受试者是成年人。
[0532]
在一个方面,提供了一种在有需要的受试者群体中治疗包括活动性as在内的as的方法,该方法包含如本文所述以一定量和/或以一定间隔向受试者施用一定剂量的jak1抑制剂,其中治疗群体中的一部分受试者(例如,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者)在施用jak1抑制剂后达到asas40反应。在一个方面,治疗群体中的受试者
在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas40应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas40应答,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas40 应答。在一个方面,治疗受试者群体中的受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas40应答,并且在第2周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的2周内、 4周内、8周内、12周内、14周内、16周内、18周内、20周内、24周内、32周内、40周内、 52周内、64周内、76周内、88周内、96周内和/或104周内(包括在第2周、第4周、第8 周、第12周、第14周、第16周、第18周、第20周、第24周、第32周、第40周、第52 周、第64周、第76周、第88周、第96周和/或第104周)达到asas40应答。在一个实施例中,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas 40应答,并维持或改善asas40应答直至施用第一剂后的至少64周内(例如,直至第 64周(包括第64周))。在一个实施例中,治疗群体中的受试者在施用第一剂后的2周内(包括在第2周)达到asas 40应答,并维持或改善asas40应答直至施用第一剂后的至少14 周(例如,直至至少第14周)。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)替代地或额外地达到选自由以下组成的组的至少一个额外结果:asas部分缓解(pr);basdai50应答;脊柱mri sparcc评分(mri-spine sparcc) 相对于基线的变化(改善);asdas相对于基线的变化(改善);basfi相对于基线的变化(改善);asdas低疾病活动性(lda);asdas非活动性疾病(id);asdas重大改善(mi);以及asdas临床重要改善(cii)。在一个方面,治疗群体中的受试者在施用第一剂jakl抑制剂后的14周内(包括在第14周)达到结果,并且在第14周后通过继续施用每日剂量的jak1 抑制剂来维持或改善结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到结果。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,以足以递送15mg的乌帕替尼游离碱当量的量向群体施用一定剂量的 jak1抑制剂。在一个方面,向群体每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,群体中的受试者未接受过bdmard。在一个方面,群体中的受试者是bdmard-ir。
[0533]
进一步提供了在有需要的受试者群体中治疗包括活动性as在内的as,该方法包含如本文所述以一定量和/或一定间隔向有需要的受试者每天一次口服施用一定剂量的jak1抑制剂,其中治疗群体中的部分受试者(例如,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者)在施用jak1抑制剂后达到asas部分缓解(pr)、asdas低疾病活动性(lda)、 asdas非活动性疾病(id)、asdas重大改善(mi)和/或asdas临床重要改善(cii)。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周) 达到asas pr、asdas lda、asdas id、asdas mi和/或asdas cii。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到每个结果 (例如,asas pr、asdas lda、asdas id、asdas mi和asdas cii)。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas pr、 asdas lda、asdas id、asdas mi和/或asdas cii。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的2周内、4周内、
8周内、12周内、14周内、16周内、18周内、20周内、24周内、32周内、40周内、52周内、64周内、76周内、88周内、96周内和 /或104周内(包括在第2周、第4周、第8周、第12周、第14周、第16周、第18周、第 20周、第24周、第32周、第40周、第52周、第64周、第76周、第88周、第96周和/ 或第104周)达到asas pr、asdas lda、asdas id、asdas mi和/或asdas cii。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周) 达到asas pr、asdas lda、asdas id、asdas mi和/或asdas cii,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到asas pr、asdas lda、 asdas id、asdas mi和/或asdas cii,并且在第2周后通过继续施用每日剂量的jak1 抑制剂来维持或改善应答。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到结果。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少2周、持续至少4周、持续至少8周、持续至少12周、持续至少14周、持续至少16周、持续至少18周、持续至少20周、持续至少 24周、持续至少32周、持续至少40周、持续至少52周、持续至少64周、持续至少76周、持续至少88周、持续至少96周和/或持续至少104周。在一个实施例中,每天一次口服施用 jak1抑制剂,持续至少14周。在一个方面,群体中的受试者未接受过bdmard。在一个方面,群体中的受试者是bdmard-ir。在一个方面,受试者是成年人。
[0534]
进一步提供了减轻as的体征和症状的方法。在一个方面,提供了一种减轻包括活动性 as在内的as的体征和症状的方法,该方法包含以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面, jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,受试者未接受过bdmard。在一个方面,受试者是bdmard-ir。
[0535]
在减轻包括活动性as在内的as的体征和症状的方法的一个方面,其中受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到选自由以下组成的组的至少一个结果: asas40应答;asdas相对于基线的变化(改善);mri-spine sparcc相对于基线的变化(改善);asas部分缓解(pr);basdai50应答;basfi相对于基线的变化(改善);asqol 相对于基线的变化(改善);asas健康指数(hi)相对于基线的变化(改善);mases(附着点炎)相对于基线的变化(改善);basmi
lin
(活动度)相对于基线的变化(改善);以及 wpai-axial spa相对于基线的变化(改善)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少 25%、至少30%、至少35%、至少40%或至少45%的受试者达到结果。在某些实施例中,对于达到的上述任何结果,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的 14周内(包括在第14周)达到一个或多个结果,并且在第14周后通过继续施用每日剂量的 jak1抑制剂来维持或改善一个或多个结果。
[0536]
在另一方面,受试者(或治疗群体中的多名受试者)在基线时具有活动性as。在一个方面,受试者(或治疗群体中的多名受试者)在基线时满足1984年修订的as纽约标准。在另一方面,受试者(或治疗群体中的多名受试者)在基线时满足2009年asas分类标准。在又
一方面,受试者(或治疗群体中的多名受试者)在基线时满足1984年修订的as纽约标准和 2009年asas分类标准。在一个实施例中,受试者(或治疗群体中的多名受试者)在基线时满足选自由以下组成的组的至少一个标准:(i)basdai评分≥4;(ii)asdas≥2.1;(iii)患者对总背痛的评估(总背痛评分)≥4(基于0至10数字评定量表)。在一个实施例中,受试者(或治疗群体中的多名受试者)在基线时具有≥4的basdai评分和≥4的患者对总背痛的评估(总背痛评分)。在另一实施例中,受试者(或治疗群体中的多名受试者)在基线时具有≥4的basdai评分和≥2.1的asdas。在某些实施例中,受试者(或治疗群体中的多名受试者)在基线时具有(i)basdai评分≥4;(ii)asdas≥2.1;(iii)患者对总背痛的评估(总背痛评分)≥4(基于0至10数字评定量表)。在一个方面,受试者(或治疗群体中的多名受试者)在基线时没有全脊柱强直。在一个方面,受试者(或治疗群体中的多名受试者)是成年受试者。在另一方面,受试者(或治疗群体中的多名受试者)是幼年受试者。
[0537]
在一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard。示范性bdmard包括但不限于生物肿瘤坏死因子抑制剂(例如,阿达木单抗、依那西普)和白细胞介素il)-17抑制剂(例如,苏金单抗(secukinumab)、依克珠单抗)。
[0538]
在一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且还具有i)对至少两种nsaid应答不足或不耐受(例如,以最大推荐或耐受剂量持续至少四周);ii)对nsaid不耐受;和/或iii)对nsaid的禁忌症,如由医生确定的。nsaid的实例包括但不限于传统的nsaid(例如,布洛芬)和水杨酸盐(例如,阿司匹林)。
[0539]
在某些方面,待治疗的受试者(或治疗群体中的多名受试者)在基线时未接受过 bdmard,对至少两种nsaids(如上所述)应答不足或不耐受,或对nsaids不耐受或有禁忌症,并且正在进一步接受至少一种额外疗法。额外疗法包括但不限于伴随施用非生物 dmard、nsaid、皮质类固醇及其组合。与本文所述的方法组合使用的合适的额外疗法包括:
[0540]
1)伴随施用非生物dmard,包括甲氨蝶呤(mtx)、来氟米特(lef)、柳氮磺胺吡啶 (ssz)和/或羟氯喹(hcq)。在一个实施例中,受试者在基线前至少28天服用稳定剂量的 mtx(≤25mg/周)、ssz(≤3g/天)、羟氯喹(≤400mg/天)和/或来氟米特(≤20mg/天)。在一些实施例中,除了mtx和来氟米特的联合用药外,允许多达两种背景非生物dmard的组合。在一个实施例中,受试者在基线前28天或五个半衰期(以较长者为准)内未接受任何非生物dmard(mtx、lef、ssz和/或hcq除外)、沙利度胺或阿普斯特。
[0541]
2)伴随施用口服皮质类固醇。在一个实施例中,受试者在基线前至少14天服用稳定剂量的泼尼松(prednisone)(≤10mg/天)或口服皮质类固醇当量。
[0542]
3)伴随施用nsaid、曲马多(tramadol)、对乙酰氨基酚和可待因(codeine)或氢可酮的联合用药,和/或非阿片类镇痛药。在一个实施例中,受试者在基线前至少14天服用稳定剂量。
[0543]
在另一方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir。在一个方面,受试者在基线时对bdmard应答不足或不耐受。bdmard-ir的受试者包括既往暴露于一种bdmard(1种肿瘤坏死因子(tnf)抑制剂(例如,阿达木单抗、依那西普)或1 种白细胞介素(il)-17抑制剂(例如,苏金单抗、依克珠单抗)),并且由于不耐受或缺乏疗效而停止bdmard(例如,由医生确定)的那些受试者。在一个实施例中,如果停药的原因不是由于缺乏疗效,则受试者(或治疗群体中的多名受试者)既往未暴露于第二次 bdmard。在一个实施例中,受
试者(或治疗群体中的多名受试者)由于缺乏疗效,尚未停用tnf抑制剂和il-17抑制剂。
[0544]
在某些实施例中,受试者(或治疗群体中的多名受试者)在接受第一剂jak1抑制剂之前已停用bdmard,对于:
[0545]
依那西普≥4周;
[0546]
阿达木单抗、英夫利昔单抗(infliximab)、赛妥珠单抗(certolizumab)、戈利木单抗 (golimumab)、阿巴西普(abatacept)、托珠单抗和依克珠单抗≥8周;
[0547]
优特克单抗≥12周;
[0548]
苏金单抗≥16周;
[0549]
利妥昔单抗(rituximab)≥1年或≥6个月,如果b细胞已经恢复到治疗前水平或如果治疗前水平不可用时则正常参考范围(中心实验室);或
[0550]
对于其它bdmard,≥12周或至少为平均终末消除半衰期的5倍,以较长者为准。
[0551]
在一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且还具有i)对至少两种nsaid的应答不足或不耐受(例如,以最大推荐或耐受剂量持续至少四个星期);ii)对nsaid不耐受;和/或iii)对nsaid的禁忌症。在一个方面,受试者(或受试者群体)是bdmard-ir,并且对至少两种nsaids应答不足或对nsaid不耐受和/或有禁忌症。nsaid的实例包括但不限于传统的nsaid(例如,布洛芬)和水杨酸盐(例如,阿司匹林)。
[0552]
在某些方面,待治疗的受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,对至少两种nsaids(如上所述)应答不足或不耐受,和/或对nsaid不耐受,和/或对nsaids 有禁忌症,并且正在进一步接受至少一种额外疗法。额外疗法包括但不限于伴随施用非生物 dmard、nsaid、皮质类固醇及其组合。与本文所述的方法组合使用的合适的额外疗法包括:
[0553]
1)伴随施用非生物dmard,包括甲氨蝶呤(mtx)、来氟米特、柳氮磺胺吡啶(ssz)、羟氯喹、氯喹和/或阿普斯特。在一个实施例中,受试者在基线前至少28天服用稳定剂量的 mtx(≤25mg/周)、ssz(≤3g/天)、羟氯喹(≤400mg/天)、氯喹(≤400mg/天);来氟米特 (≤20mg/天)或阿普斯特(≤60mg/天))。在一些实施例中,除了mtx和来氟米特的联合用药外,允许多达两种背景非生物dmard的组合。
[0554]
2)伴随施用口服皮质类固醇。在一个实施例中,受试者在基线前至少14天服用稳定剂量的泼尼松(prednisone)(≤10mg/天)或口服皮质类固醇当量。
[0555]
3)伴随施用nsaid、曲马多、对乙酰氨基酚/扑热息痛(paracetamol)和可待因的联合用药或对乙酰氨基酚/扑热息痛和氢可酮的联合用药和/或非阿片类镇痛剂。在一个实施例中,受试者在基线前至少14天服用稳定剂量。
[0556]
在一个实施例中,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,未暴露于任何jak抑制剂,并且并且在基线前的特定时间范围内未接受任何以下治疗/病症:
[0557]
1)在基线前28天内关节内注射、脊髓/椎旁注射或肠胃外施用皮质类固醇(不包括吸入或外用皮质类固醇);
[0558]
2)在基线检查前28天或5个半衰期(以较长者为准)内,包括沙利度胺在内的任何其它非生物性dmard(上述用于伴随治疗的那些除外);
[0559]
3)在基线就诊前14天内使用阿片类镇痛药(对乙酰氨基酚/扑热息痛和可待因的联合用药或对乙酰氨基酚/扑热息痛和氢可酮的联合用药除外):
[0560]
4)在第一剂jak1抑制剂之前28天(如果当地需要,或更长时间)内没有接种活疫苗,或者在用jak1抑制剂治疗期间预计需要接种活疫苗,包括在最后一剂jak1抑制剂后至少 30天(如果当地需要,或更长时间);
[0561]
5)在施用jak1抑制剂期间,未全身使用已知的强细胞色素p450 3a(cyp3a)抑制剂,或在施用jak1抑制剂前30天至治疗结束未全身使用强cyp3a诱导剂;
[0562]
6)治疗期间对cyp3a有未知影响的草药疗法或其它传统药物;
[0563]
7)在施用第一剂jak1抑制剂之前至少30天或5个药物半衰期(以较长者为准)内的化学或生物性质的研究药物;或
[0564]
8)对jak1抑制剂(及其赋形剂)和/或其它同类产品的成分有过敏反应或显着敏感性的历史。
[0565]
在一个实施例中,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard 或是bdmard-ir,并且先前未暴露于任何jak抑制剂。
[0566]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且在施用第一剂后的14周内(包括在第14周)达到asas40应答。在另一实施例中,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且在施用第一剂后的2周内(包括在第2周)达到asas40应答。在一个实施例中,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并在施用第一剂后的2周内 (包括在第2周)达到asas40应答,并维持asas40应答直至施用第一剂后的至少14周(即,包括直至至少第14周)。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂后的2周内(包括在第2周)进一步达到asas pr、asdas lda、asdas id、asdas mi 和/或asdas cii。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0567]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且在施用第一剂后的14周内(包括在第14周)在以下4个 (asas40)域的每一个中达到相对于基线的≥40%的改善和≥2个单位(范围为0到10)的绝对改善:
[0568]
a)根据数字评定量表(nrs 0-10)评估的患者对疾病活动性的总体评估(ptga);
[0569]
b)以数字评定量表(nrs 0-10)评估的患者对总背痛的评估(总背痛评分);
[0570]
c)巴氏强直性脊柱炎功能指数(basfi);和
[0571]
d)炎症,由巴氏强直性脊柱炎疾病活动指数(basdai)的问题5和6的平均值表示。
[0572]
在一个实施例中,上述改善在施用第一剂后的2周内(包括在第2周)达到。在一个实施例中,改善在施用第一剂后的14周内(包括在第14周)达到。在某些实施例中,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0573]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在
基线时未接受过bdmard,并且替代地或额外地在施用第一剂后的14周内(包括在第14周) 达到选自由以下组成的组的至少一个结果:a)asdas(crp)相对于基线的变化(改善);b) 脊柱mri sparcc评分(mri-spine sparcc)相对于基线的变化(改善);c)asas部分缓解(pr);d)basdai50应答;e)basfi相对于基线的变化(改善);f)asqol相对于基线的变化;g)asas健康指数(hi)相对于基线的变化(改善);h)mases相对于基线的变化(改善)(即,对于基线附着点炎的受试者);i)basmilin(活动度)相对于基线的变化 (改善);j)wpai-axial spa相对于基线的变化(改善)。在一个实施例中,受试者在施用第一剂后的2周内(包括在第2周)达到结果。在一个实施例中,受试者在施用第一剂后的14 周内(包括在第14周)达到结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少 30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0574]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且替代地或额外地在施用第一剂后的14周内(包括在第14周) 达到选自由以下组成的组的至少一个结果:a)asdas相对于基线的变化(改善);b)脊柱 mri sparcc评分(mri-spine sparcc)相对于基线的变化(改善);c)asas部分缓解(pr); d)basdai50应答;以及e)basfi相对于基线的变化(改善)。在一个实施例中,在施用第一剂后的14周内(包括在第14周)达到结果中的每一个。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0575]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且替代地或额外地在施用第一剂后的14周内(包括在第14周) 达到选自由以下组成的组的至少一个结果:k)asas20应答;1)骶髂(si)关节的mri sparcc 评分(mri-si关节sparcc)相对于基线的变化(改善)。在一个实施例中,施用第一剂后的14周内(包括在第14周)达到结果中的每一个。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0576]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且替代地或额外地选自由以下组成的组的至少一个结果:m) asas20应答;n)asas40应答;o)asas pr;p)asas 5/6应答;q)asdas非活动性疾病(基于asdas(crp)和asdas(esr));r)asdas低度疾病;s)asdas重大改善(基于asdas(crp)和asdas(esr));t)asdas临床重要改善(基于asdas(crp)和 asdas(esr);u)asas hi相对于基线的变化(改善);v)asdas(crp)和asdas(esr) 相对于基线的变化(改善);w)asqol相对于基线的变化(改善);x)basdai相对于基线的变化(改善);y)basfi相对于基线的变化(改善);z)
basmi
lin
相对于基线的变化(改善);aa)c反应蛋白(crp)相对于基线的变化(改善);bb)facit-f相对于基线的变化(改善);cc)isi相对于基线的变化(改善);dd)mases(基线mases>0的受试者)相对于基线的变化(改善);ee)masss(使用常规射线照片)相对于基线的变化(改善);ff)si关节的mri sparcc评分相对于基线的变化(改善);gg)脊柱的mri sparcc评分相对于基线的变化(改善);hh)患者对总背痛评分的评估(总背痛评分)相对于基线的变化(改善); ii)患者对夜间背痛的评估(夜间背痛)相对于基线的变化(改善);jj)患者对疼痛的总体评估(pt pain)相对于基线的变化(改善);kk)医生对疾病活动性的总体评估(pga-疾病活动性)相对于基线的变化(改善);11)炎症相对于基线的变化(改善),以basdai的问题5 和6的平均值相对于基线的变化(改善)表示;mm)患者对总背痛的评估相对于基线的变化 (改善),以basdai的问题2相对于基线的变化(改善)表示;nn)外周疼痛/肿胀相对于基线的变化(改善),以basdai的问题3相对于基线的变化(改善)表示;oo)晨僵持续时间相对于基线的变化(改善),以basdai的问题6相对于基线的变化(改善)表示;pp) 患者对疾病活动性的总体评估(ptga)相对于基线的变化(改善);qq)tjc68和sjc66相对于基线的变化(改善);rr)wpai-axial spa相对于基线的变化(改善);ss)在基线时存在指趾炎的受试者的指趾炎消退(改善);tt)在基线时存在指趾炎的受试者的总指趾炎计数相对于基线的变化(改善)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0577]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且替代地或额外地在施用第一剂后的14周(包括在第14周)达到选自由以下组成的组的至少一个结果:q)asdas非活动性疾病;r)asdas低度疾病;s) asdas重大改善;t)asdas临床重要改善。在一个实施例中,施用第一剂后的14周内(包括在第14周)达到结果中的每一个。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0578]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard,并且替代地或额外地达到选自由以下组成的组的至少一个结果:asdas非活动性疾病、asdas中度疾病、asdas低疾病活动性(lda)、asdas高度疾病、asdas极高度疾病、asdas重大改善和asdas临床重要改善。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0579]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且在施用第一剂后的14周内(包括在第14周)达到asas40应答。在另
一实施例中,受试者在基线时是bdmard-ir,并且在施用第一剂后的2周内(包括在第2周)达到asas40应答。在一个实施例中,受试者在施用第一剂后的2周内(包括在第 2周)达到asas40应答,并维持asas40应答直至施用第一剂后的至少14周(即,直至至少第14周)。在一个方面,受试者(或受试者群体)在施用第一剂后的2周内(包括在第2 周)进一步达到asas pr、asdas lda、asdas id、asdas mi和/或asdas cii。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0580]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且替代地或额外地在施用第一剂后的14周内(包括在第14周)达到选自由以下组成的组的至少一个结果:a)asdas相对于基线的变化(改善);b)脊柱的 mpi sparcc评分(mpi-spine sparcc)相对于基线的变化(改善);c)asas部分缓解(pr); d)basdai 50应答;e)basfi相对于基线的变化(改善);f)asqol相对于基线的变化(改善);g)asas健康指数(hi)相对于基线的变化(改善);h)mases(附着点炎)相对于基线的变化(改善);i)basmilin(活动度)相对于基线的变化(改善)。在一个实施例中,在施用第一剂后的14周内(包括在第14周)达到结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0581]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果:j)asas 20应答;k)si关节的mri sparcc评分(mri-si关节sparcc)相对于基线的变化(改善)。在一个实施例中,在施用第一剂后的14周内(包括在第14周)达到结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0582]
在本文所述的治疗as的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果:1) asas20应答;m)asas40应答;n)asas pr;o)asdas非活动性疾病;p)asdas低度疾病;q)asdas重大改善;r)asdas临床重要改善;s)在基线时使用阿片类药物的受试者中停用阿片类药物;t)asas hi相对于基线的变化(改善);u)asdas相对于基线的变化(改善);v)asqol相对于基线的变化(改善);w)basdai和basdai问题相对于基线的变化(改善),包括basdai的问题5和6的平均值相对于基线的变化(改善);x) basfi相对于基线的变化(改善);y)basmi
lin
相对于基线的变化(改善);z)高敏c反应蛋白(hscrp)相对于基线的变化(改善);aa)facit-f相对于基线的变化(改善);bb) euroqol-5d-5l(eq-5d-5l)相对于基线的变化(改善);cc)mases相对于基线的变化(改善);dd)msasss(使用常规射线照片)相对于基线的变化(改
善);ee)si关节的mri sparcc 评分相对于基线的变化(改善);ff)脊柱的mri sparcc评分相对于基线的变化(改善); gg)患者对总背痛评分的评估(总背痛评分)相对于基线的变化(改善);hh)患者对夜间背痛的评估(夜间背痛)相对于基线的变化(改善);ii)患者对背痛的总体评估(pt pain)相对于基线的变化(改善);jj)医生对疾病活动性的总体评估(pga-疾病活动性)相对于基线的变化(改善);kk)患者对疾病活动性的总体评估(ptga)相对于基线的变化(改善);11) sf-36相对于基线的变化(改善);mm)tjc68和sjc66相对于基线的变化(改善);nn) wpai-axial spa相对于基线的变化(改善);oo)nsaid评分变化相对于基线的变化(改善); pp)体力活动评估相对于基线的变化(改善)。在一个实施例中,在施用第一剂后的14周内 (包括在第14周)达到结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。
[0583]
治疗非射线学中轴型脊柱关节炎(nr-axspa)的方法
[0584]
进一步提供了治疗非射线学中轴型脊柱关节炎(nr-axspa)的方法。在一个方面,提供了治疗活动性nr-axspa的方法,其包含以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用 jak1抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52 周。在一个方面,受试者未接受过bdmard。在一个方面,受试者是bdmard-ir。
[0585]
nr-axspa的疾病活动性/严重程度可以使用多种指标来衡量,包括上述针对axspa治疗的指标。在一个特定方面,提供了一种治疗包括活动性nr-axspa在内的nr-axspa的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂,其中受试者在施用jak1抑制剂后达到国际脊柱关节炎评估协会40(asas40)应答。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52周。在一个方面,受试者未接受过bdmard。在一个方面,受试者是bdmard-ir。
[0586]
在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到 asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂后的14周内(包括在第14 周)达到asas40应答,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者在施用第一剂jakl抑制剂后的52周内(包括在第52周)达到asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂后的52周内(包括在第52 周)达到asas40应答,并且在第52周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到 asas40应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内(包括在第2周) 达到asas40应答,并且在第2周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者在施用第一剂jak1抑制剂后的2周内、4周内、8周内、12周内、 14周内、18周内、24周内、32周内、40周内和/或52周内(包括在第2周、第4周、第8 周、第12周、第14周、第18周、第24周、第32周、
第40周和/或第52周)达到asas40 应答。在一个实施例中,受试者在施用第一剂后的2周内(包括在第2周)达到asas 40应答,并维持asas40应答直至施用第一剂后的至少14周(例如,直至至少第14周)。在一个实施例中,受试者在施用第一剂后的2周内(包括在第2周)达到asas 40应答,并维持 asas40应答直至施用第一剂后的至少52周(例如,直至至少第52周)。在一个方面,jak1 抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少2周、持续至少4周、持续至少8周、持续至少12周、持续至少14周、持续至少18周、持续至少24周、持续至少32周、持续至少40周和/或持续至少52周。在一个实施例中,每天一次口服施用jak1 抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52周。
[0587]
在一个方面,提供了一种在有需要的受试者群体中治疗包括活动性nr-axspa在内的 nr-axspa的方法,该方法包含如本文所述以一定量和/或以一定间隔向受试者施用一定剂量的 jak1抑制剂,其中治疗群体中的一部分受试者(例如,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者)在施用jak1抑制剂后达到asas40应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas40应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到asas40应答,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周) 达到asas40应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到asas40应答,并且在第52周后通过继续施用每日剂量的jak1 抑制剂来维持或改善结果。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,以足以递送15mg的乌帕替尼游离碱当量的量向受试者施用一定剂量的jak1抑制剂。在一个方面,向受试者每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,向受试者每天一次口服施用jak1抑制剂,持续至少52周。在一个方面,群体中的受试者未接受过 bdmard。在一个方面,群体中的受试者是bdmard-ir。
[0588]
进一步提供了减轻nr-axspa的体征和症状的方法。在一个方面,提供了一种减轻活动性 nr-axspa的体征和症状的方法,特别是包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂的方法。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少14周。在一个方面,每天一次口服施用jak1抑制剂,持续至少52周。在一个方面,受试者未接受过bdmard。在一个方面,受试者是 bdmard-ir。
[0589]
在减轻包括活动性nr-axspa在内的nr-axspa的体征和症状的方法的一个方面,受试者 (或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的14周内(包括在第14周达到选自以下组成的组的至少一个结果:asas40应答;asdas相对于基线的变化(改善); mri-spine sparcc相对于基线的变化(改善);asas部分缓解(pr);basdai50应答; basfi相对于基线的变化(改善);asqol相对于基线的变化(改善);asas健康指数(hi) 相对于基线的变化(改善);mases(附着点炎)相对于基线的变化(改善);和basmilin (活动度)相对于基线的变化(改善);或受试者在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到
asas40应答。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的14周内(包括在第14周)达到结果,并且在第14周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的52周内(包括在第52周)达到结果,并且在第 52周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少 15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0590]
在一个方面,需要治疗的受试者(或受试者群体)在基线时患有活动性nr-axspa。在一个方面,受试者(或治疗群体中的多名受试者)在基线时满足2009年asas的axspa分类标准,但不满足1984年修订的as纽约标准的放射学标准。在一个实施例中,受试者(或治疗群体中的多名受试者)在基线时满足选自由以下组成的组的至少一个标准:(i)basdai 评分≥4;(ii)asdas≥2.1;(iii)患者对总背痛的评估(总背痛评分)≥4(基于0至10数字评定量表);(iv)炎症活动性的客观体征,选自由以下组成的组:a)骶髂(si)关节mri 上活动性炎症的客观体征;b)高敏c反应蛋白(hscrp)》正常上限(uln)。在一个方面,受试者满足标准(i)、(ii)、(iii)和(iv)。在一个方面,受试者(或治疗群体中的多名受试者)是成年受试者。在另一方面,受试者(或治疗群体中的多名受试者)是幼年受试者。
[0591]
在一个方面,受试者(或治疗群体中的多名受试者)在基线时未接受过bdmard。示范性bdmard包括但不限于生物肿瘤坏死因子抑制剂(例如,阿达木单抗、依那西普)和白细胞介素il)-17抑制剂(例如,苏金单抗、依克珠单抗)。
[0592]
在另一方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir。在一个方面,受试者(或治疗群体中的多名受试者)对bdmard应答不足或不耐受。bdmard-ir 的受试者包括既往暴露于一种bdmard(1种肿瘤坏死因子(tnf)抑制剂(例如,阿达木单抗、依那西普)或1种白细胞介素(il)-17抑制剂(例如,苏金单抗、依克珠单抗)),并且由于不耐受或缺乏疗效而停用bdmard(例如,由医生确定)的那些受试者。在一个实施例中,如果停药的原因不是由于缺乏疗效,则受试者(或治疗群体中的多名受试者)既往未暴露于第二次bdmard。在一个实施例中,受试者(或治疗群体中的多名受试者)由于缺乏疗效,尚未停用tnf抑制剂和il-17抑制剂。在某些实施例中,受试者(或治疗群体中的多名受试者)在接受第一剂jak1抑制剂之前已停用bdmard,对于:
[0593]
依那西普≥4周;
[0594]
阿达木单抗、英夫利昔单抗(infliximab)、赛妥珠单抗(certolizumab)、戈利木单抗 (golimumab)、阿巴西普(abatacept)、托珠单抗和依克珠单抗≥8周;
[0595]
优特克单抗≥12周;
[0596]
苏金单抗≥16周;
[0597]
利妥昔单抗(rituximab)≥1年或≥6个月,如果b细胞已经恢复到治疗前水平或如果治疗前水平不可用时则正常参考范围(中心实验室);或
[0598]
对于其它bdmard,≥12周或至少为平均终末消除半衰期的5倍,以较长者为准。
[0599]
在一个方面,受试者(或治疗群体中的多名受试者)在基线时还具有i)对至少两种nsaid 的应答不足或不耐受(例如,以最大推荐或耐受剂量持续至少四个星期);ii)对nsaid不耐受;和/或iii)对nsaid的禁忌症。nsaid的实例包括但不限于传统的nsaid(例如,
布洛芬)和水杨酸盐(例如,阿司匹林)。
[0600]
在某些方面,待治疗的受试者(或受试者群体)进一步接受至少一种额外疗法。额外疗法包括但不限于伴随施用非生物dmard、nsaid、皮质类固醇及其组合。与本文所述的方法组合使用的合适的额外疗法包括:
[0601]
1)伴随施用非生物dmard,包括甲氨蝶呤(mtx)、来氟米特、柳氮磺胺吡啶(ssz)、羟氯喹、氯喹,或阿普斯特。在一个实施例中,受试者在基线前至少28天服用稳定剂量的 mtx(≤25mg/周)、ssz(≤3g/天)、羟氯喹(≤400mg/天)、氯喹(≤400mg/天);来氟米特(≤20mg/天)或阿普斯特(≤60mg/天))。在一些实施例中,除了mtx和来氟米特的联合用药外,允许多达两种背景非生物dmard的组合。
[0602]
2)伴随施用口服皮质类固醇。在一个实施例中,受试者在基线前至少14天服用稳定剂量的泼尼松(prednisone)(≤10mg/天)或口服皮质类固醇当量。
[0603]
3)伴随施用nsaid、曲马多、对乙酰氨基酚/扑热息痛(paracetamol)和可待因的联合用药或对乙酰氨基酚/扑热息痛和氢可酮的联合用药和/或非阿片类镇痛剂。在一个实施例中,受试者在基线前至少14天服用稳定剂量。
[0604]
在一个实施例中,受试者(或治疗群体中的多名受试者)在基线时先前未暴露于任何jak 抑制剂。
[0605]
在本文所述的治疗nr-axspa的方法的一个方面,受试者(或治疗群体中的多名试者)替代地或额外地在施用第一剂后的14周内(包括在第14周)达到选自由以下组成的组的至少一个结果:a)asdas相对于基线的变化(改善);b)si关节的mri sparcc评分(mri-si 关节sparcc)相对于基线的变化(改善);c)asas部分缓解(pr);d)basdai50反应;e)basfi相对于基线的变化(改善);f)asqol相对于基线的变化(改善);g)asas健康指数(hi)相对于基线的变化(改善);h)mases(附着点炎)相对于基线的变化(改善); i)basmilin(活动度)相对于基线的变化(改善);在一个实施例中,受试者在施用第一剂后的2周内(包括在第2周)达到结果。在一个实施例中,受试者在施用第一剂后的14周内 (包括在第14周)达到结果中的每一个。在某些实施例中,对于达到的任何上述结果,在第 2周或第14周后通过继续施用每日剂量的jak1抑制剂来维持一个结果(或多个结果)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0606]
在本文所述的治疗nr-axspa的方法的一个方面,受试者(或治疗群体中的多名受试者) 替代地或额外地在施用第一剂后的14周内(包括在第14周)达到选自由以下组成的组的至少一个结果:j)asas 20应答;k)脊柱的mri sparcc评分(mri-spine sparcc)相对于基线的变化(改善)。在一个实施例中,在施用第一剂后的14周内(包括在第14周)达到结果中的每一个。在某些实施例中,对于达到的任何上述结果,在第14周后通过继续施用每日剂量的jakl抑制剂来维持一个结果(或多个结果)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0607]
在本文所述的治疗nr-axspa的方法的一个方面,受试者(或治疗群体中的多名受试者) 替代地或额外地达到选自由以下组成的组的至少一个结果:1)asas20应答;m)asas40 应答;n)asas pr;o)asdas非活动性疾病;p)asdas低度疾病;q)asdas重大改善; r)
asdas临床重要改善;s)在基线时使用阿片类药物的受试者停用阿片类药物;t)asas hi 相对于基线的变化(改善);u)asdas相对于基线的变化(改善);v)asqol相对于基线的变化(改善);w)basdai和basdai问题相对于基线的变化(改善),包括basdai的问题5和6的平均值相对于基线变化(改善);x)basfi相对于基线的变化(改善);y)basmi
lin
相对于基线的变化(改善);z)高敏c反应蛋白(hscrp)相对于基线的变化(改善);aa) facit-f相对于基线的变化(改善);bb)eq-5d-5l相对于基线的变化(改善);cc)mases 相对于基线的变化(改善);dd)masss(使用常规射线照片)相对于基线的变化(改善); ee)si关节的mri sparcc评分相对于基线的变化(改善);ff)脊柱的mri sparcc评分相对于基线的变化(改善);gg)患者对总背痛评分的评估(总背痛评分)相对于基线的变化 (改善);hh)患者对夜间背痛的评估(夜间背痛)相对于基线的变化(改善);ii)患者对疼痛的总体评估(pt pain)相对于基线的变化(改善);jj)医生对疾病活动性的总体评估(pga
‑ꢀ
疾病活动性)相对于基线的变化(改善);kk)患者对疾病活动性的总体评估(ptga)相对于基线的变化(改善);11)sf-36相对于基线的变化(改善);m)tjc68和sjc66相对于基线的变化(改善);nn)(wpai-axial spa)相对于基线的变化(改善);和oo)nsaid评分变化相对于基线的变化(改善)。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0608]
治疗银屑病关节炎(psa)和银屑病(pso)的方法
[0609]
进一步提供了治疗银屑病关节炎(psa)和银屑病(pso)(包括作为psa皮肤表现的pso) 的方法。
[0610]
在一个方面,提供了治疗活动性psa的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jakl抑制剂。在一个方面,jakl抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少12周。在一个方面,受试者是非生物dmard-ir。在一个方面,受试者是bdmard-ir。在一个方面,受试者是成年人。
[0611]
psa的疾病活动性/严重程度可以使用多种指标来衡量,包括美国风湿病学会应答率(例如,acr20、acr50、acr70);健康评估问卷-残疾指数(haq-di);银屑病静态研究者总体评估(siga);银屑病面积严重程度指数(pasi)(包括pasi75、pasi 90和pasi 100); sharp/van der heijde评分(shs);最小疾病活动性(mda)评估;利兹附着点炎指数(lei);利兹指趾炎指数(ldi);36项简表健康调查(sf-36);患者对疼痛的总体评估数字评定量表 (nrs);慢性病治疗疲劳功能评估(facit-f)问卷;银屑病症状自我评估(saps)问卷;压痛关节计数(tjc68);肿胀关节计数(sjc66);医生对疾病活动性的总体评估数字评定量表(pga-疾病活动性nrs)(数值评定量表);患者对疾病活动性的总体评估(ptga);高敏 c反应蛋白水平(hscrp);指趾炎计数;指趾炎的消退;lei中包括的附着点炎部位的消退;加拿大脊柱关节炎研究协会(sparcc)附着点炎指数;sparcc附着点炎指数中包括的附着点炎部位的消退;总附着点炎计数;附着点炎的消退;银屑病体表面积(bsa-ps);改良银屑病关节炎应答标准(psarc);疾病活动性评分28(das28)(crp);das28(红细胞沉降率(ers));银屑病关节炎疾
病活动性评分(pasdas);银屑病关节炎疾病活动性(dapsa) 评分;euroqol-5d-5l(eq-5d-5l)问卷;工作效率和活动障碍问卷-银屑病关节炎 (wpai-psa);卫生资源利用(hru)问卷;巴氏强直性脊柱炎疾病活动指数(basdai); basdai 50应答;晨僵(basdai问题5和6的平均值);以及强直性脊柱炎疾病活动性评分(asdas)。这些指标在临床终点定义和实例中有详细描述。
[0612]
在一个特定方面,提供了一种治疗包括活动性psa在内的psa的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂,其中受试者在施用jak1抑制剂后达到美国风湿病学会20%(acr20)应答。在一个实施例中,受试者在施用jak1抑制剂后达到acr 50%(acrs0)应答。在一个实施例中,受试者在施用jak1抑制剂后达到acr 70%(acr70)应答。在一个方面,受试者在施用第一剂jak1抑制剂后的 12周内(包括在第12周)达到acr20、acr50或acr70应答。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少12周。在一个方面,受试者是非生物dmard-ir。在一个方面,受试者是bdmard-ir。
[0613]
在另一方面,提供了一种在有需要的受试者群体中治疗包括活动性psa在内的psa的方法,该方法包含如本文所述以一定量和/或以一定间隔向受试者施用一定剂量的jak1抑制剂,其中治疗群体中的一部分受试者(例如,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者)在施用jak1抑制剂后达到acr20、acr50或acr70应答。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的12周内(包括在第12周)达到acr20、acr50 或acr70应答。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,以足以递送15mg 的乌帕替尼游离碱当量的量向受试者施用一定剂量的jak1抑制剂。在一个方面,以足以递送30mg的乌帕替尼游离碱当量的量向受试者施用一定剂量的jak1抑制剂。在一个方面,向受试者每天一次口服施用jak1抑制剂,持续至少12周。
[0614]
在某些实施例中,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的12周内(包括在第12周)达到acr20应答。在某些实施例中,在第12周后通过继续施用每日剂量的jak1抑制剂来维持或改善acr20应答。在某些实施例中,受试者(或治疗群体中的多名受试者)在施用第一剂后的2周内(包括在第2周)达到acr20应答。在某些实施例中,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的2周内(包括在第2周)达到acr20应答,并且在第2周后通过继续施用每日剂量的jak1抑制剂来维持或改善acr20应答。在某些实施例中,在基线时患有活动性psa的受试者(或治疗群体中的多名受试者)进一步达到选自由以下组成的组的至少一个结果:(a)在施用第一剂后的 12周内(包括在第12周)健康评估问卷-残疾指数(haq-di)相对于基线的变化(改善); (b)在施用第一剂后的16周内(包括在第16周)银屑病静态研究者总体评估(siga)达到 0或1,并且相对于基线改善至少2分(对于基线siga≥2的受试者);(c)在施用第一剂后的16周内(包括在第16周)达到银屑病面积严重程度指数(pasi)75应答(对于基线时 bsa银屑病≥3%的受试者);(d)在施用第一剂后的24周内(包括在第24周)sharp/van der heijde 评分(shs)相对于基线的变化(改善);(e)在施用第一剂后的24周内(包括在第24周) 达到最小疾病活动性(mda);(f)
在施用第一剂后的24周内利兹附着点炎指数(lei)相对于基线的变化(改善),优选地其中变化(改善)是在施用第一剂后的24周内(包括在第 24周)附着点炎的消退(lei=0)(对于基线存在附着点炎(lei>0)的受试者);(g)在施用第一剂后的12周内(包括在第12周)达到acr 20应答(乌帕替尼与阿达木单抗相比的非劣效性);(h)在施用第一剂后的12周内(包括在第12周)36项简表健康调查(sf-36) 相对于基线的变化(改善);(i)在施用第一剂后的12周内(包括在第12周)慢性病治疗疲劳功能评估(facit-f)问卷相对于基线的变化(改善)。在某些实施例中,在基线时患有活动性psa的受试者(或治疗群体中的多名受试者)进一步达到每个结果。在某些实施例中,在基线时患有活动性psa的受试者(或治疗群体中的多名受试者)进一步达到选自由以下组成的组的至少一个结果:(j)在施用第一剂后的12周内(包括在第12周)的acr 20应答并优于阿达木单抗(每隔一周40mg);(k)在施用第一剂后的24周内利兹指趾炎指数(ldi) 相对于基线的变化(改善),优选地其中变化(改善)是在施用第一剂后的24周内(包括在第24周)指趾炎的消退(ldi=0)(对于基线存在指趾炎(ldi>0)的受试者))。在某些实施例中,在基线时患有活动性psa的受试者(治疗群体中的多名受试者)在施用第一剂后的 12周内(包括在第12周)进一步达到acr 20应答并优于阿达木单抗(每隔一周40mg)。在一个方面,受试者(或治疗群体中的多名受试者)在基线时是非生物dmard-ir。在一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir。在一个方面,在第 12周、第16周和/或第24周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂后的16周内(包括在第16 周)进一步达到银屑病面积严重程度指数(pasi)75应答。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂后的16周内(包括在第16周)进一步达到pasi 90应答。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂后的16周内(包括在第 16周)进一步达到pasi 100应答。在一个方面,在第16周后通过继续施用每日剂量的jak1 抑制剂来维持或改善pasi应答(例如,pasi 75、pasi 90或pasi 100应答)。在某些实施例中,对于达到的任何上述结果,在达到一个结果(或多个结果)后通过继续施用每日剂量的 jak1抑制剂来维持或改善一个结果(或多个结果)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0615]
进一步提供了在有需要的受试者中减轻psa的体征和症状的方法。在一个方面,提供了一种减轻包括活动性psa在内的psa的体征和症状的方法,其包含如本文所述以一定量和/ 或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jakl抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少12周。在一个方面,受试者是非生物 dmard-ir。在一个方面,受试者是bdmard-ir。
[0616]
在另一方面,提供了一种在有需要的受试者中治疗包括活动性psa在内的psa的方法,其包含如本文所述以一定量和/或以一定间隔向受试者施用一定剂量的jak1抑制剂,其中受试者在施用jak1抑制剂后达到最小疾病活动性(mda)。在一个方面,jak1抑制剂是乌帕替尼游离碱或其药学上可接受的盐。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的
量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少24周。在一个方面,受试者在施用第一剂jak1抑制剂后的24周内(包括在第24周)达到mda。在一个方面,受试者在施用第一剂jak1抑制剂后的24周内(包括在第24周)达到mda,并且在第24周后通过继续施用每日剂量的jak1抑制剂来维持或改善mda。在一个方面,受试者在施用第一剂后的16周内进一步达到选自pasi 75应答、pasi 90应答和pasi 100应答的银屑病面积严重程度指数(pasi)应答。在一个方面,在第16周后通过继续施用每日剂量的jak1抑制剂来维持或改善pasi 75、pasi 90和/或pasi 100应答。在一个方面,受试者是非生物 dmard-ir。在一个方面,受试者是bdmard-ir。
[0617]
在另一方面,提供了一种在有需要的受试者群体中治疗包括活动性psa在内的psa的方法,该方法包含如本文所述以一定量和/或以一定间隔向受试者施用一定剂量的jak1抑制剂,其中治疗群体中的一部分受试者(例如,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者)在施用jak1抑制剂后达到最小疾病活动性(mda)。在一个方面,jak1抑制剂是乌帕替尼游离碱或其药学上可接受的盐。在一个方面,以足以递送15mg的乌帕替尼游离碱当量的量向受试者施用一定剂量的jak1抑制剂。在一个方面,以足以递送30mg的乌帕替尼游离碱当量的量向受试者施用一定剂量的jak1抑制剂。在一个方面,向受试者每天一次口服施用jak1抑制剂,持续至少24周。在一个方面,治疗群体中的受试者在施用第一剂jak1 抑制剂后的24周内(包括在第24周)达到mda。在一个方面,治疗群体中的受试者在施用第一剂jak1抑制剂后的24周内(包括在第24周)达到mda,并且在第24周后通过继续施用每日剂量的jak1抑制剂来维持或改善mda。在一个方面,治疗群体中的受试者在施用第一剂后的16周内进一步达到选自pasi 75应答、pasi 90应答和pasi 100应答的银屑病面积严重程度指数(pasi)应答。在一个方面,在第16周后通过继续施用每日剂量的jak1抑制剂来维持或改善pasi 75、pasi 90和/或pasi 100应答。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。在一个方面,治疗群体中的受试者在基线时是非生物dmard-ir。在一个方面,治疗群体中的受试者在基线时是bdmard-ir。
[0618]
在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的12 周内(包括在第12周)达到acr20、acr50或acr70应答。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的12周内(包括在第12周)达到 acr20、acr50或acr70应答,并且在第12周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂jak1 抑制剂后的2周内(包括在第2周)达到acr20、acr50或acr70应答。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂jak1抑制剂后的2周内(包括在第2周) 达到acr20、acr50或acr70应答,并且在第12周后通过继续施用每日剂量的jak1抑制剂来维持或改善应答。在一个方面,受试者(或治疗群体中的多名受试者)在施用第一剂jak1 抑制剂后的2周内、4周内、8周内、12周内、16周内、20周内、24周内、28周内、32周内、36周内、44周内和/或56周内(包括在第2周、第4周、第8周、第12周、第16周、第20周、第24周、第28周、第32周、第36周、第44周和/或第56周)达到acr20、acr50 或acr70应答。在一个实施例中,受试者(或治疗群体中的多名受试者)在
施用第一剂后的 2周内(包括在第2周)达到acr20、acr50或acr70应答,并维持acr20、acr50或 acr70应答直至施用第一剂后的至少12周(例如,直至至少第12周)。在一个方面,jak1 抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少2周、持续至少4周、持续至少8 周、持续至少12周、持续至少16周、持续至少20周、持续至少24周、持续至少28周、持续至少32周、持续至少36周、持续至少44周和/或持续至少56周。在一个实施例中,每天一次口服施用jak1抑制剂,持续至少12周。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少 25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0619]
在减轻包括活动性psa在内的psa的体征和症状的方法的一个方面,受试者(或治疗群体中的多名受试者)达到选自由以下组成的组的至少一个结果:在施用第一剂jak1抑制剂后的12周内(包括在第12周)的acr20应答;在施用第一剂jak1抑制剂后的12周内(包括在第12周)haq-di相对于基线的变化(改善);在施用第一剂jakl抑制剂后的16周内 (包括在第16周)siga为0或1,并且相对于基线改善至少2分;在施用第一剂jak1抑制剂后的16周内(包括在第16周)的pasi 75应答(对于基线时≥3%体表面积银屑病的受试者);在施用第一剂jak1抑制剂后的24周内(包括在第24周)shs相对于基线的变化(改善);在施用第一剂jak1抑制剂后的24周内(包括在第24周)的最小疾病活动性;在施用第一剂jak1抑制剂后的24周内(包括在第24周)lei相对于基线的变化(改善);在施用第一剂jak1抑制剂后的24周内(包括在第24周)ldi相对于基线的变化(改善);在施用第一剂jak1抑制剂后的12周内(包括在第12周)sf-36相对于基线的变化(改善);在施用第一剂jak1抑制剂后的12周内(包括在第12周)患者对疼痛的总体评估(pt pain)(数字评定量表)相对于基线的变化(改善);在施用第一剂jak1抑制剂后的12周内(包括在第12 周)facit-f相对于基线的变化(改善);在施用第一剂jak1抑制剂后的16周内(包括在第16周)saps问卷相对于基线的变化(改善);在施用第一剂jak1抑制剂后的12周内(包括在第12周)的acr50或acr70应答;以及在施用第一剂jak1抑制剂后的2周内(包括在第2周)的acr20应答。在一个方面,在第2周、第12周、第16周或第24周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0620]
进一步提供了在有需要的受试者中抑制psa的结构损伤进展的方法。在一个方面,提供了一种抑制包括活动性psa在内的psa的结构损伤进展的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少12周。在一个方面,受试者是非生物 dmard-ir。在一个方面,受试者是bdmard-ir。
[0621]
进一步提供了在有需要的受试者中改善身体机能的方法。在一个方面,提供了一种改善包括活动性psa在内的psa的身体机能的方法,其包含如本文所述以一定量和/或以
一定间隔向有需要的受试者施用一定剂量的jak1抑制剂。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jakl抑制剂,持续至少12周。在一个方面,受试者是非生物dmard-ir。在一个方面,受试者是bdmard-ir。
[0622]
进一步提供了在有需要的受试者中预防psa的结构进展的方法。在一个方面,提供了一种预防包括活动性psa在内的psa的结构进展的方法,其包含如本文所述以一定量和/或以一定间隔向有需要的受试者施用一定剂量的jak1抑制剂。在一个方面,jak1抑制剂是乌帕替尼游离碱。在一个方面,jak1抑制剂以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,jak1抑制剂以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用jak1抑制剂,持续至少12周。在一个方面,受试者是非生物dmard-ir。在一个方面,受试者是bdmard-ir。
[0623]
在一个方面,需要治疗的受试者(或受试者群体)在基线时患有活动性psa。在一个方面,需要治疗并且在基线时患有活动性psa的受试者(或受试者群体)进一步在基线时患有由医生确定的中度至重度活动性银屑病关节炎。在一个方面,受试者(或治疗群体中的多名受试者)临床诊断为psa,并在基线前至少6个月出现症状。在一个方面,受试者(或治疗群体中的多名受试者)在基线时满足psa分类标准(caspar)的标准。在一个实施例中,受试者(或治疗群体中的多名受试者)满足选自由以下组成的组的至少一个标准:(i)基线时≥3个压痛关节(基于68个关节计数);(ii)基线时≥3个肿胀关节(基于66个关节计数)。在一个方面,受试者(或治疗群体中的多名受试者)在基线时有≥3个压痛关节(基于68个关节计数);和(ii)≥3个肿胀关节(基于66个关节计数)。在某些实施例中,受试者(或治疗群体中的多名受试者)在基线时可能有≥5个压痛关节(基于68个关节计数)和≥5个肿胀关节(基于66个关节计数)。在一个方面,受试者(或治疗群体中的多名受试者)在基线时满足选自由以下组成的组的至少一个标准:中央成像检查确定的x光片≥1次侵蚀,以及 hs-crp》实验室定义的正常上限(uln)。在一个方面,受试者(或治疗群体中的多名受试者) 在基线时没有针对psa的最小疾病活动性(应答者和非应答者)。在一个方面,受试者(或治疗群体中的多名受试者)在基线时被诊断为活动性斑块状银屑病或有斑块状银屑病病史记录(例如,由医生确定)。在一个方面,受试者(或治疗群体中的多名受试者)是成年受试者。在另一方面,受试者(或治疗群体中的多名受试者)是幼年受试者。
[0624]
在一个方面,受试者(或治疗群体中的多名受试者)在基线时是非生物dmard-ir。在一个方面,受试者(或治疗群体中的多名受试者)对非生物dmard治疗应答不足或不耐受。非生物dmard-ir受试者包括对非生物dmard不耐受或有禁忌症(例如,由医生确定) 或对先前或伴随使用至少一种非生物dmard以最大耐受剂量或达到以下规定的剂量的治疗应答不足(即,在至少12周的治疗后缺乏疗效)的那些受试者:
[0625]
i)甲氨蝶呤(mtx):在完全滴定后对剂量≥12.5mg/周的mtx不耐受的受试者中,≤25mg/ 周,或≥15至≤25mg/周,或≥10mg/周;
[0626]
ii)柳氮磺胺吡啶(ssz):≤3000mg/天;
[0627]
iii)来氟米特(lef):≤20mg/天;
[0628]
iv)阿普斯特:≤60mg/天;
[0629]
v)羟氯喹(hcq):≤400mg/天;
[0630]
vi)布西拉明:≤300mg/天;或
[0631]
vii)艾拉莫德:≤50mg/天)
[0632]
持续时间≥12周,并且在基线前稳定剂量≥4周。在一个方面,受试者没有暴露于任何额外的非生物dmard。
[0633]
在某些方面,待治疗的受试者(或受试者群体)是非生物dmard-ir,并且正在进一步接受至少一种额外疗法。合适的额外疗法包括:
[0634]
1)稳定剂量的非生物dmard。在一个方面,受试者在基线时接受伴随的dmard治疗。在一个方面,受试者服用≤2次非生物dmard(除了mtx和来氟米特的联合用药)。 mtx、ssz、lef、阿普斯特、hcq、布西拉明和艾拉莫德的合适剂量如上所述。在一个方面,受试者未服用除mtx、ssz、lef、阿普斯特、hcq、布西拉明、艾拉莫德或mtx和 lef的联合用药之外的任何非生物dmard。在一个实施例中,受试者未接受除mtx、ssz、来氟米特、阿普斯特、hcq、布西拉明或艾拉莫德之外的任何非生物dmard的伴随治疗。
[0635]
2)稳定剂量的nsaid、对乙酰氨基酚/扑热息痛、低效阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药)、口服皮质类固醇(相当于泼尼松≤10mg/天)或吸入皮质类固醇以保持病情稳定。在一个实施例中,受试者在基线之前已经接受稳定剂量的此类额外疗法持续≥1周。
[0636]
在一个方面,待治疗的受试者(或受试者群体)是非生物dmard-ir,并且可能在基线之前停用至少一种非生物dmard治疗,以符合上述以稳定剂量伴随施用非生物dmard的方案(例如,伴随施用≤2种非生物dmard(不包括mtx和来氟米特的联合用药),选自mtx、ssz、来氟米特、阿普斯特、hcq、布西拉明和艾拉莫德)。在一个实施例中,受试者在基线之前已停用非生物dmard治疗,对于:
[0637]
如果未遵循消除程序或坚持消除程序(即,使用消胆胺11天,或活性炭冲洗30天或根据当地标签),则lef≥8周;或
[0638]
所有其它非生物dmard≥4周。
[0639]
在另一方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir。在一个方面,受试者(或治疗群体中的多名受试者)对bdmard应答不足或不耐受。bdmard-ir 受试者包括在基线之前已既往暴露于至少一种bdmard,并且由于在至少12周的治疗后缺乏疗效而导致应答不足,或对至少1种bdmard的治疗不耐受的那些受试者。在一个方面,受试者(或治疗群体中的多名受试者)在基线之前停用了所有bdmard。在某些实施例中,受试者(或治疗群体中的多名受试者)在接受第一剂jak1抑制剂之前已停用bdmard,对于:
[0640]
依那西普≥4周;
[0641]
阿达木单抗、英夫利昔单抗(infliximab)、赛妥珠单抗(certolizumab)、戈利木单抗 (golimumab)、阿巴西普(abatacept)、托珠单抗和依克珠单抗≥8周;
[0642]
优特克单抗≥12周;
[0643]
苏金单抗≥16周;或
[0644]
利妥昔单抗≥1年或≥6个月,如果b细胞已经恢复到治疗前水平或如果治疗前水平不可用时则正常参考范围(中心实验室)。
[0645]
在某些方面,待治疗的受试者(或受试者群体)是bdmard-ir,并且正在进一步接受
至少一种额外疗法。额外疗法包括但不限于伴随施用非生物dmard和/或nsaid、皮质类固醇及其组合。与本文所述的方法组合使用的合适的额外疗法包括:
[0646]
1)伴随施用≤2种非生物dmard(不包括mtx和来氟米特的联合用药),选自甲氨蝶呤(mtx)、柳氮磺胺吡啶(ssz)、来氟米特、阿普斯特、羟氯喹(hcq)、布西拉明和艾拉莫德。在一个实施例中,受试者在基线前≥12周和≥4周服用稳定剂量的mtx(≤25mg/周)、 ssz(≤3000mg/天)、来氟米特(lef)(≤20mg/天)、阿普斯特(≤60mg/天)、hcq(≤400mg/ 天)、布西拉明(≤300mg/天)或艾拉莫德(≤50mg/天)。在一个实施例中,受试者未接受除 mtx、ssz、来氟米特、阿普斯特、hcq、布西拉明或艾拉莫德之外的任何非生物dmard 的伴随治疗。
[0647]
2)在基线前≥1周伴随施用稳定剂量的nsaid、对乙酰氨基酚/扑热息痛、低效阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药)、口服皮质类固醇(相当于泼尼松≤10mg/天),或以稳定剂量吸入皮质类固醇。
[0648]
在一个方面,待治疗的受试者(或受试者群体)是bdmard-ir,并且之前已经接受过至少一种非生物dmard的治疗。在一个实施例中,这样的受试者可以在基线之前停用至少一种非生物dmard的治疗,以符合上述以稳定剂量伴随施用非生物dmard的方案(例如,伴随施用≤2种非生物dmard(不包括mtx和来氟米特的联合用药),选自mtx、ssz、来氟米特、阿普斯特、hcq、布西拉明和艾拉莫德)。在一个实施例中,受试者在基线之前已停用非生物dmard治疗,对于:
[0649]
如果未遵循消除程序或坚持消除程序(即,使用消胆胺11天,或活性炭冲洗30天或根据当地标签),则lef≥8周;或
[0650]
所有其它非生物dmard≥4周。
[0651]
在一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir或非生物 dmard-ir,并且在基线前至少1周已停用所有阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药除外)。在一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir或非生物dmard-ir,并且先前未暴露于任何jak抑制剂。在一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir或非生物dmard-ir,并且在基线时未接受>2种非生物dmard的当前治疗(即,在施用jak1抑制剂时的治疗),或使用除mtx、ssz、lef、阿普斯特、hcq、布西拉明或艾拉莫德以外的dmard治疗,或联合使用mtx与lef。在一个实施例中,受试者(或治疗群体中的多名受试者)是非生物 dmard-ir或bdmard-ir,并且没有纤维肌痛病史,没有在17岁之前发病的任何关节炎,或目前诊断出除psa以外的炎症性关节病(包括但不限于,类风湿性关节炎、痛风、重叠结缔组织病、硬皮病、多发性肌炎、皮肌炎和系统性红斑狼疮)。
[0652]
在本文所述的治疗psa的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是非生物dmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果: a)在施用第一剂后的12周内(包括在第12周)haq-di相对于基线的变化(改善);b)在施用第一剂后的16周内(包括在第16周)siga为0或1,并且相对于基线改善至少2分; c)在施用第一剂后的16周内(包括在第16周)的pasi 75应答(对于基线时体表面积(bsa) 银屑病≥3%的受试者);d)在施用第一剂后的24周内(包括在第24周)shs相对于基线的变化(改善);e)在施用第一剂后的24周内(包括在第24周)的最小疾病活动性(mda);f) 在施用第一剂后的24周内(包括在第24周)lei相对于基线的变化(改善);g)在施用第一剂后的24周内
(包括在第24周)ldi相对于基线的变化(改善);h)在施用第一剂后的 12周内(包括在第12周)sf-36相对于基线的变化(改善);i)在施用第一剂后的12周内 (包括在第12周)患者对疼痛的总体评估(pt pain)数字评定量表(nrs)相对于基线的变化(改善);j)在施用第一剂后的12周内(包括在第12周)facit-f问卷相对于基线的变化 (改善);k)在施用第一剂后的16周内(包括在第16周)saps问卷相对于基线的变化(改善)。在一个方面,在第12周、第16周或第24周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0653]
在本文所述的治疗psa的方法的一个方面,受试者(或治疗群体中的多名受试者)是非生物dmard-ir并且可以替代地或额外地达到不劣于或优于通过施用阿达木单抗达到的结果的结果。在一个方面,结果优于或不劣于当受试者每隔一周施用40mg剂量的阿达木单抗持续12周时达到的结果。实例1中描述了阿达木单抗的合适制剂的实例。在一个方面,受试者在施用第一剂后的12周内(包括在第12周)达到选自由以下组成的组的至少一个结果:i) 与阿达木单抗相比非劣效的acr20;ii)与阿达木单抗相比优效的acr20应答;与阿达木单抗相比优效的患者对疼痛的总体评估(pt pain)数值评定量表(nrs)相对于基线的变化(改善);iv)与阿达木单抗相比优效的haq-di相对于基线的变化(改善)。在一个方面,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果 (或多个结果)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0654]
在本文所述的治疗psa的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是非生物dmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果: 1)在施用第一剂后的12周内(包括在第12周)的acr50或acr70应答率;m)在施用第一剂后的2周内(包括在第2周)的acr20应答率。在一个方面,在第2周或第12周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0655]
在本文所述的治疗psa的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是非生物dmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果: n)无放射学进展,如shs基线的变化(改善)≤0所证明;o)acr应答的至少一个单独分量相对于基线的变化(改善),该acr应答选自由以下组成的组:(i)压痛关节计数(tjc68) (0至68)相对于基线的变化(改善);(ii)肿胀关节计数(sjc66)(0至66)相对于基线的变化(改善);(iii)医生总体评估-疾病活动性(pga-疾病活动性)数值评定量表(nrs)相对于基线的变化(改善);(iv)患者总体评估(ptga)-疾病活动性数值评定量表(nrs)相对于基线的变化(改善);(v)患者对疼痛的总体评估(pt pain)数字评定量表(nrs)相对于基线的变化(改善);(vi)haq-di相对于基线的变化(改善);以及(vii)hs-crp相对于基线的变化(改善);p)acr20、acr50或acr70应答;q)ldi相对于基线的变化(改善);r)指趾炎计数相对于基线的变化(改善);s)指趾炎的消退;t)lei相对于基线的变化(改善);u)lei中包括的附着点部位的
消退;v)sparcc附着点炎指数相对于基线的变化(改善);w)sparcc附着点炎指数中包括的附着点炎部位的消退;x)总附着点炎计数相对于基线的变化(改善);y)附着点炎的消退;z)基线时体表面积(bsa)银屑病≥3%的受试者的pasi 75、psa 90或psa 100应答;aa)siga评分为0或1,并且相对于基线改善至少2分;bb)银屑病体表面积(bsa-ps);cc)psarc相对于基线的变化(改善);dd) das28(crp)相对于基线的变化(改善);ee)das28(esr)相对于基线的变化(改善); ff)pasdas相对于基线的变化(改善);gg)dapsa评分相对于基线的变化(改善);hh) sf-36相对于基线的变化(改善);ii)facit-f问卷相对于基线的变化(改善);jj)eq-5d-5l 问卷相对于基线的变化(改善);kk)wpai-psa问卷相对于基线的变化(改善);11)hru问卷相对于基线的变化(改善);mm)saps问卷相对于基线的变化(改善);nn)basdai相对于基线的变化(改善);oo)basdai 50应答率;pp)晨僵相对于基线的变化(改善),以 basdai问题5和6的平均值衡量;qq)asdas相对于基线的变化(改善);rr)asdas非活动性疾病;ss)asdas重大改善;tt)asdas临床重要改善;以及uu)haq-di中有临床意义的改善(≥0.35)。在一个方面,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0656]
在一个方面在本文所述的治疗psa的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果:a)在施用第一剂后的12周内(包括在第12周)haq-di相对于基线的变化(改善); b)在施用第一剂后的16周内(包括在第16周)银屑病的siga为0或1,并且相对于基线改善至少2分;c)基线时体表面积(bsa)银屑病≥3%的受试者在施用第一剂后的16周内(包括在第16周)的pasi 75应答;d)在施用第一剂后的24周内(包括在第24周)的最小疾病活动性(mda);e)在施用第一剂后的12周内(包括在第12周)sf-36相对于基线的变化(改善);f)在施用第一剂后的12周内(包括在第12周)facit-f问卷相对于基线的变化(改善);g)在施用第一剂后的16周内(包括在第16周)saps问卷相对于基线的变化(改善)。在一个方面,在第12周、第16周或第24周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0657]
在本文所述的治疗psa的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果:h)在施用第一剂后的12周内(包括在第12周)的acr50或acr70应答;i)在施用第一剂后的 2周内(包括在第2周)的acr20应答。在一个方面,在第2周或第12周后通过继续施用每日剂量的jak1抑制剂来维持或改善结果。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少 25%、至少30%、至少35%、至少40%或至少45%的受试者达到至少一个结果。
[0658]
在本文所述的治疗psa的方法的一个方面,受试者(或治疗群体中的多名受试者)在基线时是bdmard-ir,并且替代地或额外地达到选自由以下组成的组的至少一个结果:j)acr 应答的至少一个单独分量相对于基线的变化(改善),该acr应答选自由以下组成的组:
(i) 压痛关节计数(tjc68)(0至68)相对于基线的变化(改善);(ii)肿胀关节计数(sjc66) (0至66)相对于基线的变化(改善);(iii)医生总体评估-疾病活动性(pga-疾病活动性) 数值评定量表(nrs)相对于基线的变化(改善);(iv)患者总体评估(ptga)-疾病活动性数值评定量表(nrs)相对于基线的变化(改善);(v)患者对疼痛的总体评估(pt pain)数字评定量表(nrs)相对于基线的变化(改善);(vi)健康评估问卷-残疾指数(haq-di)相对于基线的变化(改善);以及(vii)高敏c反应蛋白(hs-crp)相对于基线的变化(改善);k)acr20、acr50或acr70应答率;1)ldi相对于基线的变化(改善);m)指趾炎计数相对于基线的变化(改善);n)指趾炎的消退;n)lei相对于基线的变化(改善);o)lei 中包括的附着点炎部位的消退;p)sparcc附着点炎指数相对于基线的变化(改善);q) sparcc附着点炎指数中包括的附着点炎部位的消退;r)附着点炎总数相对于基线的变化(改善);s)附着点炎的消退;t)基线时体表面积(bsa)银屑病≥3%的受试者的pasi 75、pasi90或pasi 100应答;u)siga评分为0或1,并且相对于基线改善至少2分;v)银屑病体表面积(bsa-ps);w)psarc相对于基线的变化(改善);x)das28(crp)相对于基线的变化(改善);y)das28(esr)相对于基线的变化(改善);z)pasdas相对于基线的变化(改善);aa)dapsa评分相对于基线的变化(改善);bb)sf-36相对于基线的变化(改善);cc)facit-f问卷相对于基线的变化(改善);dd)eq-5d-5l问卷相对于基线的变化(改善);ee)wpai-psa问卷相对于基线的变化(改善);ff)hru问卷相对于基线的变化(改善); gg)saps问卷相对于基线的变化(改善);hh)basdai相对于基线的变化(改善);ii)basdai50应答;jj)晨僵相对于基线的变化(改善),以basdai问题5和6的平均值衡量;kk) asdas相对于基线的变化(改善);ll)asdas非活动性疾病;mm)asdas重大改善;nn) asdas临床重要改善;oo)haq-di中有临床意义的改善(≥0.35)。在一个方面,在达到一个结果(或多个结果)后通过继续施用每日剂量的jak1抑制剂来维持或改善一个结果(或多个结果)。在某些实施例中,对于达到的任何上述结果,治疗群体中统计学显着的受试者群体,和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少 40%或至少45%的受试者达到至少一个结果。
[0659]
在另一方面,提供了一种在有需要的受试者中治疗活动性psa的方法,该方法包含每天一次向受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16周,其量足以递送15mg的乌帕替尼游离碱当量,其中受试者在施用第一剂后的16周内(包括在第16周)达到银屑病面积严重程度指数(pasi)75应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 90应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 100应答。在某些实施例中,在第16周后通过继续施用每日剂量的乌帕替尼游离碱或其药学上可接受的盐来维持或改善pasi应答(例如,pasi 75、pasi 90和/或pasi 100应答)。在一个方面,受试者是非生物dmard-ir。在一个方面,受试者是bdmard-ir。
[0660]
在其它方面,提供了一种在有需要的受试者中治疗活动性psa的方法,该方法包含每天一次向受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16周,其量足以递送30mg的乌帕替尼游离碱当量,其中受试者在施用第一剂后的16周内(包括在第16周)达到银屑病面积严重程度指数(pasi)75应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 90应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 100应答。在某些实施例中,在第16周后通过继续
施用每日剂量的乌帕替尼游离碱或其药学上可接受的盐来维持或改善pasi应答(例如,pasi 75、pasi 90和/或pasi 100应答)。在一个方面,受试者是非生物dmard-ir。在一个方面,受试者是bdmard-ir。
[0661]
在又一方面,提供了一种在有需要的受试者群体中治疗活动性psa的方法,该方法包含根据本文所述的方法向受试者施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,其中治疗群体中的一部分受试者(例如,治疗群体中统计学显着的受试者群体和/或治疗群体中至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者) 在施用第一剂后的16周内(包括在第16周)达到pasi 75应答。在某些实施例中,治疗群体中的受试者在施用第一剂后的16周内(包括在第16周)达到pasi 90应答。在某些实施例中,治疗群体中的受试者在施用第一剂后的16周内(包括在第16周)达到pasi 100应答。在某些实施例中,在第16周后通过继续施用每日剂量的jak1抑制剂来维持或改善pasi应答(例如,pasi 75、pasi 90和/或pasi 100应答)。在一个方面,乌帕替尼游离碱或其药学上可接受的盐以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,乌帕替尼游离碱或其药学上可接受的盐以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用乌帕替尼游离碱或其药学上可接受的盐,持续至少16周。在一个方面,治疗群体中的受试者在基线时是非生物dmard-ir。在一个方面,治疗群体中的受试者在基线时是bdmard-ir。
[0662]
在某些实施例中,在基线时患有活动性psa的受试者(或治疗群体中的多名受试者)是成年受试者。在另一方面,在基线时患有活动性psa的受试者(或治疗群体中的多名受试者) 是幼年受试者。在某些实施例中,受试者(或治疗群体中的多名受试者)在基线时具有>3%的银屑病体表面积。
[0663]
进一步提供了治疗银屑病(pso)(包括作为psa皮肤表现的pso)的方法。
[0664]
例如,在一个方面,提供了一种在有需要的受试者中治疗活动性银屑病的方法,该方法包含每天一次向受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16周,其量足以递送15mg的乌帕替尼游离碱当量,其中受试者在施用第一剂后的16周内(包括在第16周)达到银屑病面积严重程度指数(pasi)75应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 90应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 100应答。在某些实施例中,在第16周后通过继续施用每日剂量的乌帕替尼游离碱或其药学上可接受的盐来维持或改善 pasi应答(例如,pasi 75、pasi 90和/或pasi 100应答)。
[0665]
在其它方面,提供了一种在有需要的受试者中治疗活动性银屑病的方法,该方法包含每天一次向受试者口服施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,持续至少16 周,其量足以递送30mg的乌帕替尼游离碱当量,其中受试者在施用第一剂后的16周内(包括在第16周)达到银屑病面积严重程度指数(pasi)75应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 90应答。在某些实施例中,受试者在施用第一剂后的16周内(包括在第16周)达到pasi 100应答。在某些实施例中,在第16 周后通过继续施用每日剂量的乌帕替尼游离碱或其药学上可接受的盐来维持或改善pasi应答(例如,pasi 75、pasi 90和/或pasi 100应答)。
[0666]
在又一方面,提供了一种在有需要的受试者群体中治疗活动性银屑病的方法,该
方法包含根据上述方法向受试者施用一定剂量的乌帕替尼游离碱或其药学上可接受的盐,其中治疗群体中的一部分受试者(例如,治疗群体中统计学显着的受试者群体和/或治疗群体中至少 10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%或至少45%的受试者) 在施用第一剂后的16周内(包括在第16周)达到pasi 75应答。在某些实施例中,治疗群体中的受试者在施用第一剂后的16周内(包括在第16周)达到pasi 90应答。在某些实施例中,治疗群体中的受试者在施用第一剂后的16周内(包括在第16周)达到pasi 100应答。在某些实施例中,在第16周后通过继续施用每日剂量的乌帕替尼游离碱或其药学上可接受的盐来维持或改善pasi应答(例如,pasi 75、pasi 90和/或pasi 100应答)。在一个方面,乌帕替尼游离碱或其药学上可接受的盐以足以递送15mg的乌帕替尼游离碱当量的量施用。在一个方面,乌帕替尼游离碱或其药学上可接受的盐以足以递送30mg的乌帕替尼游离碱当量的量施用。在一个方面,每天一次口服施用乌帕替尼游离碱或其药学上可接受的盐,持续至少16周。在一个方面,治疗群体中的受试者在基线时是非生物dmard-ir。在一个方面,治疗群体中的受试者在基线时是bdmard-ir。
[0667]
在某些实施例中,在基线时患有活动性银屑病的受试者(或治疗群体中的多名受试者) 是成年受试者。在另一方面,在基线时患有活动性银屑病的受试者(或治疗群体中的多名受试者)是幼年受试者。在某些实施例中,受试者(或治疗群体中的多名受试者)在基线时具有>3%的银屑病体表面积。
[0668]
在某些实施例中,活动性银屑病是psa的一种皮肤表现。例如,在某些实施例中,有需要的受试者(或治疗群体中的多名受试者)患有活动性psa。
[0669]
药物组合物
[0670]
可以使用一种或多种药学上可接受的载体以常规方式配制包含jak1抑制剂并且可以根据本文描述的方法口服施用的药物组合物。
[0671]
此外,如wo 2017/066775和wo 2018/165581中描述的,在组合物中包括的jak1抑制剂可以是固态形式,例如但不限于无定形游离碱、结晶溶剂化物或水合物(例如,游离碱溶剂化物形式a、游离碱水合物形式b)、结晶半水合物(例如,游离碱水合物形式c)、结晶无水物(例如,游离碱无水物形式d)或结晶酒石酸盐(例如,酒石酸盐水合物)。
[0672]
在一个实施例中,药物组合物是片剂剂型。在一个方面,一种薄膜包衣片剂。在另一个实施例中,药物组合物是胶囊剂型。
[0673]
在一个实施例中,片剂或胶囊是缓释制剂(在本文中也称为改良释放或持续释放制剂)。与允许在短的时间段内(例如,通常约60分钟或更短)释放大部分或全部活性成分的即释固体剂型相比,这种制剂允许在长的时间段内缓释活性成分。在一个方面,片剂包含jak1抑制剂和至少一种添加剂,该添加剂选自由以下组成的组:控释聚合物、填充剂、助流剂、润滑剂(例如,用于压制颗粒)、ph调节剂、表面活性剂及其组合。在一个方面,片剂包含jak1 抑制剂、控释聚合物、填充剂、助流剂和润滑剂。在一个方面,片剂包含jak1抑制剂、控释聚合物、填充剂、助流剂、润滑剂和ph调节剂。在一些实施例中,片剂进一步包括薄膜包衣。片剂上的薄膜包衣可以进一步有助于使片剂易于吞咽。薄膜包衣也可以改善口感并提供高雅外观。
[0674]
在某些实施例中,控释聚合物是亲水性聚合物。合适的控释聚合物的实例包括但不限于粘度介于1000与150,000mpa-s之间的纤维素衍生物;羟丙基甲基纤维素(例如,羟丙
基甲基纤维素2208或控释级别的羟丙基甲基纤维素,包含e、f和k系列);与聚链烯基聚醚交联的丙烯酸的共聚物(例如,聚合物);羟丙基纤维素;羟乙基纤维素;环氧乙烷的非离子均聚物(例如,polyox
tm
);多糖的水溶性天然树胶(例如,黄原胶、藻朊酸盐、刺槐豆胶等);交联淀粉;聚乙酸乙烯酯;聚乙烯吡咯烷酮;聚乙酸乙烯酯和聚乙烯吡咯烷酮的混合物及其组合。在一个实施例中,控释聚合物选自由以下组成的组:羟丙基甲基纤维素、与聚链烯基聚醚交联的丙烯酸的共聚物(例如,聚合物)及其组合。合适的填充剂 (“膨胀剂”)的实例包括但不限于微晶纤维素(例如,ph 101;ph 102;);甘露糖醇(例如,100sd或200sd);乳糖;蔗糖;山梨糖醇等。在一个实施例中,填充剂选自由微晶纤维素、甘露糖醇和其组合组成的组。合适的助流剂的实例包括但不限于二氧化硅(例如,胶体二氧化硅)、硅酸钙、硅酸镁、滑石及其组合。在一个实施例中,助流剂是胶体二氧化硅。合适的润滑剂的实例包括但不限于聚乙二醇(例如,分子量为 1000到6000)、硬脂酸镁、硬脂酸钙、硬脂酰富马酸钠、滑石等。在一个实施例中,润滑剂是硬脂酸镁。合适的ph调节剂的实例包含但不限于如酒石酸、柠檬酸、琥珀酸、富马酸等有机酸;柠檬酸钠;碳酸镁或碳酸钙或碳酸氢钙;和其组合。在一个实施例中,ph调节剂是酒石酸。合适的表面活性剂的实例包含十二烷基硫酸钠。合适的薄膜包衣的实例包括但不限于聚合成膜材料,如羟丙基甲基纤维素、羟丙基纤维素和丙烯酸酯或甲基丙烯酸酯共聚物。除了成膜聚合物之外,薄膜包衣可以进一步包含增塑剂(例如,聚乙二醇)、表面活性剂(例如,聚山梨醇酯)和任选地色素(例如,二氧化钛或氧化铁)。薄膜包衣还可以包括滑石作为抗粘合剂。在一个实施例中,薄膜包衣占药物组合物重量的小于5%。
[0675]
在一个实施例中,药物组合物包括约10w/w%到约35w/w%的ph调节剂,并且特别地为酒石酸、富马酸、柠檬酸、琥珀酸、苹果酸或其组合。在其它实施例中,调配物包括约20w/w%到约35w/w%、或约20w/w%到约30w/w%、或约20w/w%到约25w/w%、或约10w/w%、约 15w/w%、约20w/w%、约25w/w%或约30w/w%的ph调节剂。在一个实施例中,ph调节剂是酒石酸。
[0676]
在一个实施例中,片剂是压缩和/或研磨的片剂。例如,在一些实施例中,通过共混组合物组分(例如,包括活性成分和至少一种药学上可接受的载体)来形成片剂。然后可以对组合物进行直接压缩,或者可以在压缩之前对一种或多种组合物组分进行制粒。在一个实施例中,使用装配有任何合适尺寸(例如,装配有约600μm至约1400μm或约610μm或约1397 μm的筛网尺寸)的筛网的研磨机进行研磨。可以在压片机中(如在两个移动冲头之间的钢模中)完成压缩。
[0677]
在其它实施例中,使用湿法制粒方法调配压缩和/或研磨的片剂。使用湿法制粒有助于减少和/或消除当使用不含湿法制粒的压制(例如,直接压制)来配制片剂时可能发生的粘连。
[0678]
范例
[0679]
为了可以更全面地理解本公开,阐述以下实例。应理解,这些实例仅仅是出于说明性的目的,并且不应被解释为以任何方式限制本公开。
[0680]
实例1
[0681]
示范性缓释乌帕替尼制剂公开于wo2017066775和wo 2018/165581。下表提供了如本文所述的实例中使用的15mg或30mg乌帕替尼缓释(er)湿颗粒片剂。从第1天(基线)开始,
每天口服一次15mg或30mg的乌帕替尼er片剂(或匹配的安慰剂),并应在每天大约同一时间服用,无论是否进食。
[0682][0683]
*使用如wo2017066775和wo2018/1655581中公开的半水合物乌帕替尼游离碱形式c。每片 15mg和30mg的量是指片剂中无水乌帕替尼游离碱的量;**在处理过程中移除。
[0684]
阿达木单抗(ada)(或匹配的安慰剂)以皮下注射溶液形式提供,装在1ml预装注射器中,含有ada 40mg/0.8ml(或匹配的安慰剂)。ada(或匹配的安慰剂)将要每隔一周在一天中的大约同一时间皮下施用。
[0685][0686]
实例2
[0687]
2/3期强直性脊柱炎(select-axis 1)临床研究
[0688]
select-axis 1是一项乌帕替尼的多中心、随机、双盲、平行组、安慰剂对照、2/3期、两个周期的研究(图1)。15mg乌帕替尼是指如实例1中所述的每日一次(qd)15mg乌帕替尼药物产品。
[0689]
第1周期是为期14周的随机、双盲、平行组、安慰剂对照期,旨在比较乌帕替尼游离碱 15mg qd(每天一次)与安慰剂在治疗活动性as受试者的体征和症状方面的安全性和疗效,这些受试者在至少4周内以最大推荐或耐受剂量对至少两种nsaid应答不足,或对nsaid 不耐受或有禁忌症,并且未接受过bdmard。第2周期是开放标签的长期延长期,用于评估乌帕替尼游离碱15mg qd在完成第1周期的as受试者中的长期安全性、耐受性和疗效。
[0690]
在35天的筛选期内对骨盆进行x光检查,以评估si关节,以确认是否符合修订后的
as纽约标准。在35天的筛选期内还进行了脊柱的x光检查,以评估全脊柱强直;患有完全性脊柱强直的受试者不符合本研究的条件。如果受试者在筛选期的90天内曾接受过前后位(ap)骨盆x光检查和脊柱侧位x光检查,则在筛选期不需要进行脊柱和骨盆x光检查,前提是x光检查确认足以进行所需的评估,并被中央成像供应商认为是可接受的。
[0691]
符合资格标准的受试者以1∶1的比例随机分配到两个治疗组之一:
[0692]
第1组:乌帕替尼游离碱15mgqd,n=85(第1天至第14周)
→
乌帕替尼游离碱15mgqd(第14周及之后)
[0693]
第2组:安慰剂,n=85(第1天至第14周)
→
乌帕替尼游离碱15mgqd(第14周及之后)
[0694]
从第16周开始,在连续两次就诊中未达到至少asas20应答的受试者可以选择在第20周或之后添加或修改nsaid、对乙酰氨基酚/扑热息痛、低效阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药)的剂量和/或修改mtx或ssz的剂量。
[0695]
从第24周开始,在两次连续就诊中仍未达到至少asas20应答的受试者停用研究药物治疗。
[0696]
完成第14周就诊(第1周期结束)的受试者进入研究的开放标签长期延长部分,第2周期(90周)。在第1周期分配到乌帕替尼的受试者继续以开放标签方式接受乌帕替尼游离碱15mgqd。在基线时被随机分配到安慰剂的受试者也在第14周接受了开放标签乌帕替尼游离碱15mgqd。
[0697]
主要入选标准:
[0698]
1.男性或女性≥18岁。
[0699]
2.临床诊断为as并符合修订后的as纽约标准的受试者。
[0700]
3.在筛选和基线就诊时,受试者必须具有由基于0至10数字评定量表(nrs)的巴氏强直性脊柱炎疾病活动指数(basdai)评分≥4和患者对总背痛评分的评估(总背痛评分)≥4所定义的基线疾病活动性。
[0701]
4.受试者对至少两种nsaid在最大推荐剂量或耐受剂量下累计至少4周应答不足,或受试者对nsaid不耐受或有禁忌症。
[0702]
5.如果参与伴随甲氨蝶呤(mtx)、来氟米特、柳氮磺胺吡啶(ssz)和/或羟氯喹的研究,受试者必须在基线就诊前至少28天内服用稳定剂量的mtx(≤25mg/周)和/或ssz(≤3g/天)和/或羟氯喹(≤400mg/天)或来氟米特(≤20mg/天)。除了mtx和来氟米特的联合用药外,最多允许联合使用两种背景的常规合成疾病改善抗风湿药(csdmard)。
[0703]
6.如果参与伴随口服皮质类固醇的研究,受试者必须在基线就诊前至少14天服用稳定剂量的泼尼松(≤10mg/天)或口服皮质类固醇等效物。
[0704]
7.如果参与使用nsaid、曲马多、对乙酰氨基酚和可待因或氢可酮联合用药和/或非阿片类镇痛药的研究,受试者必须在基线就诊前至少14天服用稳定剂量。
[0705]
主要排除标准:
[0706]
1.全脊柱强直的患者不合格。
[0707]
2.既往暴露于任何janus激酶(jak)抑制剂(包括但不限于托法替尼(tofacitinib)、巴瑞克替尼(baricitinib)和非戈替尼)。
[0708]
3.先前暴露于任何对脊柱关节炎(spa)有潜在治疗影响的生物疗法。
[0709]
4.基线就诊前28天内关节内注射、脊髓/椎旁注射或肠胃外施用皮质类固醇。允许吸入或外用皮质类固醇。
[0710]
5.受试者在基线就诊前28天或药物的五个半衰期(以较长者为准)内服用任何其它dmard(允许的药物除外)、沙利度胺或阿普斯特。
[0711]
6.受试者在基线就诊前14天内服用阿片类镇痛剂(除了允许的对乙酰氨基酚/可待因或对乙酰氨基酚/氢可酮联合用药)或使用吸入大麻。
[0712]
7.受试者有除中轴型spa以外的不同病因的炎性关节炎病史(包括但不限于类风湿性关节炎[ra]、银屑病关节炎[psa]、混合性结缔组织病、系统性红斑狼疮、反应性关节炎、硬皮病、多发性肌炎、皮肌炎、纤维肌痛),或任何在17岁之前发病的关节炎。
[0713]
主要终点
[0714]
主要疗效终点是第14周的asas 40应答。
[0715]
关键次要终点
[0716]
第14周的关键多重性调整次要疗效终点是:
[0717]
1.强直性脊柱炎疾病活动性评分(asdas(crp))相对于基线的变化;
[0718]
2.mri加拿大脊柱关节炎研究协会(sparcc)评分(脊柱)相对于基线的变化;
[0719]
3.basdai 50应答的受试者比例(定义为巴氏as疾病活动指数改善50%);
[0720]
4.as生活质量(asqol)相对于基线的变化;
[0721]
5.具有asas部分缓解(pr)的受试者比例(定义为asas 40中确定的四个域中的每一个的绝对分数≤2个单位);
[0722]
6.basfi相对于基线的变化;
[0723]
7.basmi
lin
相对于基线的变化;
[0724]
8.马斯特里赫特强直性脊柱炎附着点炎评分(mases)相对于基线的变化(即,对于患有基线附着点炎的受试者);
[0725]
9.工作效率和活动障碍(wpai)相对于基线的变化(由于spa导致的总体工作障碍);
[0726]
10.asas hi相对于基线的变化。
[0727]
其它关键次要终点是:
[0728]
1.第14周的asas 20应答。
[0729]
2.第14周的mri sparcc评分(si关节)相对于基线的变化。
[0730]
其它终点是在除主要和关键次要变量指定的时间点之外的预定时间点,在接受乌帕替尼治疗的受试者与安慰剂治疗的受试者中评估的以下测量值:
[0731]
1.具有asas 20应答的受试者比例;
[0732]
2.具有asas 40应答的受试者比例;
[0733]
3.具有asas pr应答的受试者比例;
[0734]
4.具有asas 5/6的受试者比例(在以下六个域中的五个相对于基线改善20%:basfi、患者对总背痛的评估、ptga、炎症[basdai的问题5和6的平均值]、basmilin的腰椎侧屈和高敏crp[hscrp]);
[0735]
5.基于asdas(crp)和asdas(esr)(asdas评分<1.3),患有非活动性疾病的受试者比例;
[0736]
6.基于asdas(crp)和asdas(esr)具有重大改善的受试者比例(相对于基线的变化≤-2.0);
[0737]
7.基于asdas(crp)和asdas(esr)具有临床重要改善的受试者比例(相对于基线的变化≤-1.1);
[0738]
8.具有指趾炎消退的受试者比例(即,对于基线存在指趾炎的受试者);
[0739]
9.asas hi相对于基线的变化;
[0740]
10.asdas(crp)和asdas(esr)分别相对于基线的变化;
[0741]
11.asqol相对于基线的变化;
[0742]
12.basdai相对于基线的变化;
[0743]
13.basfi相对于基线的变化;
[0744]
14.basmilin相对于基线的变化;
[0745]
15.crp相对于基线的变化;
[0746]
16.总指趾炎计数相对于基线的变化(即,对于基线存在指趾炎的受试者);
[0747]
17.facit-f相对于基线的变化;
[0748]
18.isi相对于基线的变化;
[0749]
19.mases相对于基线的变化(即,对于基线mases>0的受试者);
[0750]
20.在常规射线照片下改良stoke强直性脊柱炎脊柱评分(msasss)评分相对于基线的变化;
[0751]
21.si关节的mri sparcc评分相对于基线的变化;
[0752]
22.脊柱的mri sparcc评分相对于基线的变化;
[0753]
23.患者对总背痛的评估nrs评分0至10相对于基线的变化;
[0754]
24.患者对夜间背痛的评估nrs评分0至10相对于基线的变化;
[0755]
25.患者对疼痛的总体评估nrs评分0至10相对于基线的变化;
[0756]
26.医生对疾病活动性的总体评估nrs评分0至10相对于基线的变化;
[0757]
27.炎症(basdai的问题5和6的平均值nrs评分0至10)相对于基线的变化;
[0758]
28.患者对总背痛的评估(basdai问题2nrs评分0至10)相对于基线的变化;
[0759]
29.外周疼痛/肿胀(basdai问题3nrs评分0至10)相对于基线的变化;
[0760]
30.晨僵持续时间(basdai问题6nrs评分0至10)相对于基线的变化;
[0761]
31.患者对疾病活动性的总体评估nrs评分0至10相对于基线的变化;
[0762]
32.tjc68和sjc66相对于基线的变化;
[0763]
33.wpai相对于基线的变化(所有4个维度评分);
[0764]
34.isi类别相对于基线的变化。
[0765]
参见,例如,machado等人,《风湿病学年鉴》(2018)77:1539-40;maksymowych等人,《关节炎和风湿病》(2005)53:502-9。
[0766]
分析窗口
[0767]
对于每个方案指定的研究就诊,将确定一个目标研究日以代表相应的就诊以及目标日周围的窗口。将以非重叠方式选择窗口,以便收集日期不会落入多个就诊窗口。如果受试者在一个就诊窗口中有两次或更多处的实际就诊,则将使用最接近目标日期的就诊进行分析。如果两次就诊与目标日期的距离相等,则后一次的就诊将用于分析。
[0768]
统计分析
[0769]
本研究计划的170个样本量(随机化比为1∶1)被确定为检测asas40应答率的26%差异提供≥90%的疗效(假设安慰剂asas40应答率为20%)。考虑到10%的退出率,在0.05的双侧显着性水平下进行疗效和样本量计算。完整分析集包括所有接受至少一剂研究药物的随机患者。安全性分析集包括接受至少一剂研究药物的所有患者。在统计分析计划中预先指定了 sparcc mri评估群体(基线包括第一剂研究药物后≤3天的mri数据,第14周包括直至第 2周期研究药物第一剂前的mri数据;第2周期的第一剂是在第14周)。进行了补充的事后 sparcc mri分析,以包括在基线和第14周的名义就诊时收集的所有mri数据。生成累积概率图以说明患者水平上的mri sparcc评分变化。
[0770]
在完整分析集中,使用cochran-mantel-haenszel检验比较乌帕替尼和安慰剂组之间的主要终点,并调整筛选hscrp水平的分层因素。无应答者插补用于处理缺失数据。对次要疗效二元终点进行了与主要终点类似的分析。对于连续的次要疗效终点,使用混合模型对乌帕替尼和安慰剂组进行比较,以重复测量,以治疗组、就诊和逐次就诊交互作为固定效应和相应的基线值和筛选hscrp水平的分层因子作为协变量。为了将总体i型错误率保持在α=0.05水平,采用降压方法测试主要和多重性对照的关键次要终点。测试序列包括一组通过hochberg 程序测试的端点,包括basdai50、asqol、asas pr、basfi、basmi、mases和wpai (图2)。参见,例如,hochberg y,tamhane ac.多重比较程序:约翰威立国际出版公司(johnwiley&sons,inc.),1987。
[0771]
第14周结果
[0772]
在2017年10月24日至2018年9月10日期间,对395名患者的资格进行了评估,其中 187名患者被纳入研究。在筛选的395名患者中,208名(52.7%)不符合资格标准并被排除在研究之外(筛选失败的主要原因是不符合修订后的as纽约标准的放射学标准)。其余187 名符合资格标准的患者被随机分配至安慰剂(n=94)或乌帕替尼(n=93)。总体而言,95.2%的患者完成了第1周期至第14周的研究药物治疗(安慰剂,89/94[94.7%];乌帕替尼,89/93 [95.7%]);安慰剂组中的一名患者停用研究药物,但完成了第1周期就诊。到第14周停用研究药物的最常见主要原因是安慰剂组的不良事件(n=3[3.2%])和乌帕替尼组不良事件(n=2 [2.2%])和撤回同意(n=2[2.2%])。
[0773]
平均年龄为45.4岁,从出现症状的平均持续时间为14.4年,自诊断以来的平均持续时间为6.9年。大多数患者为男性(132[70.6%]),人类白细胞抗原(hla)b27阳性(143[76.5%]) 并且在基线时接受了伴随的nsaid(150[80.2%])。两组之间的基线疾病特征基本平衡。下表总结了患者的主要人口统计数据和基线特征。
[0774][0775][0776]
a.韩国和日本。
[0777]
b.新西兰和澳大利亚。
[0778]
c.对基线时存在附着点炎的受试者进行总结。
[0779]
d.对其基线mri数据在研究药物首次施用后长达3天的受试者进行总结。
[0780]
e.对在基线雇用的受试者进行总结。
[0781]
该研究达到了其主要终点,在第14周,接受乌帕替尼治疗的患者与安慰剂相比,在统计学上显着更多的患者达到了asas40应答(48/93[51.6%]与24/94[25.5%]相比;p=0.0003),治疗差异(95%ci)为26.1%(12.6至39.5%)(图3a)。早在第一次基线后就诊(第2周)就观察到乌帕替尼与安慰剂在asas40(图4a)及其四个单独域中的每一个的平均变化(图4b 至4e)的显着差异,并且这差异一直保持到第14周,第14周在多重性对照分析中实现了统计学上的显着差异。考虑多重性调整,asdas(crp)(图3c)、sparcc mri脊柱(图3b) 和
basfi(图3c)从基线到第14周的变化以及达到basdai50(图3a)和asas pr(图 3a)的患者比例对于乌帕替尼与安慰剂相比具有统计学意义。乌帕替尼与安慰剂相比,从基线到第14周的平均(95%ci)变化对于asdas(crp)为-1.45(-1.62至-1.28)与-0.54(
‑ꢀ
0.71至-0.37;治疗差异为-0.91[-1.14至-0.68;p<0.0001]),对于basfi为-2.29(-2.73至
‑ꢀ
1.85)与-1.30(-1.74至-0.86);治疗差异为-1.00[-1.60至-0.39;p=0.0013])。在接受乌帕替尼治疗的患者中有42/93(45.2%)达到了basdai50,而安慰剂组为22/94(23.4%)名患者(治疗差异为21.8%[8.5-35.0%;p=0.0016])并且在乌帕替尼组中有18/93(19.4%)的患者达到了 asas pr,而安慰剂组为1/94(1.1%)(治疗差异为18.3%[10.0-26.6%;p<0.0001])。
[0782]
对于其它多重性对照的关键疗效终点,根据hochberg程序未达到基于多重性调整的统计显着性。在接受乌帕替尼治疗的患者中观察到与安慰剂相比的持续改善,在第14周,除了 wpai(图3c),mases(p=0.0488)、basmi(p=0.0296)、asqol(p=0.0156)和asas hi (p=0.0073)的标称p值<0.05。
[0783]
基于标称p值,乌帕替尼与安慰剂相比,其它关键次要疗效终点asas20和sparcc mri si联合评分也有所改善(图3a和3b)。在第14周,接受乌帕替尼治疗的患者中有60/93(64.5%) 达到了asas20,而安慰剂组有38/94(40.4%)的患者达到了asas20(治疗差异为24.1%[10.2
‑ꢀ
38.0%;p=0.0010])。
[0784]
对于sparcc mri结果,从基线到第14周,乌帕替尼的sparcc mri脊柱变化为-6.93 (-8.58至-5.28),而安慰剂为-0.22(-2.01至1.57)(治疗差异为-6.71[-9.01至-4.41;在多重性对照分析中显着,p<0.0001])并且从基线到第14周,乌帕替尼的sparcc mri si关节变化为-3.91(-5.05至-2.77),而安慰剂组为-0.22(-1.47至-1.04)(治疗差异为-3.69[-5.31 至-2.08;p<0.0001];图3b)。在所有具有可用数据的患者中进行的补充mri分析证实了脊柱和si关节的主要sparcc mri分析的结果(图5)。sparcc评分变化的累积概率图表明,与安慰剂相比,接受乌帕替尼的患者的sparcc mri脊柱和si关节评分从基线到第14周的改善程度更大;主要mri分析(图6a至6b)和补充mri分析(图6b至6c)的结果是一致的。下表d总结了第14周时的主要和关键次要疗效终点。下表e提供了在第14周的额外疗效测量。
[0785][0786]
在第14周,乌帕替尼与安慰剂相比,达到asdas lda、asdas id、asdas cii和asdasmi的患者比例更高(标称p<0.0001)(图7a)。这些结果总结在下表中。早在使用乌帕替尼的第2周就观察到平均asdas(图7b)和个别asdas分量(图8a至8d)的改善,并持续改善直至第14周。
[0787][0788]a事后分析
[0789]
在第14周,与安慰剂相比,接受15mg qd剂量乌帕替尼治疗的患者在背痛方面表现出更大的改善,这是通过asas应答的总背痛成分评估的。使用basdai问题2证明了颈部、背部或臀部疼痛总体水平的改善。外周疼痛和肿胀(由basdai问题3关于除颈部、背部或臀部以外的关节的整体疼痛评估)和夜间背痛也得到了改善。早在第2周就观察到了总背痛和夜间背痛的改善。
[0790]
在第1周期未报告有严重感染、带状疱疹、恶性肿瘤、静脉血栓栓塞事件,或死亡。与安慰剂组的52/94(55.3%)相比,乌帕替尼组出现不良事件的患者比例58/93(62.4%)较高。在第1周期,每组均报告了一次严重不良事件:安慰剂组的心血管疾病/循环失调(患者感觉不适并住院,但未获得显着发现)和乌帕替尼组有颈椎病和椎间盘突出病史的患者的脊柱骨关节炎恶化。出现导致研究药物停用的不良事件(乌帕替尼,2/93[2.2%]);安慰剂,
3/94[3.2%]) 和感染(乌帕替尼,19/93[20.4%]);安慰剂,26/94[27.7%])的患者比例在两个治疗组中相似。乌帕替尼组中最常见的不良事件是血肌酸磷酸激酶(cpk)升高(8/93[86%]患者与安慰剂组中的2/94[2.1%]患者),有4起事件(与安慰剂中的1起相比)经研究者评估可能与研究药物有关;所有患者均无症状,升高<4
×
uln,除了安慰剂组中的一名患者出现肌肉疼痛并升高至4.3
×
uln。这些事件中的大多数是可逆的,无需中断研究药物(乌帕替尼为6/8,安慰剂为1/2)。乌帕替尼组中的一名患者在基线时已经患有2级中性粒细胞减少症,出现了2级中性粒细胞减少症的轻度不良事件。
[0791]
七名患者报告了肝功能紊乱不良事件(乌帕替尼,5/93[5.4%];安慰剂,2/94[2.1%]);均未导致研究药物停用,并且所有患者均为无症状的丙氨酸氨基转移酶或天冬氨酸氨基转移酶升高,其中6/7患者的相关升高<2
×
uln,其余患者升高<3
×
uln。两组的平均血红蛋白水平在整个14周期间均未观察到差异。
[0792]
从基线到第14周,在乌帕替尼组观察到低密度脂蛋白胆固醇(0.318mmol/l)和高密度脂蛋白(hdl)胆固醇(0.263mmol/l)与安慰剂组(分别为-0.083和0.010mmol/l)相比有所增加;然而,没有观察到总胆固醇/hdl比率的变化(乌帕替尼,-0.071mmol/l;安慰剂,-0.083mmol/l)。
[0793]
第14周结果讨论
[0794]
select-axis 1是乌帕替尼在as中的首个临床试验,并证明了多重性控制终点支持的一致疗效结果。该研究在第14周达到了asas40应答的主要终点(51.6%与25.5%相比)以及反映疾病活动性(asas pr、basdai50、asdas)、功能(basfi)和mri结果(sparccmri脊柱)在统计学上显着改善的几个多重性对照次要终点。其它多重性控制的次要终点在多重性测试中没有达到显着性,但显示出与安慰剂相比,乌帕替尼在asqol、basmi、mases 和asas hi方面的持续改善(标称p<0.05),wpai除外。
[0795]
对于asas40和asdas综合评分及其疾病活动性的各个域(例如,背痛、ptga、晨僵、功能和炎症血清标志物[hscrp]),观察到对乌帕替尼游离碱15mg qd治疗的快速应答,最早在第2周(第一次基线后就诊)观察到应答,并一直维持到第14周。乌帕替尼在改善as 体征和症状方面的结果通过mri显示脊柱和si关节的活动性炎症显着减少得到进一步证实。
[0796]
另外,还达到了与缓解或低疾病活动性的临床相关治疗目标相关的结果,如asdas id 或lda,50%的患者达到asdas lda(与安慰剂相比差异为39%)。参见,例如,smolen等人,《风湿病学年鉴》2018;77:3-17。值得注意的是,本研究中asas20和asas40的安慰剂应答率与近期as临床研究中观察到的应答率相似;早在第2周,使用乌帕替尼与安慰剂的asas40应答差异为p<0.05(基于标称p值)并持续14周。参见例如,van der heijde等人,《风湿病学年鉴》2017;76:1340-47;van der heijde人,《柳叶刀杂志》2018;392:2378-87; van der heijde等人,《柳叶刀杂志》2018;392:2441-51;landewe等人,《风湿病学年鉴》2014; 73:39-47。有趣的是,安慰剂组脊柱和si关节的mri sparcc评分从基线到第14周的平均变化非常小。
[0797]
该研究结果与在活动性as患者中进行的两项2/3期jak抑制剂研究的结果一致。参见,例如,van der heijde等人,《风湿病学年鉴》2017;76:1340-47;van der heijde等人,《柳叶刀杂志》2018;392:2378-87。结合这些发现,select-axis 1结果进一步支持jak抑制剂可能是as的有效治疗选择。目前,只有tnf-α和il-17抑制已被证明对axspa有效;但是,这
些细胞因子不能被包括乌帕替尼在内的jak抑制剂直接阻断。参见,例如,furst和louie,《关节炎研究与治疗(arthritisresther)》2019;21:135。然而,来自乌帕替尼ra研究的新数据表明,对jak1的选择性抑制可能导致不依赖于jak1信号传导的其它途径的二次抑制,如tnf-α和il-12。参见,例如,sornasse等人,《风湿病学年鉴》2019;78:365-66。此外,其它jak1相关途径,包括il-7和il-22,已在临床前研究中进行了描述,但需要进一步研究来评估jak抑制剂在axspa中的作用机制。参见,例如,veale等人,《风湿病学(rheumatology)》(牛津)2019;58:197-205;gracey等人,《风湿病学年鉴》2016;75:2124-32。
[0798]
在乌帕替尼组和安慰剂组中,出现不良事件的患者比例基本相似,与之前的乌帕替尼3期ra研究相比,未观察到新的安全性发现。参见,例如,burmester等人,《柳叶刀杂志》2018;391:2503-12;genovese等人,《柳叶刀杂志》2018;391:2513-24;fleischmannr,panganal,songi等人,乌帕替尼与安慰剂或阿达木单抗治疗类风湿性关节炎和对甲氨蝶呤应答不足的患者:一项3期双盲随机对照试验的结果,《关节炎和风湿病》2019;doi:10.1002/art.41032。[电子版提前印刷];科恩等人,《风湿病学年鉴》(2019)78。未报告严重感染、恶性肿瘤、贫血、淋巴细胞减少症、带状疱疹、肾功能不全、已裁定的主要不良心血管事件、静脉血栓栓塞事件或死亡,并且在整个研究过程中血红蛋白水平保持一致。
[0799]
乌帕替尼组中有较高比例的患者出现cpk升高的不良事件,所有这些都是无症状的,并且大多数是轻度和可逆的,无需中断研究药物。安慰剂组中的一名患者在cpk升高和永久停用研究药物的情况下出现症状(肌肉疼痛)。在之前的两项jak抑制剂研究中,还观察到cpk升高。参见例如,vanderheijde等人,《风湿病学年鉴》2017;76:1340-47;vanderheijde等人,《柳叶刀杂志》2018;392:2378-87。需要更多数据来更好地了解乌帕替尼在axspa中的安全性。
[0800]
jak抑制剂,如乌帕替尼,可以帮助解决axspa治疗中未满足的需求,因为只有大约一半未接受过bdmard的患者达到了asas40应答,甚至更少的患者通过tnf或il-17抑制剂治疗达到了缓解。参见,例如,sieper等人,《风湿病学年鉴》2017;76:571-92;deodhar等人,《关节炎和风湿病》2019;71:599-611;vanderheijde等人,《柳叶刀杂志》2018;392:2441-51;landewe等人,《风湿病学年鉴》2014;73:39-47;liee等人,《风湿病学年鉴》2011;70:157-63;glintborg等人,《风湿病学年鉴》2013;72:1149-55。此外,预计达到持续缓解/lda治疗目标的患者更少,而对bdmard疗法无应答的as患者的应答率甚至更低。参见,例如,sieper等人,《柳叶刀杂志》2017;390:73-84;sieper等人,《风湿病学年鉴》2017;76:571-92;deodhar等人,《关节炎和风湿病》2019;71:599-611。此外,一些axspa患者可能不符合il-17和tnf抑制剂疗法的条件,或者可能有il-17和tnf抑制剂疗法的常见禁忌症,如过敏反应和注射部位疼痛,或tnf抑制剂的特异性禁忌症,如充血性心力衰竭和伴随的脱髓鞘疾病。参见,例如,cortese等人,《multsclerrelatdisord》2019;35:193-95。也不建议同时患有炎症性肠病的患者使用il-17抑制剂。参见,例如,vanderheijde等人,《风湿病学年鉴》2017;76:978-91;fragoulis等人,《世界胃肠病学杂志(worldjgastroenterol)》2019;25:2162-76。由于as患者通常更年轻,并且可能有更积极的生活方式,因此口服施用的治疗方案对于这类患者群体中可能尤为重要。参见例如,alten等人,《患者偏好依从性(patientpreferadherence)》2016;10:2217-28。考虑到这些未满足的需求,select-axis1研究的结果表明乌帕替尼治疗效果在bdmard和其它jak抑制剂对as观
察到的范围内,支持对乌帕替尼治疗as的进一步研究。参见,例如,sieper等人,《风湿病学年鉴》2017;76:571-92;deodhar 等人,《关节炎和风湿病》2019;71:599-611;van der heijde等人,《风湿病学年鉴》2017;76: 1340-47;van der heiide等人,《柳叶刀杂志》2018;392:2378-87;van der heijde等人,《柳叶刀杂志》2018;392:2441-51;landewe等人,《风湿病学年鉴》2014;73:39-47。
[0801]
这项研究并非没有局限性。对未接受过bdmard的as患者的关注允许在同质人群中对效益和风险进行集中评估,但乌帕替尼在bdmard-ir的as患者或非放射学axspa患者中的安全性和疗效尚未进行评估,需要在这些患者群体中进行进一步研究。此外,本研究仅评估了一个剂量的乌帕替尼,因此没有数据证实更高剂量是否会产生更好的疗效。最后,本文只报告了14周的短期数据,但乌帕替尼的长期疗效和安全性将在正在进行的长达2年的 select-axis 1延长期内收集。
[0802]
总之,口服乌帕替尼游离碱15mg qd在治疗14周后可显着改善活动性as患者的疾病活动性、功能和mri检测到的轴性炎症。乌帕替尼和安慰剂的不良事件发生率相似,与之前的ra研究相比,未观察到新的安全性信号。总体而言,这些结果支持对乌帕替尼治疗 as/axspa的进一步研究。
[0803]
第一年结果
[0804]
2/3期select-axis 1研究包括一项随机、安慰剂对照、为期14周的研究,然后是90 周的开放标签延长期;这里报告的是第64周的数据。
[0805]
这些研究招募患有活动性as的成年人(≥18岁),他们对≥2种非甾体抗炎药疗法应答不足(或对nsaid不耐受或有禁忌症)并且未接受过生物疾病改善抗风湿药,并且根据对骶髂关节射线照片的独立中心读数符合修订后的纽约标准,并且在基线时患有活动性疾病,定义为在筛选和基线就诊时巴氏强直性脊柱炎疾病活动指数(basdai)评分≥4,患者对背痛评分的评估≥4(数字评定量表[nrs],0至10)。接受稳定剂量的伴随常规合成疾病改善抗风湿药(dmard)、口服糖皮质激素、nsaid和镇痛药的患者符合条件;既往暴露于jak抑制剂或生物dmard对脊柱关节炎有潜在影响的患者被排除在外。在随机分配到第1周期的187 名患者中,有178名(持续服用乌帕替尼,n=89;安慰剂转换,n=89)完成了第14周的研究药物治疗并进入了开放标签延长期;有160名患者(持续服用乌帕替尼,n=78[83.9%];安慰剂转换,n=82[87.2%])完成第64周的治疗。缺乏疗效(n=10)和ae(n=4)是在第14周至第64周期间停用研究药物的最常见原因。在持续服用乌帕替尼和安慰剂到乌帕替尼转换组中,自as症状发作以来的平均持续时间分别为14.8年和14.0年,自诊断以来的平均持续时间分别为7.8年和6.0年,平均asdas分别为3.5和3.7,平均hscrp水平分别为9.6mg/l 和11.4mg/l。伴随用药包括nsaid(76%和86%)、常规合成dmard(14%和18%)和糖皮质激素(分别为6%和13%)。
[0806]
基于64周内达到asas20应答、asas40应答、asas部分缓解、basdai50和强直性脊柱炎疾病活动性评分(asdas)非活动性疾病(id;<1.3)、低疾病活动性(lda;<2.1)、重大改善(mi;相对于基线下降≥2.0)和临床重要改善(cii;相对于基线下降≥1.1)的患者比例评估疗效。另外,对64周内基于c反应蛋白的asdas(asdas-crp)、巴氏强直性脊柱炎功能指数(basfi)和线性巴氏强直性脊柱炎计量指数(basmi)相对于基线的变化以及52周内马斯特里赫特强直性脊柱炎附着点炎评分(mases)、工作效率和活动障碍(wpai;范围为0至100)、
(asdas、basdai、asas及其分量)、炎症(hscrp)、身体机能(basfi)、生活质量(asqol、 asas hi)和疾病的其它方面(basmi、mases)均有所改善。在第14周从安慰剂转为乌帕替尼的患者中,与从第0周开始持续接受乌帕替尼的患者相比,观察到直至第64周(包括第64周)的起效速度和疗效应答程度相似。值得注意的是,基于更难达到的终点asas部分缓解(pr)或asdas非活动性疾病(id),大约40%至45%的接受乌帕替尼的患者直至第64 周(包括第64周)达到了缓解,并且>80%处于asdas低疾病活动性(lda)状态。
[0813]
下表提供了第14周的乌帕替尼、第16周的生物制剂依克珠单抗和阿达木单抗,以及第 12周的jak小分子抑制剂托法替尼和非戈替尼的安慰剂校正数据,用于主要和次要终点。虽然该数据不是直接比较,但针对更难难达到的终点asas pr、asdas id和asdas lda计算的乌帕替尼的安慰剂校正应答显示出优于其它两种jak小分子抑制剂所显示的疗效的明确前景,其显著疗效仅与生物制剂所显示的疗效相当。此外,这种疗效一旦在第14周达到,就会随着时间的推移而持续或改善,在这些难以达到的终点(包括asdas重大改善(mi)和 asdas临床重要改善(cii))具有长期疗效,持续或改善直至第64周(包括第64周)。再加上乌帕替尼耐受性良好且没有新的或意外的安全性发现(特别是与其它jak抑制剂相比) 这一事实,数据表明乌帕替尼将成为一种有前途的新型安全口服疗法,用于as患者,尤其是那些患有活动性疾病且对nsaid应答不足的患者。
[0814]
[0815]
ns:不显着;na:不可用;upa p值是标称值,除非*在多重性调整后显着;未接受过依克珠单抗bdmard as coast-v研究。van der heijde等人,《柳叶刀杂志》2018;392:2441
‑ꢀ
51;托法替尼研究:van der heijde等人,ard 2017;0:1-8;非戈替尼研究:van der heijde等人,《柳叶刀杂志》2018;392:2378-87。
[0816]
实例3
[0817]
一项3期随机、安慰剂对照、双盲计划,用于评估乌帕替尼在患有中轴型脊柱关节炎的成年受试者中的疗效和安全性(selectaxis 2)
[0818]
来自2/3期研究的安全性和疗效数据(如实例2中所述)显示出有利的益处:乌帕替尼的风险概况,并支持对患有活动性axspa且对生物疾病改善抗风湿药疗法应答不足 (bdmard-ir)的成年受试者(研究1)和患有活动性nr-axspa的成年受试者(研究2)继续进行乌帕替尼研究。
[0819]
3期临床研究计划如图9所示。
[0820]
患有活动性强直性脊柱炎(as)且对生物疾病改善抗风湿药疗法应答不足(bdmard-ir)的成年受试者(研究1)
[0821]
评估乌帕替尼与安慰剂相比在减轻对生物疾病改善抗风湿药疗法应答不足 (bdmard-ir)的活动性强直性脊柱炎(as)成年受试者的体征和症状方面的安全性、耐受性和疗效。
[0822]
研究1(主要研究)由35天的筛选期;为期14周的随机、双盲、平行组、安慰剂对照期(双盲期);90周的开放标签长期延长期(open-label extension period);和30天的随访就诊(f/u就诊)组成。第1组:乌帕替尼游离碱15mg qd;第2组:安慰剂qd。在研究1 的开放标签延长期的第14周,安慰剂组的受试者将被转为乌帕替尼游离碱15mg qd。
[0823]
符合条件的受试者将是成年女性和男性,在筛选时至少18岁,临床诊断为as,符合修订后的as纽约标准,并且没有全脊柱强直(研究1,bdmard-ir as)。在筛选和基线就诊时,符合条件的研究受试者必须具有基于0至10的数字评定量表的巴氏强直性脊柱炎疾病活动指数评分≥4和患者对总背痛评分的评估(总背痛评分)≥4。
[0824]
对于研究1(bdmard-ir as),可以招募既往暴露于1种生物疾病改善抗风湿药 (bdmard)(1种肿瘤坏死因子[tnf]抑制剂或1种白细胞介素[il]-17抑制剂)的受试者,并且受试者必须由于不耐受或缺乏疗效而停用bdmard。如果停药的原因不是由于缺乏疗效,则允许不超过30%的受试者既往暴露于第2次bdmard。对tnf抑制剂和il-17抑制剂均无效的受试者不符合条件。
[0825]
研究1主要终点
[0826]
主要终点是在第14周达到asas40应答的受试者比例。研究1的次要终点如下所述。
[0827]
患有活动性非放射学中轴型脊柱关节炎(nr-axspa)的成年受试者(研究2)
[0828]
评估乌帕替尼与安慰剂相比在减轻患有活动性非放射学中轴型脊柱关节炎(nr-axspa) 成年受试者的体征和症状方面的安全性、耐受性和疗效。
[0829]
研究2(nr-axspa)由35天的筛选期;为期52周的随机、双盲、平行组、安慰剂对照期 (双盲期);52周的开放标签长期延长期(open-label extension period);和30天的f/u就诊组成。第1组:乌帕替尼游离碱15mg qd;第2组:安慰剂qd。
[0830]
在研究2的开放标签延长期(nr-axspa)的第52周,安慰剂组的受试者将被转为乌
帕替尼游离碱15mg qd。
[0831]
符合条件的受试者将是成年女性和男性,在筛选时至少18岁,临床诊断为nr-axspa,符合2009年asas的axspa分类标准,但不符合修订后的as纽约标准的放射学标准,并且骶髂关节磁共振成像或基于高敏c反应蛋白>正常上限的活动性炎症的客观征象(研究2, nr-axspa)。在筛选和基线就诊时,符合条件的研究受试者必须具有基于0至10的数字评定量表的巴氏强直性脊柱炎疾病活动指数评分≥4和患者对总背痛评分的评估(总背痛评分)≥4。受试者在筛选时必须在si关节的mri或hscrp>uln上有活动性炎症的客观征象。
[0832]
对于研究2(nr-axspa),可以招募既往使用非甾体抗炎药(nsaid)失败的受试者,并且允许受试者子集(至少25%,但不超过总入选受试者的35%)在之前使用最多1种bdmard (1种tnf抑制剂或1种il-17抑制剂)进行治疗。可以招募既往暴露于1种bdmard(1 种肿瘤坏死因子[tnf]抑制剂或1种白细胞介素[il]-17抑制剂)的受试者,并且受试者必须由于不耐受或缺乏疗效而已经停用bdmard。如果停药的原因不是由于缺乏疗效,则允许不超过30%的受试者既往暴露于第2次bdmard。对tnf抑制剂和il-17抑制剂均无效的受试者不符合条件。
[0833]
研究2主要终点
[0834]
欧盟[eu]/欧洲药品管理局[ema]监管目的的主要终点是在第14周达到asas40应答的受试者比例。
[0835]
美国/美国食品和药物管理局[fda]监管目的的主要终点是在第52周达到asas40应答的受试者比例。
[0836]
研究2的次要终点如下所述。
[0837]
研究1:bdmard-ir as具体标准
[0838]
1.受试者必须临床诊断为as并且受试者必须符合修订后的as纽约标准。
[0839]
2.受试者不得患有全脊柱强直。
[0840]
3.可以招募既往暴露于1种bdmard(1种肿瘤坏死因子[tnf]抑制剂或1种白细胞介素[il]-17抑制剂)的受试者,并且受试者必须由于不耐受或缺乏疗效而已经停用bdmard。如果停药的原因不是由于缺乏疗效,则允许不超过30%的受试者既往暴露于第2次bdmard。对tnf抑制剂和il-17抑制剂均无效的受试者不符合条件。
[0841]
研究2:nr-axspa-具体标准
[0842]
1.受试者必须临床诊断为nr-axspa,满足2009年asas的axspa分类标准但不满足修订后的as纽约标准的放射学标准。
[0843]
2.既往暴露于或未暴露于bdmard的受试者均可招募。
[0844]
对于既往暴露于bdmard的受试者子集(至少25%,但不超过总入选受试者的35%),允许之前使用最多1种bdmard(1种肿瘤坏死因子抑制剂或1种il-17抑制剂)进行治疗,并且受试者必须由于不耐受或缺乏疗效而已经停用bdmard。对tnf抑制剂和il-17抑制剂均无效的受试者不符合条件。
[0845]
3.受试者在筛选时必须在si关节的mri或hscrp>uln上有活动性炎症的客观征象。
[0846]
以下资格标准适用于研究1和研究2:
[0847]
1.受试者必须是成年男性或女性,筛选时至少18岁。
[0848]
2.受试者必须在筛选和基线就诊中达到以下评分:基于0至10nrs,basdai评分≥4 和总背痛评分≥4。
[0849]
3.受试者对至少两种nsaid在最大推荐剂量或耐受剂量下累计至少4周应答不足,或受试者对nsaid不耐受或有禁忌症。
[0850]
4.第一剂研究药物前的bdmard清除期规定如下:
[0851]
·
依那西普≥4周;
[0852]
·
阿达木单抗、英夫利昔单抗、赛妥珠单抗、戈利木单抗、阿巴西普、托珠单抗和依克珠单抗≥8周;
[0853]
·
优特克单抗≥12周;
[0854]
·
苏金单抗≥16周;
[0855]
·
利妥昔单抗≥1年或≥6个月,如果b细胞已经恢复到治疗前水平或如果治疗前水平不可用时则正常参考范围(中心实验室);
[0856]
·
对于其它bdmard,≥12周或至少为平均终末消除半衰期的5倍,以较长者为准。
[0857]
5.如果参与以下伴随csdmard(甲氨蝶呤(≤25mg/周);或柳氮磺胺吡啶(ssz)(≤3g/ 天);或羟氯喹(≤400mg/天);或氯喹(≤400mg/天);或来氟米特(≤20mg/天);或阿普斯特(≤60mg/天))的研究,受试者必须在基线就诊前至少28天服用如下所示的稳定剂量。除了甲氨蝶呤(mtx)和来氟米特的联合用药外,最多允许2种背景csdmard的联合用药。
[0858]
6.如果参与伴随口服皮质类固醇的研究,受试者必须在基线就诊前至少14天服用稳定剂量的泼尼松(≤10mg/天)或口服皮质类固醇等效物。
[0859]
7.如果参与伴随使用nsaid、曲马多、对乙酰氨基酚/扑热息痛和可待因的联合用药或对乙酰氨基酚/扑热息痛和氢可酮的联合用药和/或非阿片类镇痛药的研究,受试者必须在基线就诊前至少14天服用稳定剂量。
[0860]
8.受试者不得暴露于任何jak抑制剂。
[0861]
9.受试者在基线就诊之前的指定时间范围内不得使用以下禁止的伴随治疗:
[0862]
·
基线就诊前28天内关节内注射、脊髓/椎旁注射或肠胃外施用皮质类固醇。允许吸入或外用皮质类固醇;
[0863]
·
基线就诊前28天或药物的5个半衰期(以较长者为准)内服用任何其它csdmard (根据资格标准允许的除外),包括沙利度胺;
[0864]
·
在基线就诊前14天内使用阿片类镇痛药(除了允许的对乙酰氨基酚/扑热息痛和可待因的联合用药或对乙酰氨基酚/扑热息痛和氢可酮的联合用药除外)。
[0865]
10.受试者在接受第一剂研究药物前的28天内(如果当地需要,或更长时间)不得接种活疫苗,或在参与研究期间,包括接受最后一剂研究药物后的至少30天内(如果当地需要,或更长时间)预期需要接种活疫苗。
[0866]
11.受试者从筛选到研究药物施用结束不得全身使用已知的强细胞色素p450 3a (cyp3a)抑制剂,或在研究药物施用前30天到研究药物施用结束期间不得使用强cyp3a 诱导剂。从筛选到研究药物施用结束,受试者不得使用对cyp3a有未知影响的草药疗法或其它传统药物。
[0867]
12.受试者在接受第一剂研究药物之前至少30天或药物的5个半衰期(以较长者为准) 内不得接受任何化学或生物性质的研究药物治疗,或目前正在参加另一项介入性研
究。
[0868]
13.受试者不得对研究药物(及其赋形剂)和/或同类其它产品的成分有过敏反应或显着敏感性。
[0869]
研究1和研究2次要终点
[0870]
第14周的关键多重性对照的次要终点如下:
[0871][0872]
第14周的其它关键次要终点包括:
[0873][0874]
其它研究1和研究2终点
[0875]
其他终点是在除主要和关键次要变量指定的时间点之外的时间点评估的以下测量值,如下所示:
[0876]
[0877]
[0878][0879]
实例4
[0880]
患有活动性银屑病关节炎且先前对至少一种非生物疾痛改善抗风湿药(非生物dmard) 应答不足的受试者的第3期方案(select-psa 1)
[0881]
select-psa1第3期研究设计在图10中提供。
[0882]
15mg或30mg乌帕替尼是指如实例1中所述的每天一次(qd)15mg和30mg乌帕替尼药物产品。
[0883]
这是一项3期多中心研究,包括两个周期。第1周期为期56周,包括24周的随机、双盲、平行组、安慰剂对照和活性比较对照期,随后是另外32周的盲法、活性比较对照治疗(第 24至56周)。第1周期旨在比较乌帕替尼游离碱15mg qd和30mg qd与安慰剂和阿达木单抗每隔一周(eow)40mg在中度至重度活动性psa且对非生物dmard应答不足 (dmard-ir)的受试者中的安全性、耐受性和疗效。第1周期还旨在比较乌帕替尼游离碱 15mg qd和30mg qd与安慰剂在预防结构进展方面的疗效。第2周期是开放标签(盲法直到最后一名受试者完成第1周期的最后一次就诊),长期延长至总治疗时间约3年,以评估乌帕替尼游离碱15mg qd和30mg qd在完成第1周期的中度至重度活动性psa受试者的安全性、耐受性和疗效。
[0884]
研究期限包括35天的筛选期;56周的盲期,其中包括24周的双盲、安慰剂对照和活性比较对照治疗,随后是32周的活性比较对照治疗(第1周期);长达约3年的总治疗持续时间的长期延长期([在最后一名受试者完成第1周期的最后一次就诊之前设盲]第2周期);30 天的随访电话或就诊;以及70天的随访电话。
[0885]
符合资格标准的受试者按银屑病程度(≥3%体表面积[bsa]或<3%bsa)、当前使用至少1 种dmard、存在指趾炎和存在附着点炎进行分层,来自中国和日本的受试者除外,其中每个国家的随机化仅根据银屑病程度(≥3%体表面积[bsa]或<3%bsa)进行分层,然后以2∶2∶2∶1∶1 的比例随机分配到五个治疗组中的一个:
[0886]
第1组:乌帕替尼15mg qd(n=430)
[0887]
第2组:乌帕替尼游离碱30mg qd(n=423)
[0888]
第3组:阿达木单抗(ada)(40mg eow)(n=429)
[0889]
第4组:安慰剂,然后是乌帕替尼游离碱15mg qd(n=211)
[0890]
第5组:安慰剂,然后是乌帕替尼游离碱30mg qd(n=212)
[0891]
不超过约15%的受试者入选时伴随使用羟氯喹、柳氮磺胺吡啶、布西拉明或艾拉莫德。
[0892]
受试者接受口服研究药物qd(乌帕替尼游离碱15mg、乌帕替尼游离碱30mg或匹配的安慰剂)和皮下研究药物eow(ada40mg或匹配的安慰剂),直到所有受试者完成第1周期 (第56周),并且研究中心和受试者对研究治疗不设盲。
[0893]
在基线时被分配到安慰剂的受试者被预先分配以1∶1的比例从第24周开始接受乌帕替尼游离碱15mg qd或乌帕替尼游离碱30mg qd。完成第56周就诊(第1周期结束)的受试者将进入研究的长期延长部分,即第2周期(总研究持续时间长达约3年)。受试者将按照第 1周期的分配继续研究治疗。分配到乌帕替尼游离碱15mg qd、乌帕替尼游离碱30mg qd 或阿达木单抗40mg eow的受试者将继续以盲法方式分别接受乌帕替尼游离碱15mg qd、乌帕替尼游离碱30mg qd或阿达木单抗40mg eow。当最后一名受试者完成第1周期(第56 周)的最后一次就诊时,两个周期的研究药物分配将向各研究中心揭盲,并且受试者将以开放标签的方式分配研究药物,直到第2周期完成。
[0894]
受试者必须在筛选就诊前对≥1种非生物dmard(mtx、ssz、lef、阿普斯特、布西拉明或艾拉莫德)应答不足,或对dmard不耐受或有禁忌症。在参与本研究期间不需要背景非生物dmard治疗。对于在基线时正在接受非生物dmard治疗(mtx、ssz、lef、阿普斯特、羟氯喹(hcq)、布西拉明或艾拉莫德)的受试者,非生物dmard应在基线就诊前≥12周开始,必须在第一剂研究药物前以稳定剂量≥4周,并保持稳定剂量至研究的第36 周;非生物dmard剂量可能仅出于安全原因而减少。另外,所有服用mtx的受试者应在整个研究参与期间服用口服叶酸(或等效物)的膳食补充剂。
[0895]
在第16周,将向分类为无应答者(定义为在第12周和第16周的压痛关节计数(tjc) 和肿胀关节计数(sjc)之一或两者均未达到至少20%的改善)的受试者提供救援治疗,如下:1)添加或调整非生物dmard、nsaid、对乙酰氨基酚/扑热息痛、低效阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药)、口服皮质类固醇的剂量和/或2)对于1个外周关节、1个触发点、1个压痛点、1个滑囊或1个附着点,接受1次关节内、触发点或压痛点、滑囊内或腱鞘内皮质类固醇注射(抢救治疗)。
[0896]
在最后一名受试者完成第24周的研究就诊后,将进行非盲分析,以用于初始监管提交。为了在盲法的56周期间保持试验的完整性,研究中心和受试者将保持盲法,直到所有受试者都达到第56周。在所有受试者完成第1周期后,可以出于监管目的进行第二次非盲分析。在所有受试者完成第2周期后,将进行最终分析。
[0897]
主要和次要终点
[0898]
主要疗效终点是在第12周达到美国风湿病学会(acr)20%应答率的受试者比例。本研究(select-psa1临床研究;实例4)和select-psa2临床研究(实例5)的主要和次要临床终点在实例6中进一步描述。
[0899]
研究群体
[0900]
患有活动性银屑病关节炎且先前对至少一种非生物疾病改善抗风湿药(非生物dmard 或dmard)应答不足的患者。
[0901]
主要纳入标准包括:
[0902]
1.成年男性或女性,筛查时≥18岁。
[0903]
2.在筛选就诊前至少6个月临床诊断出psa症状发作并满足psa分类标准(caspar) 的标准。
[0904]
3.受试者在基线时患有活动性疾病,定义为在筛选和基线就诊时≥3个压痛关节(基于 68个关节计数)和≥3个肿胀关节(基于66个关节计数)。
[0905]
4.在筛选时出现:
[0906]
≥1次x光片侵蚀,由中央成像检查确定,或;
[0907]
hs-crp>实验室定义的正常上限(uln)。
[0908]
5.活动性斑块状银屑病的诊断或有斑块状银屑病病史记录。
[0909]
6.受试者对先前或当前使用至少1种非生物dmard以最大耐受剂量或高达纳入标准7 定义的剂量的治疗应答不足(在至少12周的治疗持续时间后缺乏疗效)[(对mtx应答不足定义为≥15至≤25mg/周;或在完全滴定后对剂量≥12.5mg/周的mtx不耐受的受试者,≥10mg/ 周;对于中国、日本的受试者对mtx应答不足定义为≥7.5mg/周)、ssz、lef、环孢素、阿普斯特、布西拉明或艾拉莫德)],或受试者对dmard不耐受或有禁忌症。
[0910]
7.在研究开始时正在伴随使用非生物dmard治疗的受试者必须在基线就诊前≥12周和≥4周以稳定剂量使用≤2种非生物dmard(mtx和来氟米特的联合用药除外),剂量如下: mtx(≤25mg/周)、ssz(≤3000mg/天)、来氟米特(lef)(≤20mg/天)、阿普斯特(≤60mg/ 天)、hcq(≤400mg/天)、布西拉明(≤300mg/天)或艾拉莫德(≤50mg/天)。研究期间不允许使用其它dmard。需要在基线就诊之前停用dmard以符合此纳入标准的受试者必须遵循以下指定的程序或至少五倍于药物的平均终末消除半衰期:
[0911]
如果未遵循消除程序或坚持消除程序(即,使用消胆胺11天,或活性炭冲洗30天或根据当地标签),则lef≥8周;
[0912]
对于所有其它,≥4周
[0913]
8.稳定剂量的非甾体抗炎药(nsaid)、对乙酰氨基酚/扑热息痛、低效阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药)、口服皮质类固醇(相当于泼尼松≤10mg/ 天)或吸入皮质类固醇允许用于稳定病情,但必须在基线就诊前≥1周保持稳定剂量。
[0914]
9.受试者必须在第一剂研究药物之前至少1周停用所有阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药除外)以及至少4周停止口服中药。
[0915]
10.仅在当地要求规定的情况下,需要使用至少一种以下背景药物进行治疗:非生物 dmard、nsaid、对乙酰氨基酚、低效阿片类药物(曲马多或对乙酰氨基酚可待因或氢可酮的联合用药)或口服皮质类固醇。
[0916]
主要排除标准包括:
[0917]
1.既往暴露于任何janus激酶(jak)抑制剂(包括但不限于鲁索替尼(ruxolitinib)、托法替尼、巴瑞替尼和非戈替尼)。
[0918]
2.目前使用>2种非生物dmard,或使用除mtx、ssz、lef、阿普斯特、hcq、布西拉明或艾拉莫德以外的dmard进行治疗,或在基线时使用mtx与lef联合用药。
[0919]
3.纤维肌痛病史、在17岁之前发病的任何关节炎,或目前诊断出除psa以外的炎症性关节病(包括但不限于类风湿性关节炎、痛风、重叠结缔组织病、硬皮病、多发性肌炎、皮肌炎、系统性红斑狼疮)。如果对psa的诊断改变或对psa的附加诊断有记录,则允许有反应性关节炎或中轴型脊柱关节炎(包括强直性脊柱炎和非放射学中轴型脊柱关节炎)的既往史。如果有psa诊断改变的记录或纤维肌痛诊断错误的记录,则允许有纤维肌痛的既往史。
[0920]
分析窗口
[0921]
将应用以下规则将实际受试者就诊分配给方案指定的就诊。对于每个方案指定的研究就诊,将确定一个目标研究日以代表相应的就诊以及目标日周围的窗口。将以非重叠方式选择窗口,以便收集日期不会落入多个就诊窗口。如果受试者在一个就诊窗口中有两次或更多处的实际就诊,则将使用最接近目标日期的就诊进行分析。如果两次就诊与目标日期的距离相等,则后一次的就诊将用于分析。
[0922]
结果直至第24周(包括第24周)
[0923]
如上所述,在最后一位受试者完成第24周的研究就诊后,为了初始监管提交的目的,进行了非盲分析。
[0924]
该研究在第12周达到了主要终点acr20。在第12周,与接受乌帕替尼15mg的303名 (70.6%;差异:34.5%;95%ci:28.2至40.7;p<0.001)和接受乌帕替尼30mg的332名(78.5%;差异:42.3%;95%ci:36.3至48.3;p<0.001)相比,接受安慰剂的153名(36.2%)患者达到了acr20应答的主要结果;与接受乌帕替尼15mg(差异:5.6%;95%ci:-0.6至11.8)和 30mg(差异:13.5%;95%ci:7.5至19.4;p<0.001)相比,接受阿达木单抗治疗的279名(65.0%) 患者达到了acr20应答率。乌帕替尼30mg优于阿达木单抗;15mg剂量并不优于阿达木单抗,这阻止了对测试层次较低的次要终点的显着性测试。无论评估的亚组如何,包括基线bmi、基线hscrp和既往非生物dmard的数量(≤1或>1),都证明了乌帕替尼游离碱15mg的疗效。
[0925]
该研究还显示,与阿达木单抗相比,两种乌帕替尼游离碱剂量在第12周时acr20的非劣效性(ni)具有统计学意义。在完成非劣效性(ni)比较后,进行了与阿达木单抗相比的优效性测试。乌帕替尼游离碱30mg在第12周的acr20中显示出优于阿达木单抗的优势。
[0926]
与安慰剂相比,关键次要终点的结果与主要终点的结果一致。在第12周,乌帕替尼15mg 和30mg的acr50应答与安慰剂的治疗差异(95%ci)分别为24.3%(187至29.9)和38.5% (32.8至44.3),与阿达木单抗相比分别为0.0%(-6.5至6.5)和14.2%(7.6至20.9)。乌帕替尼15mg和30mg的acr70应答与安慰剂的治疗差异(95%ci)分别为13.3%(9.5至17.0) 和22.9%(18.5至27.3),与阿达木单抗相比分别为1.9%(-2.9至6.6)和11.5%(6.3至16.8)。在第24周,与安慰剂和阿达木单抗相比,两种剂量乌帕替尼达到acr20/50/70应答的患者比例与主要终点方向相同;不能从这些数据中得出任何临床推论。在第24周,与安慰剂相比,两种剂量的乌帕替尼达到最小疾病活动性(mda)的患者比例显着更高(两次比较p<0.001),并且与阿达木单抗的主要结果方向相同。
[0927]
在第16周,两种剂量乌帕替尼与安慰剂相比,显着更高比例的患者达到至少75%的银屑病面积严重程度指数(pasi 75)和0或1的银屑病静态研究者总体评估,并且相对于基线改善至少2分(siga 0/1)应答(每次比较p<0.001)。两种剂量乌帕替尼与阿达木单抗相比,达到pasi 75应答的患者比例与主要终点方向相同,以及siga 0/1应答与乌帕替尼30mg的方向相同;乌帕替尼15mg与阿达木单抗相比,siga 0/1应答者比例没有显着差异。通过银屑病症状自我评估(saps)衡量,与安慰剂相比,在第16周,接受乌帕替尼游离碱15mg qd 治疗的患者也观察到患者报告的银屑病症状有更大的改善。在第12周,观察到每个乌帕替尼剂量与安慰剂相比,haq-di、sf-36pcs和facit-f相对于基线的变化具有统计学上更大的疗效(每次比较p<0.001)。在第12周,接受乌帕替尼游离碱15mg qd的患者和接受安慰剂
的患者中,haq-di应答者的比例(haq-di评分相对于基线改善≥0.35)为58%和33%。
[0928]
sf-36评估了与健康相关的生活质量。在第12周,与安慰剂相比,接受乌帕替尼游离碱 15mg qd的患者在身体分量综合评分方面比基线有显着改善。与安慰剂相比,在心理分量综合评分和sf-36的所有8个域(身体功能、身体疼痛、活力、社会功能、身体角色、一般健康、情绪角色和心理健康)方面也观察到了更大的改善。
[0929]
在第24周,与安慰剂相比,明显更多的接受乌帕替尼治疗的患者达到附着点炎的消退(每次比较p<0.001);乌帕替尼15mg与阿达木单抗在附着点炎消退方面没有显着差异,乌帕替尼30mg的结果与主要终点方向相同。下表列出了指趾炎的消退率。
[0930]
与安慰剂相比,该研究还证明了在psa体征和症状以及抑制放射学进展方面的疗效显着提高。例如,在第24周,与安慰剂治疗的患者相比,乌帕替尼治疗的患者表现出显着更少的放射学进展,这可以从改良的sharp/van der heijde总评分(mtss;p<0.001)(mtss相当于 shs)相对于基线的变化证明;乌帕替尼和阿达木单抗的平均进展没有差异。对于15mg qd 剂量的侵蚀和关节间隙狭窄评分,也达到了统计学上显着的结果。在第24周,与安慰剂(89%) 相比,使用乌帕替尼游离碱15mg qd(93%)的无放射学进展(mtss变化≤0)的患者比例更高。这些结果总结在下表中。
[0931]
下表(第1部分和第2部分)中提供了主要和关键次要疗效结果的摘要。参见图11a至 11b,列出了排序次要终点,第1部分和第2部分。acr20应答的时间进程和相关联的95%置信区间(ci)直至第24周(包括第24周)在如图12a所示,其它关键次要终点的结果如图12b至12bb所示。据观察,这种临床疗效通过持续每日给药得以维持。
[0932]
[0933][0934]
备注:标称p值在括号中提供。*使用图形多重测试程序实现了统计学显着性,将总体i型错误率控制在双侧0.0499水平。请注意,0.0001用于中期无效分析。
[0935]
a.二元终点的结果基于nri分析。第24周的mda和附着点炎消退的结果基于无应答者估算和额外的救援处理,其中第16周获救的受试者被估算为无应答者。连续终点的结果基于 mmrm模型,具有固定的治疗效果、就诊、逐次就诊交互作用、当前dmard使用的分层因素(是/否)和基线测量。shs的结果基于对缺失数据和救援处理进行线性外推的ancova。
[0936]
b.对基线siga≥2的受试者进行总结;n
(pbo)
=313,n
(ada)
=330,n
(upa15)
=322,n
(upa30)
=324。
[0937]
c.对基线bsa受银屑病影响≥3%的受试者进行总结;n(pbo)=211,n(ada)=211,n(upa15) =214,n(upa30)=210。
[0938]
d.对基线lei>0的受试者进行总结;n(pbo)=241,n(ada)=265,n(upa15)=270,n(upa30) =267。
[0939]
e.乌帕替尼与阿达木单抗相比的非劣效性测试,保留了50%的阿达木单抗效果。
[0940][0941]
备注:标称p值在括号中提供。*使用图形化多重测试程序控制总体i型错误率的统计学显着性。
[0942]
a.二元终点的结果基于nri分析。第24周的指趾炎消退结果基于无应答者估算和额外的救援处理,其中第16周获救的受试者被估算为无应答者。连续终点的结果基于mmrm模型,具有固定的治疗效果、就诊、逐次就诊交互作用、当前dmard使用的分层因素(是/否)和基线测量。
[0943]
b.乌帕替尼与阿达木单抗的优效性测试。
[0944]
c.对基线ldi>0的受试者进行总结;n(pbo)=126,n(ada)=127,n(upa15)=136,n(upa30) =127。
[0945][0946]
a.完整分析集:n(安慰剂)=423,n(ada)=429,n(upa15)=429,n(upa30)=423。
[0947]
第24周的acr20、acr50和acr70应答以及第12周的mda、附着点炎消退(lei=0) 和指趾炎消退(ldi=0)的其它数据列于下表。
[0948][0949]
在评估周停止随机治疗或缺失数据的患者在分析中被归为无应答者。对于第24周的mda、附着点炎的消退和指趾炎的消退,在第16周获救的受试者在分析中被归为无应答者。
[0950]a在基线时患有附着点炎的患者中(分别为n=241和270)
[0951]b在基线时患有指趾炎的患者中(n=126和136)
[0952]
与安慰剂相比,使用乌帕替尼游离碱15mg治疗可改善单个acr分量,包括压痛/疼痛和肿胀关节计数、患者和医生对疾病活动性的总体评估、haq-di、疼痛评估和hscrp。这些结果示于图12f至12k和12q。早在第2周就看到acr20及其分量的疗效开始。
[0953]
在第24周,对结构损伤进展的抑制进行了放射学评估,并表示为改良的总sharp评分 (mtss)及其分量、侵蚀评分和关节间隙变窄评分相对于基线的变化。在第24周,与安慰剂相比,使用乌帕替尼游离碱15mg qd治疗可显着抑制结构关节损伤的进展。侵蚀和关节间隙变窄评分也取得了统计学上显着的结果。在第24周,与安慰剂(89%)相比,使用乌帕替尼游离碱15mg qd(93%)的无放射学进展(mtss变化≤0)的患者比例更高。这些结果总结在图12z中。
[0954]
该试验中乌帕替尼的安全性概况与其它乌帕替尼临床试验中观察到的安全性概况大致相似。到第24周,安慰剂、阿达木单抗和乌帕替尼15mg的治疗出现不良事件和严重不良事件(包括严重感染)的发生率相似,但乌帕替尼30mg的发生率更高。最常见的ae是上呼吸道感染。安慰剂、阿达木单抗、乌帕替尼15mg和30mg的严重感染率分别为0.9%、0.7%、1.2%和2.6%。直至第24周,治疗出现的机会性感染包括1例使用乌帕替尼15mg的念珠菌性尿道炎事件,以及使用乌帕替尼30mg的耶氏肺孢子菌肺炎和巨细胞病毒事件各1例。安慰剂和乌帕替尼15mg和30mg分别报告了3、4和5例带状疱疹病例。安慰剂和乌帕替尼15mg 各发生1例恶性肿瘤,乌帕替尼30mg和阿达木单抗组各报告3例恶性肿瘤。使用乌帕替尼未报告主要不良心血管事件(mace)。安慰剂报告了1起深静脉血栓形成事件,阿达木单抗组报告了2起事件;使用乌帕替尼30mg报告了1起肺栓塞事件。安慰剂报告了1例死亡,原因不明。
[0955]
在所有治疗组中,从基线到第24周,平均血红蛋白、中性粒细胞、淋巴细胞和血小板水平保持在正常范围内。乌帕替尼15mg和安慰剂报告的贫血和淋巴细胞减少症不良事件发生率相似,而乌帕替尼30mg的不良事件发生率更高。与安慰剂相比,乌帕替尼的中性粒细胞减少症不良事件更常见。总体而言,≥3级实验室异常的频率≤2.1%;大多数在不中断治疗的情况下得到解决。没有患者的血液学参数出现4级下降。一名患者在停用乌帕替尼30mg后分别报告了血红蛋白或血小板的3级单独下降。与其它组相比,乌帕替尼30mg的3级中性粒细胞和淋巴细胞减少症的频率更高。
[0956]
安慰剂、阿达木单抗、乌帕替尼15mg和30mg组分别报告了3.8%、15.6%、9.1%和
pj等人,《风湿病学年鉴》2017;76:79-87。
[0962]
实例5
[0963]
患有活动性银屑病关节炎且先前对至少一种生物疾痛改善抗风湿药(bdmard)应答不足的受试者的第3期方案(select psa2)
[0964]
select-psa2第三期研究设计在图12中提供。
[0965]
15mg或30mg乌帕替尼是指如实例1中所述的每天一次(qd)15mg和30mg乌帕替尼药物产品。
[0966]
这是一项3期多中心研究,包括两个周期。第1周期为期56周,包括24周的随机、双盲、平行组、安慰剂对照期,随后是另外32周的盲法治疗(第24至56周)。第1周期旨在比较乌帕替尼游离碱15mg qd和30mg qd与安慰剂在对至少一种生物疾病改善抗风湿药 (bdmard)应答不足(psabio-ir)的中度至重度活动性psa受试者中的安全性、耐受性和疗效。第2周期是开放标签(盲法直到最后一名受试者完成第1周期的最后一次就诊)长期延长至总治疗时间约3年,以评估乌帕替尼游离碱15mg qd和30mg qd在完成第1周期的 psa受试者的安全性、耐受性和疗效。
[0967]
研究期限包括35天的筛选期;为期56周的盲期,其中包括24周的双盲、安慰剂对照治疗,随后是32周对乌帕替尼剂量盲法的治疗(第1周期);长达约3年的总治疗持续时间的长期延长期([在最后一名受试者完成第1周期的最后一次就诊之前设盲]第2周期);以及30 天的随访电话或就诊。
[0968]
符合资格标准的受试者按银屑病程度(≥3%体表面积[bsa]或<3%bsa)、当前使用至少1 种dmard(是或否)和先前失败(应答不足)生物dmard(1与>1相比)的次数进行分层,来自日本的受试者除外,其随机化仅按银屑病程度(≥3%体表面积[bsa]或<3%bsa)进行分层。受试者以2∶2∶1∶1的比例随机分配到四个治疗组之一:
[0969]
第1组:乌帕替尼游离碱15mg qd(n=211)
[0970]
第2组:乌帕替尼游离碱30mg qd(n=219)
[0971]
第3组:安慰剂,然后是乌帕替尼游离碱15mg qd(n=106)
[0972]
第4组:安慰剂,然后是乌帕替尼游离碱30mg qd(n=106)
[0973]
不超过约40%的入选受试者具有bsa程度<3%的银屑病,不超过约30%的入选受试者具有先前失败的超过1次生物dmard。
[0974]
受试者接受口服研究药物qd(乌帕替尼游离碱15mg、乌帕替尼游离碱30mg或匹配的安慰剂)直到研究结束或直到他们停用研究药物。
[0975]
在基线时被分配到安慰剂的受试者被预先分配以1∶1的比例从第24周开始接受乌帕替尼游离碱15mg qd或乌帕替尼游离碱30mg qd。完成第56周就诊(第1周期结束)的受试者进入研究的长期延长部分,即第2周期(总治疗长达约3年)。受试者按照第1周期的分配继续研究治疗。受试者继续以盲法方式分别接受乌帕替尼游离碱15mg qd或乌帕替尼游离碱30mg qd,直到最后一名受试者完成第1周期(第56周)的最后一次就诊,此时两个周期的研究药物分配将向各研究中心揭盲,并且受试者将以开放标签的方式分配研究药物,直到第2周期完成。
[0976]
受试者必须在筛选就诊之前对≥1种bdmard应答不足,并且必须在第一剂研究药物剂量之前停用所有bdmard。在参与本研究期间不需要背景非生物dmard治疗。对于在基线
时接受非生物dmard治疗(甲氨蝶呤(mtx)、柳氮磺胺吡啶(ssz)、来氟米特(lef)、阿普斯特、羟氯喹(hcq)、布西拉明或艾拉莫德)的受试者,非生物dmard应在基线就诊前≥12周开始,必须在第一剂研究药物前以稳定剂量≥4周,并保持稳定剂量至研究的第36 周;非生物dmard剂量可能仅出于安全原因而减少。另外,所有服用mtx的受试者应在整个研究参与期间服用口服叶酸(或等效物)的膳食补充剂。
[0977]
在第16周,将向分类为无应答者(定义为在第12周和第16周的压痛关节计数(tjc) 和肿胀关节计数(sjc)之一或两者均未达到至少20%的改善)的受试者提供救援治疗,如下: 1)添加或调整非生物dmard、nsaid、对乙酰氨基酚/扑热息痛、低效阿片类药物(曲马多或对乙酰氨基酚和可待因或氢可酮的联合用药)、口服皮质类固醇的剂量和/或2)对于1个外周关节、1个触发点、1个压痛点、1个滑囊或1个附着点,接受1次关节内、触发点或压痛点、滑囊内或腱鞘内皮质类固醇注射(抢救治疗)。
[0978]
在第24周,无论临床应答如何,在基线时分配到安慰剂的所有受试者都将转为盲法乌帕替尼(在基线时随机分配至15mg qd或30mg qd)治疗。
[0979]
在最后一名受试者完成第24周的研究就诊后,将进行非盲分析,以用于初始监管提交。为了在盲法的56周期间保持试验的完整性,研究中心和受试者将保持盲法,直到所有受试者都达到第56周。在所有受试者完成第1周期后,可以出于监管目的进行第二次非盲分析。在所有受试者完成第2周期后,将进行最终分析。
[0980]
主要和次要终点
[0981]
主要疗效终点是在第12周达到美国风湿病学会(acr)20%应答率的受试者比例。 select-psa1临床研究(实例4)和本研究(select-psa2临床研究;实例5)的主要和次要临床终点在实例6中进一步描述。
[0982]
研究群体
[0983]
患有活动性银屑病关节炎且先前对至少一种生物dmard应答不足的患者。
[0984]
纳入标准包括:
[0985]
1.成年男性或女性,筛选时至少≥18岁。
[0986]
2.在筛选就诊前至少6个月临床诊断出psa症状发作并满足psa分类标准(caspar)。
[0987]
3.受试者在基线时患有活动性疾病,定义为在筛选和基线就诊时≥3个压痛关节(基于 68个关节计数)和≥3个肿胀关节(基于66个关节计数)。
[0988]
4.活动性斑块状银屑病的诊断或有斑块状银屑病病史记录。
[0989]
5.受试者对至少1种bdmard的治疗应答不足(在至少12周的治疗持续时间后缺乏疗效)或不耐受。
[0990]
6.受试者必须在第一剂研究药物之前停用所有bdmard。需要在基线就诊之前停用 bdmard以符合此纳入标准的受试者必须遵循以下指定的程序或至少五倍于药物的平均终末消除半衰期:
[0991]
·
依那西普≥4周;
[0992]
·
阿达木单抗、英夫利昔单抗(infliximab)、赛妥珠单抗(certolizumab)、戈利木单抗 (golimumab)、阿巴西普(abatacept)、托珠单抗和依克珠单抗≥8周;
[0993]
·
苏金单抗≥16周;
qd 在psa中的疗效显着提高。参见下表t、u和v1,以及图14a至bb。在图14a中提供了主要终点acr20直到第24周(包括第24周)的时间进程,并且在图14b至14d中提供了在 acr50、acr70的关键次要终点以及在24周内达到最小疾病活动性(mda)的患者比例。利兹附着点炎指数(lei)和附着点炎消退(lei=0)的受试者直到第24周(包括第24周) 相对于基线变化的时间进程如图14e至14g所示。利兹指趾炎指数(ldi)和指趾炎消退(ldi =0)的受试者直到第24周(包括第24周)相对于基线变化的时间进程如图14h至14i所示。图14j中提供了其它关键次要终点的数据。
[1012]
与安慰剂相比,两种乌帕替尼剂量的所有p值均具有统计学意义(图形化多重测试程序将总体i型错误率控制在0.05水平)。
[1013]
主要分析显示,与安慰剂组相比,乌帕替尼治疗组的主要终点应答率显着提高。在第12 周,乌帕替尼15mg和30mg组与安慰剂组相比,有显着更多的患者达到acr20应答(分别为56.9%、63.8%和24.1%;乌帕替尼两组与安慰剂组相比p<0.001)。到第2周,更多乌帕替尼 15mg和30mg治疗的患者达到acr20应答(标称p<0.001)。随着时间的推移,两个治疗组中具有acr20应答的患者比例持续增加,在第12周乌帕替尼30mg组中观察到反应平稳,而在第20周乌帕替尼15mg组中具有acr20应答的患者比例增加,接近安慰剂对照期结束时30mg剂量组的应答率。乌帕替尼15mg和乌帕替尼30mg的应答率在超过1次生物 dmard失败的患者亚组中分别为44.9%和64.8%,在单药治疗患者亚组中为55.8%和66.7%;这些应答与总群体中的结果相似。另外,在第12周观察到两种乌帕替尼剂量与安慰剂相比 acr50和acr70均有所改善。从第2周到第24周,与安慰剂相比,使用乌帕替尼15mg或 30mg观察到acr应答的所有分量均较基线有所改善。无论评估的亚组如何,包括基线bmi、基线hscrp和既往非生物dmard的数量(≤1或>1),都证明了乌帕替尼游离碱15mg的疗效。
[1014]
与安慰剂相比,15mg和30mg剂量的乌帕替尼在所有关键次要终点方面表现出更大的改善。到第12周和第24周,通过pasi 75/90/100测量,两种乌帕替尼剂量与安慰剂相比均观察到银屑病改善(在第16周,pasi 75的p<0.001,pasi 90/100的标称p<0.001;所有其它时间点的标称p<0.001)和siga0/1(第16周的p<0.001;第12周和第24周的标称p<0.001)。在第16周(p<0.001)和第24周(标称p<0.001),乌帕替尼组与安慰剂相比,saps相对于基线的变化更大。根据从第2周到第24周的haq-di(第12周的p<0.001)和第12周(p<0.001) 和第24周(标称p<0.001)的sf-36 pcs相对于基线的平均变化,观察到两种剂量的乌帕替尼与安慰剂相比患者的身体功能均有改善。在第12周,接受乌帕替尼游离碱15mg qd的患者和接受安慰剂的患者中,haq-di应答者的比例(haq-di评分相对于基线改善≥0.35)为 45%和27%。在第12周(p<0.001)和第24周(标称p<0.001),与安慰剂相比,使用两种剂量乌帕替尼的患者报告通过facit-f评估的疲劳改善。在第12周和第24周观察到晨僵相对于基线的平均改善(标称p<0.001)。从第12周到第24周,使用乌帕替尼的任一剂量的患者与安慰剂相比,都有较高比例的患者报告使用lei和sparcc附着点炎指数的附着点炎和指趾炎消退(标称p<0.001)。与安慰剂相比,接受乌帕替尼的任一剂量的患者在第24周达到mda 的比例更高(第24周的p<0.001;第12周和第16周的标称p<0.001)。到第24周,与安慰剂相比,两种乌帕替尼剂量与安慰剂相比,dapsa评分相对于基线的平均变化更大(所有时间点的标称p<0.001)。
[1015]
sf-36评估了与健康相关的生活质量。在第12周,与安慰剂相比,接受乌帕替尼游
离碱 15mg qd的患者在身体分量综合评分方面比基线有显着改善。与安慰剂相比,在心理分量综合评分和sf-36的所有8个域(身体功能、身体疼痛、活力、社会功能、身体角色、一般健康、情绪角色和心理健康)方面也观察到了更大的改善。
[1016]
[1017][1018]
[1019]
备注:标称p值在括号中提供。*统计显着性使用图形化多重测试程序将总体i型错误率控制在0.05水平。
[1020]
a.二元端点的结果基于nri(净重分类改善)分析。第24周的最小疾病活动性(mda)的结果基于无应答者估算和额外的救援处理,其中第16周获救的受试者被估算为无应答者。连续终点的结果基于重复测量的混合模型(mmrm)模型,该模型具有固定的治疗效果、就诊、逐次就诊交互作用、当前dmard使用的分层因素(是/否)和基线测量。
[1021]
b.对基线siga≥2的受试者进行总结。
[1022]
c.对基线bsa受银屑病影响≥3%的受试者进行总结。
[1023][1024]
a.完整分析集:n(pbo)=212,n(upa15)=211,n(upa30)=218
[1025][1026]
第24周的acr20、acr50和acr70应答、第12周的mda和第24周的附着点炎消退 (lei=0)和指趾炎消退(ldi=0)的其它数据列于下表。
[1027][1028]
在评估周停止随机治疗或缺失数据的患者在分析中被归为无应答者。对于第24周的mda、附着点炎的消退和指趾炎的消退,在第16周获救的受试者在分析中被归为无应答者。
[1029]a在基线时患有附着点炎的患者中(分别为n=144和133)
[1030]b在基线时患有指趾炎的患者中(n=64和55)
[1031]
与安慰剂相比,使用乌帕替尼游离碱15mg qd治疗可改善单个acr分量,包括压痛/ 疼痛和肿胀关节计数、患者和医生对疾病活动性的总体评估、haq-di、疼痛评估和hscrp。这些结果总结在图14n至14s和14v中。早在第2周就看到acr20及其分量的疗效开始。
[1032]
到第24周,乌帕替尼30mg组的整体治疗突发ae(teae)率较高,两种乌帕替尼剂量与安慰剂相比,严重ae(sae)和导致试验药物停用的teae率都较高。
[1033]
在乌帕替尼治疗的患者中,最常见的teae是上呼吸道感染和鼻咽炎。安慰剂有四名(1.9%),而乌帕替尼15mg有十二名(5.7%),乌帕替尼30mg有十八名(8.3%)患者报告了sae。安慰剂和乌帕替尼15mg组各有一名(0.5%)患者出现严重感染,乌帕替尼30mg有六名(2.8%)患者出现严重感染。肺炎是最常报告的严重感染(一名患者服用乌帕替尼15mg,三名患者服用乌帕替尼30mg)。直到第24周,治疗出现的机会性感染(不包括肺结核和带状疱疹)包括气管念珠菌病和口咽念珠菌病各1例,均使用乌帕替尼30mg。在安慰剂、乌帕替尼15mg和30mg组中,分别有两、三和八名患者报告了带状疱疹;这些病例都不严重。一名服用乌帕替尼15mg的患者和两名服用乌帕替尼30mg的患者患有皮肤播散性带状疱疹。没有观察到中枢神经系统受累的带状疱疹病例。安慰剂有三名(1.4%)患者报告肝功能紊乱,乌帕替尼15mg有4名(1.9%),乌帕替尼30mg有18名(8.3%);大多数是无症状的肝酶升高。
[1034]
每个乌帕替尼组的三名患者报告了恶性肿瘤(乌帕替尼15mg:一例基底细胞癌、一例前列腺癌、一例直肠癌;乌帕替尼30mg:一例直肠腺癌、一例卵巢癌和子宫内膜癌以及一例基底细胞癌),而在安慰剂组中无一例。这些恶性事件的事件发生时间<6个月。
[1035]
到第24周没有报告经裁定的胃肠道穿孔。乌帕替尼15mg组报告了一例主要不良心血管事件(mace;0.5%,非致命性心肌梗死)和一例静脉血栓栓塞事件(vte;0.5%;肺栓塞);两名患者分别有至少一个mace或vte的危险因素(例如,肥胖、高血压或高胆固醇血症)。在24周期间,安慰剂组报告了一例与机动车事故有关的死亡。
[1036]
一般来说,在所有治疗组中,从基线到第24周,平均血红蛋白、中性粒细胞、淋巴细胞和血小板水平保持在正常范围内。在乌帕替尼30mg组中,有两名患者的血红蛋白值出现3级下降。据报道,一名服用安慰剂的患者(0.5%)、两名服用乌帕替尼15mg的患者(1.0%)和四名服用乌帕替尼30mg的患者(1.8%)报告了3级中性粒细胞减少。没有患者的血小板、白细胞、中性粒细胞或淋巴细胞出现4级下降。
[1037]
在治疗组中,≤1%的患者观察到丙氨酸氨基转移酶或天冬氨酸氨基转移酶的孤立性3级升高,未观察到4级升高。没有报道海氏法则案例。据报道,安慰剂组和乌帕替尼15mg和30mg组分别有1名(0.5%)、1名(0.5%)和5名(2.3%)患者出现cpk值3级升高。两名服用安慰剂的患者和一名服用乌帕替尼15mg的患者报告了cpk值的4级升高。没有导致试验药物停药,也没有横纹肌溶解事件。与安慰剂组相比,在乌帕替尼组中观察到低密度脂蛋白胆固醇(ldl-c)和高密度脂蛋白胆固醇(hdl-c)的平均轻度增加。ldl-c:hdl-c和总胆固醇:hdl-c的比率通常在第24周保持不变。
[1038]
乌帕替尼作为治疗psa的一种有前途的疗法
[1039]
对乌帕替尼和jak小分子抑制剂托法替尼(批准用于治疗psa的剂量较低(5mgbid))的关键主要和次要终点的安慰剂校正数据进行了审查,虽然不是直接比较,但表明乌帕替尼15mgqd和30mgqd对更难达到的最小疾病活动性(mda)的终点,以及银屑病终点pasi75或pasi90,以及某些acr分量(例如,acr20/50/70)显示出明确的前景。此外,观察到这种疗效一旦达到,就会随着时间的推移而持续或改善。
[1040][1041]
托法替尼研究:gladman d等人,《新英格兰医学杂志》2017;377:1525-1536(5mg bid剂量在美国获准用于治疗psa;10mg bid剂量在美国未获批准);依克珠单抗研究:nash p等人,《柳叶刀杂志》2017;389:2317-2327。
[1042]
实例6
[1043]
select-psa1(实例4)和select-psa2(实例5)临床终点
[1044]
psa1和psa2研究的主要终点
[1045]
主要疗效终点是在第12周达到美国风湿病学会(acr)20%应答率的受试者比例。acr20 应答率是基于压痛和肿胀关节计数(tjc和sjc)和患者对疼痛的总体评估(pt pain)数值评定量表(nrs)、患者总体评估(ptga)-疾病活动性数值评定量表(nrs)、医生总体评估(pga)
ꢀ‑
疾病活动性数值评定量表(nrs)、健康评估问卷-残疾指数(haq-di)或高敏c反应蛋白(hs-crp)这5项测量中的≥3项改善20%或更高来确定的。
[1046]
psa1和psa2研究的关键次要终点
[1047]
关键多重性调整的次要疗效终点(除非另有说明,每种剂量的乌帕替尼与安慰剂相比) 是:
crp 的5项测量中的≥3项确定的。
[1053]
psa1和psa2研究的其它次要终点
[1054]
与安慰剂和阿达木单抗相比,在除主要和关键次要变量指定的时间点外,在接受乌帕替尼治疗的受试者中评估了以下结果测量:
[1055]
[1056]
[1057][1058]
其它实施例
[1059]
本技术涉及各种已发布的专利、已公开的专利申请、期刊文章和其它出版物,其各自通过引用并入本文。
[1060]
上文已经对本公开的某些非限制性实施例进行了描述。本领域普通技术人员将了解,在不背离如所附权利要求所限定的本公开的精神或范围的情况下,可以对该描述作出各种修改和改变。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1