一种活性成分为草酸纳洛解的药物组合物及制备方法与流程
1.本发明属于医药技术领域,涉及到草酸纳洛解口服片剂的制备方法。
背景技术:
2.草酸纳洛解(naloxegol oxalate)是纳洛酮的聚乙二醇化衍生物,是p~糖蛋白转运蛋白的底物。与纳洛酮比较,草酸纳洛解中聚乙二醇基团的存在降低了其被动通透性,通过血脑屏障的渗透性降低,在推荐剂量水平,草酸纳洛解的cns穿透性可忽略不计,从而不干扰中枢性介导的阿片类镇痛作用。
3.草酸纳洛解是一种与μ
‑
阿片受体结合的阿片类药物拮抗剂。当在推荐剂量水平给药时,草酸纳洛解作为一种外周μ
‑
阿片受体拮抗剂作用于胃肠道等组织,因此减低阿片类药物的便秘效应。
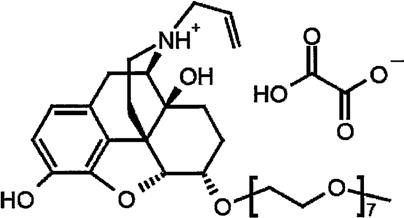
4.草酸纳洛解的化学名:(5α,6α)
‑
17
‑
烯丙基
‑6‑
(2,5,8,11,14,17,20
‑
七氧杂二十二烷
‑
22
‑
基氧基)
‑
4,5
‑
环氧基吗啡喃
‑
3,14
‑
二醇草酸盐,cas登记号:1354744
‑
91
‑
4,分子式:c
34
h
53
no
11
.c2h2o4,其分子结构式如下:
[0005][0006]
草酸纳洛解片由阿斯利康(astrazeneca)制药有限公司研发,于2014年9月由fda批准上市,用于伴有慢性非癌性疼痛的阿片药物诱导的成人便秘患者的治疗。
[0007]
草酸纳洛解片按纳洛解(c
34
h
53
no
11
)计算,规格为12.5mg、25mg/片,规格较小,临床使用需要达到快速溶出,同时产品稳定性一般。
[0008]
在制药行业中,原料的颗粒特性已成为口服固体制剂开发和质量控制中至关重要的的因素之一。原料药的粒度分布会对制剂产品的性能产生显著的影响,如溶解度、生物利用度、含量均匀度和稳定性等。此外,原料的粒度分布也会影响药物的可生产性如流动性、总混均匀度、可压性等,最终可能影响药物的安全性、有效性和质量。粉体的粒度分布对口服固体制剂生产过程的每一步都有很大影响,包括预混、混合、干燥、整粒、压片。
[0009]
目前,有必要提供一种新的解决方案克服草酸纳洛解制备过程中原料粘度大、物料混合均匀度不好、制剂稳定性差的缺点,使得制备的草酸纳洛解混合物料具有均匀度好,产品质量稳定,有利于商业化生产,且具有快速溶出的特点。
技术实现要素:
[0010]
本发明解决了草酸纳洛解片在生产中因草酸纳洛解粘度大,物料混合后含量均匀度差,产品质量不稳定的问题,同时采用粉末直压技术降低了生产成本和能耗,产品稳定性
可控,有利于用药安全、商业化生产。
[0011]
本发明人经研究惊奇地发现:将处方中粘合剂羧甲基纤维素钠、稳定剂棓丙酯混合后与粒径分布d(v,0.9)30~200μm的草酸纳洛解混合,再与适量的填充剂、崩解剂、润滑剂混合,粉末混合均匀度好,流动性、可压性好,采用粉末直压技术压片后包衣制得的产品可以快速溶出,相对原研产品稳定性得到显著提升。
[0012]
本发明提供一种活性成分为草酸纳洛解的药物口服制剂,其特征在于:
[0013]
本制剂所采用的草酸纳洛解原料为6α构型;粒度分布为d90(μm)为30~200,优选为d90(μm)为50~150。
[0014]
片芯由以下重量百分比的药物组分组成:
[0015]
组分重量百分比(%)功能草酸纳洛解5~10活性成分喷雾干燥甘露醇40~90填充剂微晶纤维素20~50填充剂、崩解剂羧甲基纤维素钠0.02~2粘合剂棓丙酯0.05~0.5稳定剂硬脂酸镁0.5~3润滑剂
[0016]
本发明采用的粘合剂羧甲基纤维素钠与稳定剂棓丙酯预混,后与粒度分布为d90为50~150μm的草酸纳洛解混合,最后再与填充剂微晶纤维素、甘露醇,润滑剂硬脂酸镁混合能得到含量均一、流动性极佳的粉末,可满足粉末直压的技术要求。
[0017]
药物颗粒度采用激光粒度仪进行测定,d90/d(v,0.9)表示一个样品的累计粒度分布数达到90%时所对应的粒度分布。它的物理意义是粒度分布小于它的颗粒占90%。
[0018]
本发明采用激光衍射法测定草酸纳洛解粒度分布,检测方法如下:
[0019]
(1)仪器:马尔文master sizes
[0020]
(2)方法:激光粒度法干法
[0021]
(3)方法验证:选择分散压力为2.0bar、进样速度为50%、重复性试验、中间精密度验证结果良好。
[0022]
(4)样品测定:分别取三批原料进行测定。
[0023]
本发明采用的草酸纳洛解的粒度分布d90为30~200,优选为d90(μm)为50~150。影响因素(高温60℃、光照4500lx
±
500lx、高湿75%条件下10天)、加速试验(40℃
±
2℃,rh75%)、长期试验(25℃
±
2℃,rh60%
±
5%)6个月后原料粒度分布未发生明显变化。
[0024]
根据本发明,药学可接受的添加剂包括填充剂、粘合剂、崩解剂、稳定剂、助流剂、润滑剂等。
[0025]
根据本发明,粘合剂选自羧甲基纤维素钠、羟丙甲纤维素、羟丙纤维素、聚乙烯吡咯烷酮、海藻糖盐、淀粉中的一种或其组合物。优选的是羧甲基纤维素钠。羧甲基纤维素钠其用量为占药物组合物重量的0.02%~2%,优选为0.2~1%。
[0026]
根据本发明,填充剂选自甘露醇、碳酸氢钙、预胶化淀粉、糊精、乳糖、淀粉、微晶纤维素、优化微晶纤维素中的一种或其组合物,优选喷雾干燥甘露醇和微晶纤维素两种的组合物。微晶纤维素选自微晶纤维素ph101、微晶纤维素ph102,微晶纤维素101、微晶纤维素102中的一种或多种;更优选为微晶纤维素102。其中,喷雾干燥甘露醇用量
为占药物组合物重量的40~90%,优选为50~75%;微晶纤维素用量为20~50%,优选为25~35%;其填充剂总量为占药物组合物重量的60%~95%,更优选为75%~90%。
[0027]
根据本发明,崩解剂选自微晶纤维素、优化微晶纤维素、羧甲基纤维素钠、交联羧甲淀粉钠、羧甲淀粉钙、羧甲淀粉钠、低取代羟丙纤维素、交联聚乙烯吡咯烷酮中的一种或其组合物。优选为微晶纤维素,其用量为占药物组合物重量的20~40%,优选为25~35%。
[0028]
根据本发明,稳定剂选自没食子酸丙酯(即棓丙酯)、亚硫酸钠、亚硫酸氢钠、焦亚硫酸钠中的一种或其组合物。优选为棓丙酯,其用量为占药物组合物重量的0.05%~0.5%,优选为0.05%~0.2%。
[0029]
根据本发明,润滑剂选自硬脂酸镁、滑石粉、二氧化硅、硬脂富马酸钠、巴西棕榈蜡、硬脂酸钙、棕榈酸、硬脂酸中的一种或其组合物。优选为硬脂酸镁,其用量为占药物组合物重量的0.5%~3%。
[0030]
根据本发明,包衣粉选自欧巴代胃溶型薄膜薄膜包衣粉。包衣增重为0.5~4%,优选为2%~3%。
[0031]
根据本发明,提供一种优选的技术方案:
[0032]
根据处方比例称量活性成分和辅料,首先将羧甲纤维素钠和棓丙酯进行混合均匀,其次加入粒度分布为d90(μm)为50~150的草酸纳洛解原料进行混合,再加入微晶纤维素、甘露醇、硬脂酸镁混合,压片、包衣。发明人惊奇的发现,采用该方法混合的物料草酸纳洛解分散均匀,无需将草酸纳洛解研磨或控制较小粒度,获得物料流动性好、含量均匀、可压性好,包衣后药物快速溶出。
[0033]
本发明所述的药物组合物的根据中国药典2020年版四部通则0931溶出度与释放度测定法第二法(桨法)进行测定,以0.1mol/l盐酸、水、ph6.8磷酸盐缓冲液、ph4.5磷酸盐缓冲液溶液为溶出介质,优选500ml的0.1mol/l盐酸溶液,并在37℃
±
5℃下以50rpm的转速对发明组合物进行溶出试验,经5、10、15、30、45、60分钟时,取溶出液滤过,取续滤液作为供试品溶液(12.5mg规格);或精密量取续滤液适量,用溶出介质稀释一倍,作为供试品溶液(25mg规格);另取草酸纳洛解对照品适量,加0.1mol/l盐酸溶液溶解并制成每1ml中含25μg的溶液,照含量测定项下的方法测定,计算每片的溶出量,以时间为横坐标,溶出量为纵坐标,绘制溶出曲线,结果显示两个规格片剂在四种介质中15min溶出均大于85%,均为快速溶出。
[0034]
根据本发明的技术方案制备的包衣片剂,在加速试验(40℃
±
2℃,rh75%)6个月、长期试验(25℃
±
2℃,rh60%
±
5%)后溶出曲线未发生明显变化,15min均大于85%,其溶出受温度和湿度的影响小,产品质量稳定;并且稳定性过程中最大单个杂质不超过0.10%,总杂质不超过0.5%;稳定性试结果证实杂质几乎不增长,与原研品相比杂质具有明显的优势,产品稳定性得到显著提升,具有良好的安全性。
[0035]
本发明所述的含量、溶出度通过hplc检测获得;检测条件为:用十八烷基硅烷键合硅胶为填充剂;以0.05%三氟乙酸水溶液~乙腈(75∶25)为流动相;流速为每分钟1.0ml;检测波长为210nm;柱温为40℃。
附图说明
[0036]
图1:自制品(规格:25mg)在四种介质中溶出曲线
[0037]
图2:自制品(规格:12.5mg)在四种介质中溶出曲线
具体实施方式
[0038]
以下将结合实施例更详细地解释本发明,本发明的实施例仅用于说明本发明的技术方案,并非限定本发明的实质和范围。
[0039]
实施例1
[0040]
对甘露醇和微晶纤维素用量进行选择,具体处方见表1。
[0041]
表1:
[0042] 实验例1实验例2实验例3成分重量比(%)重量比(%)重量比(%)草酸纳洛解8.18.18.1喷雾干燥甘露醇59.230.744.95微晶纤维素10230.759.244.95羧甲基纤维素钠1.01.01.0硬脂酸镁1.01.01.0总计100100100
[0043]
制备工艺:将羧甲基纤维素钠与活性药物成分草酸纳洛解预混,再与喷雾干燥甘露醇、微晶纤维素102、硬脂酸镁混合均匀,调整压片机装量及硬度进行压片。
[0044]
将实验例1~3制备得到的片剂,照溶出度与释放度测定法第二法,取6片,以水500ml为溶出介质,转速为50转/分,分别在5、10、15、30、45分钟取样测定本品溶出度,测定结果见表2。
[0045]
表2:
[0046][0047]
上述实验结果表明,实验例2样品溶出最快,实验例1、实验例3样品溶出稍慢,三个处方样品溶出度在15分钟内均达到了85%以上,不同的喷雾干燥甘露醇和微晶纤维素102用量,对样品溶出时速度有影响,微晶纤维素102用量越大,样品溶出速度越快;同时随着水不溶性物质微晶纤维素用量的加大,溶出杯堆积的物料越多,综合考虑,选择实施例1做进一步考察。
[0048]
实施例2
[0049]
对羧甲基纤维素钠用量进行筛选,具体处方见表3。
[0050]
表3:
[0051] 实验例4实验例5实验例6成分重量比(%)重量比(%)重量比(%)草酸纳洛解8.18.18.1
喷雾干燥甘露醇59.260.058.2微晶纤维素10230.730.730.7羧甲基纤维素钠1.00.22.0硬脂酸镁1.01.01.0总计100100100
[0052]
制备工艺:将活性药物草酸纳洛解与羧甲基纤维素钠预混,再与甘露醇、微晶纤维素、硬脂酸镁混合均匀,调整压片机装量及硬度进行压片。
[0053]
将实验例4~6制备得到的片剂,照溶出度与释放度测定法第二法以0.1mol/l盐酸溶液500ml为溶出介质,转速为50转/分,分别在5、10、15、30、45、60分钟取样测定本品溶出度,测定结果见表4。
[0054]
表4:
[0055][0056]
上述实验结果表明,羧甲基纤维素钠用量为0.2%和1%时,处方样品溶出趋势基本一致;羧甲基纤维素钠用量增加到2%时,样品溶出有明显变慢的趋势,因此羧甲基淀粉钠的用量定为0.2%。
[0057]
实施例3
[0058]
对稳定剂用量进行筛选,具体处方见表5。
[0059]
表5:
[0060][0061]
制备工艺:将羧甲基纤维素钠、棓丙酯(如有)混合后与活性药物成分草酸纳洛解混合均匀;再与处方量的喷雾干燥甘露醇和微晶纤维素102、硬脂酸镁混合均匀,调整压片机装量及硬度进行压片;对片剂进行包衣,增重范围2%~3%。
[0062]
将实验例7~10处方样品置于4500lx
±
500lx的光照、60℃
±
2℃高温、高湿75%
±
1%及高湿92.5%的条件下,分别于0天、10天对其性状、有关物质以及含量进行检查,结果见表6。
[0063]
表6:
[0064][0065]
上述结果表明,实验例7样品在光照、高温60℃条件下放置10天有关物质增长明显,在高湿75%
±
5%条件下略有增长;添加棓丙酯的实施例8、9和10处方样品在影响因素各条件下,有关物质无显著变化。综上,0.05~0.5%范围内棓丙酯作为抗氧剂对处方样品的稳定性作用明显。
[0066]
实施例4
[0067]
进行两个规格(25mg/片、12.5mg/片)中试批样品制备,采用等比放大处方,处方信息见表7。
[0068]
表7:
[0069][0070]
制备工艺:
[0071]
1)领料、称重:按处方称取处方量的各原辅料。
[0072]
2)混料:先将羧甲基纤维素钠与棓丙酯预混;后依次将草酸纳洛解(d90为50~150μm)、微晶纤维素和喷雾干燥甘露醇、硬脂酸镁加入混合机中混合。
[0073]
3)中间体检测:取样进行中间体检测,水分≤3%,含量95.0%~105.0%。
[0074]
4)压片:选择冲模型号根据理论片重调整压片机片重及硬度参数,控制硬度范围为5~7kg,进行压片。
[0075]
5)包衣:根据要求配制包衣液,对本品进行包衣,包衣增重控制为2%~3%。
[0076]
6)包装:内包装采用口服固体药用高密度聚乙烯瓶,内置口服固体药用袋装硅胶干燥剂。
[0077]
将中试样品分别置于光照4500lx
±
500lx、高温60℃、相对湿度75%
±
1%,温度25℃条件下于0天、10天对其性状、有关物质、缩水甘油醛、溶出度及含量等进行测定。自制25mg规格样品影响因素结果见表8,自制12.5mg规格样品结果见表9,原研品(规格:25mg)结果见表10。
[0078]
表8:
[0079][0080][0081]
表9:
[0082][0083]
表10:
[0084][0085]
上述影响因素对比试验结果表明,原研药品在光照、高温、高湿10天条件下,有关物质总杂从0.85增长到0.97,有所增加,且起点较高;在光照10天时缩水甘油醛从1.2增长到3.3,增长明显,其他指标无显著变化。自制制剂在光照、高温、高湿10天条件下,有关物质总杂从0.11增长到0.15,无显著变化;在光照10天时缩水甘油醛从1.1增长到1.7(12.5mg规格),略有增加,其他指标无显著变化。自制制剂起始产品质量明显优于原研品,自制制剂杂质涨幅明显低于原研品,影响因素稳定性优于原研品。
[0086]
实施例5
[0087]
按照实施例4处方分别制备两个规格各三批产业化样品,采用口服固体药用高密度聚乙烯瓶包装,进行加速(40℃
±
2℃、rh75
±
5%)试验,并与原研品进行对比,自制制剂加速试验结果见表11(规格:25mg)、表12(规格:12.5mg),原研品(规格:25mg)加速实验结果见表13。
[0088]
表11:
[0089][0090][0091]
表12:
[0092][0093]
表13:
[0094][0095]
上述结果表明,自研产品与原研产品(阿斯利康生产的草酸纳洛解片)0天质量对比研究的结果表明,自研产品质量不低于阿斯利康原研上市样品。在加速6月期间,自制制剂有关物质和缩水甘油醛杂质量和增长幅度均明显小于原研品。性状、溶出度、稳定剂棓丙酯含量和活性成分含量等指标无显著变化,均符合规定。
[0096]
实施例6
[0097]
按照实施例4处方分别制备两个规格各三批产业化样品,采用口服固体药用高密度聚乙烯瓶包装,进行长期(25℃,rh60%)试验,自制制剂长期试验结果见表14(规格:25mg)、表15(规格:12.5mg)。
[0098]
表14:
[0099]
[0100][0101]
表15:
[0102][0103]
结果显示,两种规格各三批的自研草酸纳洛解片在长期稳定性条件下,杂质涨幅较小,各指标均符合限度要求,表明本处方工艺可行,产品质量可靠,稳定性良好。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1