纤维束的制作方法
背景技术:
已做出重大努力来生产低密度聚烯烃纤维,以改善成品中天然资源的使用和降低碳排放量。生产低密度聚烯烃纤维的典型方法是使用物理或化学发泡剂来使聚合物发泡,所述发泡剂产生穿过本体的气室。化学发泡剂是进行释放气体的化学反应的化合物,该气体穿过聚合物本体产生室结构。物理发泡剂通常是分散在聚合物中并膨胀产生室的压缩气体。无论如何,典型的发泡方法引起低的分子取向,因为室的形成发生在聚合物处于熔融状态时。这阻止了聚合物发生通常发生在远高于聚合物熔融温度或玻璃化转变温度的温度下的应变硬化,从而产生具有低机械强度的产品。此外,典型的发泡方法产生大的室尺寸,诸如大于100μm。这降低了熔体强度,由此导致纤维在纺丝过程中的断裂。
因此,目前需要在聚烯烃纤维中产生多孔结构的改进的技术,使得它们可具有较低的密度。
技术实现要素:
根据本发明的一个实施例,公开了一种纤维束,该纤维束包含围绕纵向轴线扭转的多根纤维。纤维的至少一部分由热塑性组合物形成,该热塑性组合物包含含有聚烯烃基体聚合物的连续相和以离散结构域的形式分散在连续相中的纳米包合物添加剂。在所述组合物中限定包括多个纳米孔的多孔网络。
根据本发明的另一个实施例,公开了一种形成多孔纤维的方法,该方法包括在低于基体聚合物的熔融温度的温度下牵伸如上所述的纤维束,从而形成包括多个纳米孔的多孔网络。
本发明的其他特征和方面在下文更详细地讨论。
附图说明
针对本领域普通技术人员的本发明的完整且能够实现的公开内容(包括其最佳模式)在说明书的剩余部分中参照附图更具体地阐述,在附图中:
图1是可采用本发明的纤维的吸收制品的一个实施例的透视图;
图2是可用于本发明的一个实施例以形成纤维的方法的示意图;
图3是可根据本发明的一个实施例牵伸的一束纤维的示意图;以及
图4-5是实例1的纤维的sem显微照片。
具体实施方式
现在将详细参照本发明的各种实施例,其一个或多个例子在下文示出。每个例子都以解释本发明而不是限制本发明的方式提供。事实上,对于本领域技术人员显而易见的是,在不背离本发明的范围或精神的情况下,可以在本发明中做出各种修改和变化。例如,作为一个实施例的一部分而说明或描述的特征,可以用于另一个实施例以产生另一个实施例。因此,本发明打算覆盖落入所附权利要求书及其等同物的范围内的这样的修改和变化。
一般来讲,本发明涉及包含多根纤维的纤维束。该纤维由热塑性组合物形成,该热塑性组合物包含连续相和纳米包合物添加剂,该连续相含有聚烯烃基体聚合物,该纳米包合物添加剂至少部分地与聚烯烃基体聚合物不相容以使得其变成作为离散的纳米级相结构域分散在连续相中。在某些条件下牵伸时,本发明人已发现,这些纳米级相结构域能够以独特的方式相互作用而在纤维的内壁中形成孔的网络。也就是说,据信,在牵伸过程中所经历的伸长应变由于材料的不相容性引起的应力集中可以在离散相结构域附近形成强局部剪切区和/或应力密集区(例如,法向应力)。这些剪切和/或应力密集区导致在邻近结构域的聚烯烃基体中的一些初始剥离。一旦形成了初步的孔,位于结构域之间的基体便可塑性变形以形成局部变窄(或颈缩)和应变硬化的内部拉伸区域。该过程使得可以形成穿过内壁本体的以拉伸方向生长的孔,从而导致形成多孔网络同时分子取向导致应变硬化,该应变硬化增加机械强度。
本发明人还已发现,在形成多孔网络的过程中纤维的断裂程度可通过选择性控制纤维束的性质而最大程度降低。例如,纤维束通常包含约5根或更多根纤维,在一些实施例中约50根或更多根纤维,在一些实施例中从约100至约1000根纤维以及在一些实施例中从约200至约800根纤维。无论所用的数量如何,束中的纤维基本上沿着纵向轴线取向并围绕纵向轴线扭转在一起。参见图3,例如,在一个实施例中,示出了包含围绕纵向轴线“l”扭转在一起的三(3)根单独纤维310的束300。扭转的性质可根据需要而变化。在某些实施例中,例如,可以采用螺旋扭转(例如,圆锥螺旋、圆柱螺旋等)来形成纤维束。当采用时,螺旋扭转角可在约0.1°至约20°、在一些实施例中从约0.2°至约10°以及在一些实施例中从约0.5°至约5°之间变化。作为束围绕纵向轴线完成一个完整旋转的线性距离的间距可同样在从约1至约300转/米、在一些实施例中从约2至约200转/米以及在一些实施例中从约5至约100转/米之间变化。束的总纤维纤度也可在从约1至约30千克每9000米、在一些实施例中从约2至约20千克每9000米以及在一些实施例中从约5至约15千克每9000米的范围内。
一旦形成后,纤维束之后便可被牵伸以在纤维中形成稳定的多孔网络。例如,在给定的单位体积的纤维中由孔占据的平均体积百分比可为从约15%至约80%每cm3,在一些实施例中从约20%至约70%以及在一些实施例中从约30%至约60%每立方厘米纤维。通过这样的孔体积,纤维可具有相对低的密度,诸如约0.90克/立方厘米(“g/cm3”)或更低,在一些实施例中约0.85g/cm3或更低,在一些实施例中约0.80g/cm3或更低,在一些实施例中从约0.10g/cm3至约0.75g/cm3以及在一些实施例中从约0.20g/cm3至约0.70g/cm3。该多孔网络中的大部分孔还具有“纳米级”尺寸(“纳米孔”),诸如具有约800纳米或更小,在一些实施例中从约5至约700纳米以及在一些实施例中从约10至约500纳米的平均横截面尺寸的那些。术语“横截面尺寸”通常是指孔的特性尺寸(例如宽度或直径),该特性尺寸与其主轴(例如长度)基本正交并且通常还与在拉伸过程中施加的应力的方向基本正交。纳米孔还可例如具有从约100至约5000纳米,在一些实施例中从约50至约2000纳米以及在一些实施例中从约100至约1000纳米范围内的平均轴向尺寸。“轴向尺寸”是主轴(例如,长度)方向上的尺寸,该方向通常为牵伸方向。这样的纳米孔可例如占纤维中的总孔体积的约15vol.%或更多,在一些实施例中约20vol.%或更多,在一些实施例中从约30vol.%至100vol.%以及在一些实施例中从约40vol.%至约90vol.%。
现在将更详细地描述本发明的各种实施例。
i.热塑性组合物
a.聚烯烃基体
聚烯烃通常占热塑性组合物的从约60wt.%至约99wt.%,在一些实施例中从约60wt.%至约98wt.%以及在一些实施例中从约80wt.%至约95wt.%。聚烯烃可以具有从约100℃至约220℃,在一些实施例中从约120℃至约200℃以及在一些实施例中从约140℃至约180℃的熔融温度。熔融温度可以根据astmd-3417采用差示扫描量热法("dsc")来测定。适合的聚烯烃可以例如包括乙烯聚合物(例如,低密度聚乙烯(“ldpe”)、高密度聚乙烯(“hdpe”)、线性低密度聚乙烯“lldpe”)等)、丙烯均聚物(例如,间同立构、无规立构、全同立构等)、丙烯共聚物等等。在一个特定的实施例中,聚合物为丙烯聚合物,诸如均聚丙烯或丙烯共聚物。丙烯聚合物可以例如由基本上全同立构的聚丙烯均聚物或含有等于或低于约10wt.%的其他单体(即,按重量计至少约90%的丙烯)的共聚物形成。这样的均聚物可以具有从约140℃至约170℃的熔点。
当然,其他聚烯烃也可用在本发明的组合物中。在一个实施例中,例如,聚烯烃可以是乙烯或丙烯与另一种α-烯烃诸如c3-c20α-烯烃或c3-c12α-烯烃的共聚物。适合的α-烯烃的具体例子包括1-丁烯;3-甲基-1-丁烯;3,3-二甲基-1-丁烯;1-戊烯;具有一个或多个甲基、乙基或丙基取代基的1-戊烯;具有一个或多个甲基、乙基或丙基取代基的1-己烯;具有一个或多个甲基、乙基或丙基取代基的1-庚烯;具有一个或多个甲基、乙基或丙基取代基的1-辛烯;具有一个或多个甲基、乙基或丙基取代基的1-壬烯;乙基、甲基或二甲基取代的1-癸烯;1-十二碳烯以及苯乙烯。特别期望的α-烯烃共聚单体是1-丁烯、1-己烯和1-辛烯。这样的共聚物的乙烯或丙烯含量可以为从约60摩尔%至约99摩尔%,在一些实施例中从约80摩尔%至约98.5摩尔%以及在一些实施例中从约87摩尔%至约97.5摩尔%。α-烯烃的含量可同样在从约1摩尔%至约40摩尔%,在一些实施例中从约1.5摩尔%至约15摩尔%以及在一些实施例中从约2.5摩尔%至约13摩尔%的范围内。
用于本发明的示例性烯烃共聚物包括可以以名称exacttm得自texas的houston的exxonmobilchemicalcompany的基于乙烯的共聚物。其他适合的乙烯共聚物可以以名称engagetm、affinitytm、dowlextm(lldpe)和attanetm(uldpe)得自michigan的midland的dowchemicalcompany。其他适合的乙烯聚合物在授予ewen等人的美国专利号4,937,299、授予tsutsui等人的美国专利号5,218,071、授予lai等人的美国专利号5,272,236和授予lai等人的美国专利号5,278,272中有所描述。适合的丙烯共聚物也可以从texas的houston的exxonmobilchemicalco.以名称vistamaxxtm;从belgium的feluy的atofinachemicals以名称finatm(例如,8573);从mitsuipetrochemicalindustries以tafmertm以及从michigan的midland的dowchemicalco.以versifytm商购获得。适合的聚丙烯均聚物可包括exxonmobil3155聚丙烯、exxonmobilachievetm树脂和totalm3661pp树脂。适合的丙烯聚合物的其他例子在授予datta等人的美国专利号6,500,563、授予yang等人的美国专利号5,539,056和授予resconi等人的美国专利号5,596,052中有所描述。
多种已知技术中的任一种通常都可以用于形成烯烃共聚物。例如,烯烃聚合物可以使用自由基或配位催化剂(例如,齐格勒-纳塔(ziegler-natta))形成。优选地,烯烃聚合物由单中心配位催化剂诸如茂金属催化剂形成。这样的催化剂体系产生这样的乙烯共聚物,其中共聚单体在分子链内无规分布而在不同分子量的部分中均匀分布。茂金属催化的聚烯烃在例如授予mcalpin等人的美国专利号5,571,619、授予davis等人的美国专利号5,322,728、授予obijeski等人的美国专利号5,472,775、授予lai等人的美国专利号5,272,236和授予wheat等人的美国专利号6,090,325中有所描述。茂金属催化剂的例子包括双(正丁基环戊二烯基)二氯化钛、双(正丁基环戊二烯基)二氯化锆、双(环戊二烯基)氯化钪、双(茚基)二氯化锆、双(甲基环戊二烯基)二氯化钛、双(甲基环戊二烯基)二氯化锆、二茂钴、环戊二烯基三氯化钛、二茂铁、二氯二茂铪、异丙基(环戊二烯基-1-芴基)二氯化锆、二氯二茂钼、二茂镍、二氯二茂银、二茂钌、二氯二茂钛、氢氯二茂锆、二氯二茂锆等。用茂金属催化剂制得的聚合物通常具有窄分子量范围。例如,茂金属催化的聚合物可以具有4以下的多分散性数值(mw/mn)、受控的短链支化分布以及受控的全同立构规整度。
b.纳米包合物添加剂
如本文所用,术语“纳米包合物添加剂”通常是指能够以纳米级大小的离散结构域的形式分散在聚合物基体内的材料。例如,在牵伸之前,结构域可以具有从约1至约1000纳米,在一些实施例中从约5至约800纳米,在一些实施例中从约10至约500纳米以及在一些实施例中从约20至约200纳米的平均横截面尺寸。结构域可具有多种不同的形状,诸如椭圆形、球形、圆柱形、板状、管状等。在一个实施例中,例如,结构域具有大致椭圆形的形状。按连续相聚烯烃基体的重量计,纳米包合物添加剂通常的用量为热塑性组合物的从约0.05wt.%至约20wt.%,在一些实施例中从约0.1wt.%至约10wt.%以及在一些实施例中从约0.5wt.%至约5wt.%。纳米包合物添加剂在整个热塑性组合物中的浓度可同样为热塑性组合物的从约0.01wt.%至约15wt.%,在一些实施例中从约0.05wt.%至约10wt.%以及在一些实施例中从约0.3wt.%至约6wt.%。
纳米包合物添加剂在可基本上均匀地分散在聚合物基体中但以离散结构域的形式这个意义上来说部分地与聚烯烃不相容。这样的部分不相容性可以多种方式实现。在某些实施例中,例如,纳米包合物添加剂可具有与聚烯烃基体相容且使其变得均匀分散在该基体中的非极性组分(例如,烯烃)。然而,添加剂还可以包含与聚烯烃基体不相容的极性组分,从而使其聚结或离析成离散结构域。这样的组分可包括低或高分子量极性分子区段或区块、离子基团、带电或不带电的极性结构域和/或极性分子基团。作为另外一种选择,添加剂在性质上可以为完全非极性的,但具有仍允许形成离散结构域的某些物理性质。例如,在某些实施例中,纳米包合物添加剂可在某一温度之上与聚烯烃相容或混溶,但在低于临界溶液温度的温度下发生相分离。这样,纳米包合物添加剂可在熔融相中与聚烯烃形成稳定的共混物,但随着温度增加,连续相发生结晶并离析使得纳米包合物添加剂可以发生相分离、聚结和形成分开的纳米级结构域。
纳米包合物添加剂的特定状态或形式不是关键性的,只要可以形成所需的结构域即可。例如,在一些实施例中,纳米包合物添加剂在室温(例如,25℃)下可以是液体或半固体的形式。这样的液体可以容易地分散在基体中以形成亚稳定分散体,然后通过降低共混物的温度而猝灭以保持结构域大小。这样的液体或半固体材料的运动粘度在40℃下测定时通常为从约0.7至约200厘沲(“cs”),在一些实施例中从约1至约100cs以及在一些实施例中从约1.5至约80cs。适合的液体或半固体可包括例如硅氧烷、硅氧烷-聚醚共聚物、脂族聚酯、芳族聚酯、亚烷基二醇类(例如乙二醇、二甘醇、三甘醇、四甘醇、丙二醇、聚乙二醇、聚丙二醇、聚丁二醇等)、链烷二醇(例如,1,3-丙二醇、2,2-二甲基-1,3-丙二醇、1,3-丁二醇、1,4-丁二醇、1,5-戊二醇、1,6-己二醇、2,2,4-三甲基-1,6-己二醇、1,3-环己烷二甲醇、1,4-环己烷二甲醇、2,2,4,4-四甲基-1,3-环丁二醇等)、氧化胺(例如,辛基二甲基氧化胺)、脂肪酸酯、脂肪酸酰胺(例如,油酸酰胺、芥酸酰胺、硬脂酰胺、亚乙基双(十八酰胺)等)、矿物和植物油等。一种特别适合的液体或半固体是聚醚多元醇,诸如可以从basfcorp.以商品名
在另外其他实施例中,纳米包合物添加剂为固体的形式,其可以是无定形、结晶或半结晶的。例如,纳米包合物添加剂在性质上可以是聚合的,并且具有相对高的分子量以帮助改善热塑性组合物的熔体强度和稳定性。如上所指出的那样,纳米包合物添加剂与聚烯烃基体部分地不相容。这样的添加剂的一个例子是微晶聚烯烃蜡,其通常衍生自乙烯和/或c3-c101-烯烃,诸如丙烯、1-丁烯、1-戊烯、1-己烯、1-庚烯、1-辛烯、1-壬烯和1-癸烯。微晶蜡通常具有相对低的熔融温度,诸如从约30℃至约150℃,在一些实施例中从约50℃至约140℃以及在一些实施例中从约80℃至约130℃。在这样低的熔融温度下,蜡可在熔融相时与聚烯烃形成可混溶的共混物,但随着温度增加且聚合物发生结晶或固化,蜡将离析并聚结,从而形成分开的纳米级结构域。
聚合型纳米包合物添加剂的另一个例子是包含极性和非极性组分的官能化聚烯烃。极性组分可以例如由一个或多个官能团提供,并且非极性组分可以由烯烃提供。纳米包合物添加剂的烯烃组分通常可由任何直链或支链α-烯烃单体、源自烯烃单体的低聚物或聚合物(包括共聚物)形成,如上所述。纳米包合物添加剂的官能团可以是任何基团、分子链段和/或嵌段,其向分子提供极性组分并且与聚烯烃基体聚合物是不相容的。与聚烯烃不相容的分子链段和/或嵌段的例子可以包括丙烯酸酯、苯乙烯、聚酯、聚酰胺等。官能团可以具有离子特性并包括带电的金属离子。特别适合的官能团是马来酸酐、马来酸、富马酸、马来酰亚胺、马来酰肼、马来酸酐和二胺的反应产物、甲基降冰片烯二酸酐、二氯马来酸酐、马来酸酰胺等。马来酸酐改性的聚烯烃特别适用于本发明。这样的改性的聚烯烃通常通过将马来酸酐接枝到聚合物主链材料上形成。这样的马来酸化的聚烯烃以名称
在某些实施例中,聚合型纳米包合物添加剂还可以是反应性的。这样的反应性纳米包合物添加剂的一个例子是聚环氧化物,其每分子平均含有至少两个环氧乙烷环。无意受理论的限制,据信,这样的聚环氧化物分子可与组合物的某些组分发生反应(例如,链延伸、侧链支化、接枝、共聚物形成等)以改善熔体强度,而不明显降低玻璃化转变温度。反应性添加剂还可以在聚烯烃与其他极性更高的添加剂(诸如微米包合物添加剂)之间提供相容化,并可改善分散体的均匀性并降低微米包合物添加剂的大小。例如,如下文将更详细地描述的那样,本发明的某些实施例可采用聚酯作为微米包合物添加剂。在这样的实施例中,反应性纳米包含物添加剂可使得能够通过聚酯的羧基末端基团实现亲核开环反应(酯化)或通过羟基实现亲核开环反应(醚化)。可以同样发生噁唑啉副反应以形成酯酰胺部分。通过这样的反应,聚醚纳米包合物添加剂的分子量可以增加以抵消在熔融加工过程中通常观察到的降解。本发明人已发现,过多的反应可导致聚合物主链之间的交联。如果允许这样的交联进行到一定程度,所得的聚合物共混物可以变脆并难以加工成具有期望强度和伸长特性的纤维。
就这一点而言,本发明人已经发现具有相对低环氧官能度的聚环氧化物可以是特别有效的,环氧官能度可以通过“环氧当量”定量。环氧当量反映含有一分子环氧基团的树脂量,并且可以通过将改性剂的数均分子量除以分子中环氧基团的数量来计算。本发明的聚环氧化物通常具有从约7,500至约250,000克每摩尔,在一些实施例中从约15,000至约150,000克每摩尔以及在一些实施例中从约20,000至100,000克每摩尔的数均分子量,而多分散指数通常在从2.5至7的范围内。聚环氧化物可以含有小于50,在一些实施例中从5至45以及在一些实施例中从15至40个环氧基团。反过来,环氧当量可以小于约15,000克每摩尔,在一些实施例中从约200至约10,000克每摩尔以及在一些实施例中从约500至约7,000克每摩尔。
聚环氧化物可以是包含末端环氧基团、骨架环氧乙烷单元和/或环氧侧基的直链或支链均聚物或共聚物(例如,无规、接枝、嵌段等)。用于形成这样的聚环氧化物的单体可以有所不同。在一个特定的实施例中,例如,聚环氧化物含有至少一种环氧官能化(甲基)丙烯酸单体组分。如本文所用,术语“(甲基)丙烯酸”包括丙烯酸和甲基丙烯酸单体,及其盐或酯,诸如丙烯酸酯和甲基丙烯酸酯单体。例如,适合的环氧官能化(甲基)丙烯酸单体可以包括但不限于含有1,2-环氧基团的那些,诸如丙烯酸缩水甘油酯和甲基丙烯酸缩水甘油酯。其他适合的环氧官能化单体包括烯丙基缩水甘油醚、乙基丙烯酸缩水甘油酯和衣康酸缩水甘油酯。
如上所指出的那样,聚环氧化物通常具有相对高的分子量,以使其不仅可以导致链延伸,还可以帮助实现期望的共混物形态。因此,在2160克的负荷和190℃的温度下测定时,产生的聚合物的熔体流动速率通常在从约10至约200克每10分钟,在一些实施例中从约40至约150克每10分钟以及在一些实施例中从约60至约120克每10分钟的范围内。
聚环氧化物通常还包含至少一种直链或支链α-烯烃单体,诸如具有从2至20个碳原子优选地从2至8个碳原子的那些。具体的例子包括乙烯、丙烯、1-丁烯;3-甲基-1-丁烯;3,3-二甲基-1-丁烯;1-戊烯;具有一个或多个甲基、乙基或丙基取代基的1-戊烯;具有一个或多个甲基、乙基或丙基取代基的1-己烯;具有一个或多个甲基、乙基或丙基取代基的1-庚烯;具有一个或多个甲基、乙基或丙基取代基的1-辛烯;具有一个或多个甲基、乙基或丙基取代基的1-壬烯;乙基、甲基或二甲基取代的1-癸烯;1-十二烯以及苯乙烯。特别期望的α-烯烃共聚单体是乙烯和丙烯。另一种适合的单体可以包括非环氧官能化的(甲基)丙烯酸单体。这样的(甲基)丙烯酸单体的例子可以包括丙烯酸甲酯、丙烯酸乙酯、丙烯酸正丙酯、丙烯酸异丙酯、丙烯酸正丁酯、丙烯酸仲丁酯、丙烯酸异丁酯、丙烯酸叔丁酯、丙烯酸正戊酯、丙烯酸异戊酯、丙烯酸异冰片酯、丙烯酸正己酯、丙烯酸-2-乙基丁酯、丙烯酸-2-乙基己酯、丙烯酸正辛酯、丙烯酸正癸酯、丙烯酸甲基环己酯、丙烯酸环戊酯、丙烯酸环己酯、甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸-2-羟乙酯、甲基丙烯酸正丙酯、甲基丙烯酸正丁酯、甲基丙烯酸异丙酯、甲基丙烯酸异丁酯、甲基丙烯酸正戊酯、甲基丙烯酸正己酯、甲基丙烯酸异戊酯、甲基丙烯酸仲丁酯、甲基丙烯酸叔丁酯、甲基丙烯酸-2-乙基丁酯、甲基丙烯酸甲基环己酯、甲基丙烯酸肉桂酯、甲基丙烯酸巴豆酯、甲基丙烯酸环己酯、甲基丙烯酸环戊酯、甲基丙烯酸-2-乙氧基乙酯、甲基丙烯酸异冰片酯等以及它们的组合。
在本发明的一个尤其期望的实施例中,聚环氧化物是由环氧官能化(甲基)丙烯酸单体组分、α-烯烃单体组分和非环氧官能化(甲基)丙烯酸单体组分形成的三元共聚物。例如,聚环氧化物可以是乙烯-丙烯酸甲酯-甲基丙烯酸缩水甘油酯共聚物,其具有以下结构:
其中x、y和zζ为1或更大。
环氧官能化单体可以使用多种已知的技术形成聚合物。例如,含有极性官能团的单体可以接枝到聚合物主链上以形成接枝共聚物。这样的接枝技术在本领域中是熟知的并例如在美国专利号5,179,164中有所描述。在其他实施例中,可以使用已知的自由基聚合技术,诸如高压反应、齐格勒-纳塔催化剂反应体系、单位点催化剂(例如,金属茂)反应体系等使含有环氧官能团的单体与单体共聚以形成嵌段或无规共聚物。
可以选择一种或多种单体组分的相对部分以实现环氧反应性和熔体流动速率之间的平衡。更具体地讲,高环氧单体含量可以导致良好的反应性,但是太高的含量可以使熔体流动速率降低到这样的程度,该程度使得聚环氧化物不利地影响聚合物共混物的熔体强度。因此,在大多数实施例中,一种或多种环氧官能化(甲基)丙烯酸单体占共聚物的从约1wt.%至约25wt.%,在一些实施例中从约2wt.%至约20wt.%以及在一些实施例中从约4wt.%至约15wt.%。一种或多种α-烯烃单体可同样占共聚物的从约55wt.%至约95wt.%,在一些实施例中从约60wt.%至约90wt.%以及在一些实施例中从约65wt.%至约85wt.%。当采用时,其他单体组分(例如,非环氧官能化(甲基)丙烯酸单体)可以占共聚物的从约5wt.%至约35wt.%,在一些实施例中从约8wt.%至约30wt.%以及在一些实施例中从约10wt.%至约25wt.%。可以用于本发明的适合的聚环氧化物的一个具体例子可以以名称
除了控制用于形成聚环氧化物的单体的类型和相对含量以外,还可以控制总重量百分比以实现期望的益处。例如,如果改性水平太低,则可能无法实现熔体强度和机械特性的期望的提高。然而,本发明人还发现如果改性水平太高,则加工可由于强的分子相互作用(例如,交联)和环氧官能团引起的物理网络形成而受限。因此,按组合物中采用的聚烯烃的重量计,聚环氧化物通常的用量为从约0.05wt.%至约10wt.%,在一些实施例中从约0.1wt.%至约8wt.%,在一些实施例中从约0.5wt.%至约5wt.%以及在一些实施例中从约1wt.%至约3wt.%。按组合物的总重量计,聚环氧化物还可以占从约0.05wt.%至约10wt.%,在一些实施例中从约0.05wt.%至约8wt.%,在一些实施例中从约从0.1wt.%至约5wt.%以及在一些实施例中从约0.5wt.%至约3wt.%。
还可以在本发明中采用其他反应性纳米包合物添加剂,诸如噁唑啉官能化的聚合物、氰化物官能化的聚合物等。当采用时,这样的反应性纳米包合物添加剂可以在上文针对聚环氧化物所述的浓度内采用。在一个特定的实施例中,可以采用噁唑啉接枝的聚烯烃,其为使用含噁唑啉环的单体接枝的聚烯烃。噁唑啉可以包括2-噁唑啉,诸如2-乙烯基-2-噁唑啉(例如,2-异丙烯基-2-噁唑啉)、2-脂肪烷基-2-噁唑啉(例如,可得自油酸、亚油酸、棕榈油酸、鳕肝油酸、芥酸和/或花生四烯酸的乙醇酰胺)以及它们的组合。在另一个实施例中,例如,噁唑啉可以选自蓖麻油酸噁唑啉马来酸酯、十一基-2-噁唑啉、大豆-2-噁唑啉、蓖麻-2-噁唑啉以及它们的组合。在另一个实施例中,噁唑啉选自2-异丙烯基-2-噁唑啉、2-异丙烯基-4,4-二甲基-2-噁唑啉以及它们的组合。
在本发明的某些实施例中,多种纳米包合物添加剂可以按组合方式使用。例如,第一纳米包合物添加剂(例如,聚环氧化物)可以按平均横截面尺寸为从约50至约500纳米,在一些实施例中从约60至约400纳米以及在一些实施例中从约80至约300纳米的结构域的形式分散。第二纳米包合物添加剂还可以按比第一纳米包合物添加剂更小的结构域,诸如平均横截面尺寸为从约1至约50纳米,在一些实施例中从约2至约45纳米以及在一些实施例中从约5至约40纳米的结构域的形式分散。当采用时,按连续相(一种或多种基体聚合物)的重量计,第一和/或第二纳米包合物添加剂通常占热塑性组合物的从约0.05wt.%至约20wt.%,在一些实施例中从约0.1wt.%至约10wt.%以及在一些实施例中从约0.5wt.%至约5wt.%。第一和/或第二纳米包合物添加剂在整个热塑性组合物中的浓度可同样为热塑性组合物的从约0.01wt.%至约15wt.%,在一些实施例中从约0.05wt.%至约10wt.%以及在一些实施例中从约0.1wt.%至约8wt.%。
纳米填料可任选地用于第二纳米包合物添加剂,其例子可包括炭黑、碳纳米管、碳纳米纤维、纳米粘土、金属纳米粒子、纳米二氧化硅、纳米氧化铝等。纳米粘土是特别适合的。术语“纳米粘土”一般是指粘土材料的纳米颗粒(天然存在的矿物质、有机改性的矿物质或合成纳米材料),其通常具有薄片结构。纳米粘土的例子包括例如蒙脱石(2:1层状蒙皂石粘土结构)、膨润土(主要由蒙脱石形成的层状硅酸铝)、高岭土(具有板状结构和经验式al2si2o5(oh)4的1:1铝硅酸盐)、埃洛石(具有管状结构和经验式al2si2o5(oh)4的1:1铝硅酸盐)等。适合的纳米粘土的例子是
如果需要,纳米粘土可以包括表面处理以帮助改进与基体聚合物(例如,聚酯)的相容性。表面处理可以是有机或无机的。在一个实施例中,采用通过有机阳离子与粘土的反应获得的有机表面处理。适合的有机阳离子可以包括例如能够与粘土交换阳离子的有机季铵化合物,诸如二甲基双[氢化牛脂]氯化铵(2m2ht)、甲基苄基双[氢化牛脂]氯化铵(mb2ht)、氯化甲基三[氢化牛脂烷基](m3ht)等。可商购获得的有机纳米粘土的例子可以包括例如
无论所采用的材料如何,纳米包合物添加剂通常被选择成具有某一粘度(或熔体流动速率)以确保可充分地维持离散结构域和所得的孔。例如,如果纳米包合物添加剂的粘度过低(或熔体流动速率过高),则其倾向于在连续相中不可控地流动和分散。这导致难以维持层状、板状结构域或共连续相结构,并且还可能过早断裂。相反地,如果粘度过高(或熔体流动速率过低),则其倾向于聚集在一起并形成非常大的椭圆形结构域,这些结构域在共混期间是难以分散的。这可导致纳米包合物添加剂在整个连续相中的不均匀分布。例如,聚烯烃的熔体流动速率与聚合型纳米包合物添加剂的熔体流动速率之比例如可以为从约0.2至约8,在一些实施例中从约0.5至约6以及在一些实施例中从约1中约5。纳米包合物添加剂可例如在2160克的负荷和高于熔融温度至少约40℃的温度(例如,在190℃)下根据astmd1238测定时具有从约0.1至约100克每10分钟,在一些实施例中从约0.5至约50克每10分钟以及在一些实施例中从约5至约15克每10分钟的熔体流动速率(基于干燥状态)。聚烯烃可同样在2160克的负荷和高于熔融温度至少约40℃的温度(例如,在230℃)下根据astmd1238测定时具有从约0.5至约80克每10分钟,在一些实施例中从约1至约40克每10分钟以及在一些实施例中从约5至约20克每10分钟的熔体流动速率(基于干燥状态)。
c.微米包合物添加剂
虽然不是必需的,但是本发明的组合物也可采用微米包合物添加剂。如本文所用,术语“微米包合物添加剂”通常是指能够以微米级大小的离散结构域的形式分散在聚合物基体内的材料。例如,在牵伸前,结构域可具有从约0.1μm至约25μm,在一些实施例中从约0.5μm至约20μm以及在一些实施例中从约1μm至约10μm的平均横截面尺寸。当采用时,本发明人已发现,微米级和纳米级相结构域能够在受到变形和伸长应变(例如,牵伸)时以独特的方式相互作用以形成孔的网络。也就是说,据信,伸长应变由于材料的不相容性引起的应力集中可以在离散相结构域附近形成强局部剪切区和/或应力密集区(例如,法向应力)。这些剪切和/或应力密集区导致在邻近微米级结构域的聚烯烃基体中的一些初始剥离。然而,值得注意的是,在纳米级离散相结构域附近形成的局部剪切和/或应力密集区可与微米级区重叠而导致在聚合物基体中发生甚至进一步的剥离,从而在纳米级结构域和/或微米级结构域附近形成大量的纳米孔。
微米包合物添加剂的特定性质不是关键性的,并且可以包括液体、半固体或固体(例如,无定形、结晶或半结晶的)。在某些实施例中,微米包合物添加剂通常在性质上是聚合的并具有相对高的分子量,以帮助改善热塑性组合物的熔体强度和稳定性。通常,微米包合物添加剂聚合物一般不能与基体聚合物相容。以此方式,添加剂可以作为离散的相结构域在基体聚合物的连续相内更好地分散。离散结构域能够吸收由外力产生的能量,这增加了所得纤维的整体韧度和强度。结构域可具有多种不同的形状,诸如椭圆形、球形、圆柱形、板状、管状等。在一个实施例中,例如,结构域具有大致椭圆形的形状。各个结构域的物理尺寸通常足够小以使当施加外部应力时穿过纤维的裂纹传播减至最低限度,但又足够大以引发微观塑性变形并且允许在颗粒包合物处和周围出现剪切区。
微米包合物添加剂可以具有某一熔体流动速率(或粘度)以确保离散结构域和所得的孔可被适当地维持。例如,如果添加剂的熔体流动速率过高,则其倾向于在连续相中不可控地流动和分散。这导致难以维持层状、板状结构域或共连续相结构,并且还可能过早断裂。相反地,如果添加剂的熔体流动速率过低,则其倾向于聚集在一起并形成非常大的椭圆形结构域,这些结构域在共混期间是难以分散的。这可导致添加剂在整个连续相中的不均匀分布。就这一点而言,本发明人已经发现,微米包合物添加剂的熔体流动速率与基体聚合物的熔体流动速率之比通常为从约0.5至约10,在一些实施例中从约1至约8以及在一些实施例中从约2至约6。微米包合物添加剂可例如在2160克的负荷和高于其熔融温度至少约40℃的温度(例如,210℃)下测定时具有从约5至约200克每10分钟,在一些实施例中从约20至约150克每10分钟以及在一些实施例中从约40至约100克每10分钟的熔体流动速率。
除了以上所述的特性以外,也可选择微米包合物添加剂的机械性能,以实现期望的多孔网络。例如,施加外力时,可以在离散相结构域处和周围引起应力集中(例如包括法向应力或剪切应力)和剪切和/或塑性屈服区,这是因为由添加剂和基体聚合物的弹性模量的差异所引起的应力集中。较大的应力集中促进了在结构域中更强的局部塑性流动,这使得这些结构域在被赋予应力时变得明显伸长。这些伸长的结构域可使得组合物表现出更加柔韧和柔软的行为。为了增强应力集中,可将微米包合物添加剂选成具有比聚烯烃基体相对高的杨氏弹性模量。例如,添加剂的弹性模量与聚烯烃基体的弹性模量之比通常为从约1至约250,在一些实施例中从约2至约100以及在一些实施例中从约2至约50。微米包合物添加剂的弹性模量可以例如在从约200至约3,500兆帕(mpa),在一些实施例中从约300至约2,000mpa以及在一些实施例中从约400至约1,500mpa的范围内。相反,聚烯烃的弹性模量可例如在从约100至约1,500mpa以及在一些实施例中从约200至约1000mpa的范围内。作为另外一种选择,微米包合物添加剂的弹性模量可低于聚烯烃基体的弹性模量。该弹性模量可例如在从约10mpa至约100mpa以及任选地从约20mpa至约80mpa的范围内。
虽然可以采用具有上文所确定的性质的多种多样的微米包合物添加剂,但是这样的添加剂的尤其适合的例子可包括苯乙烯共聚物(例如,苯乙烯-丁二烯-苯乙烯、苯乙烯-异戊二烯-苯乙烯、苯乙烯-乙烯-丙烯-苯乙烯、苯乙烯-乙烯-丁二烯-苯乙烯等);含氟聚合物,诸如聚氯乙烯(pvc)、聚四氟乙烯(ptfe)、聚氯三氟乙烯(pctfe)等;聚乙烯醇;聚乙酸乙烯酯;聚酯,诸如脂族聚酯,诸如聚己内酯、聚酯酰胺、聚乳酸(pla)及其共聚物、聚乙醇酸、聚碳酸亚烷基酯(例如,聚碳酸亚乙酯)、聚-3-羟基丁酸酯(phb)、聚-3-羟基戊酸酯(phv)、3-羟基丁酸酯与4-羟基丁酸酯的共聚物、3-羟基丁酸酯与3-羟基戊酸酯的共聚物(phbv)、3-羟基丁酸酯与3-羟基己酸酯的共聚物、3-羟基丁酸酯与3-羟基辛酸酯的共聚物、3-羟基丁酸酯与3-羟基癸酸酯的共聚物、3-羟基丁酸酯与3-羟基十八烷酸酯的共聚物和基于琥珀酸酯的脂族聚合物(例如,聚琥珀酸丁二醇酯、聚琥珀酸己二酸丁二醇酯、聚琥珀酸乙二醇酯等)、脂族-芳族共聚酯(例如,聚己二酸对苯二酸丁二醇酯、聚己二酸对苯二酸乙二醇酯、聚己二酸间苯二酸乙二醇酯、聚己二酸间苯二酸丁二醇酯等)、芳族聚酯(例如,聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯等)等等。
尤其适合的是在性质上通常为刚性达到具有相对高玻璃化转变温度的程度的微米包合物添加剂。例如,玻璃化转变温度(“tg”)可为约0℃或更高,在一些实施例中从约5℃至约100℃,在一些实施例中从约30℃至约80℃以及在一些实施例中从约50℃至约75℃。玻璃化转变温度可以根据astme1640-09通过动态机械分析来测定。
一种特别适合的刚性聚酯是聚乳酸,其通常可源自乳酸的任何同分异构体的单体单元,诸如左旋乳酸(“l-乳酸”)、右旋乳酸(“d-乳酸”)、内消旋乳酸或它们的混合物。单体单元也可以由乳酸的任何同分异构体的酸酐形成,包括l-丙交酯、d-丙交酯、内消旋丙交酯或它们的混合物。这样的乳酸和/或丙交酯的环状二聚体也可以被采用。任何已知的聚合方法,诸如缩聚或开环聚合,均可以用来聚合乳酸。少量的扩链剂(例如,二异氰酸酯化合物、环氧化合物或酸酐)可以被采用。聚乳酸可以是均聚物或共聚物,诸如含有源自l-乳酸的单体单元和源自d-乳酸的单体单元的那些。虽然不要求,但源自l-乳酸的单体单元和源自d-乳酸的单体单元中之一的含量比率优选为约85摩尔%或更高,在一些实施例中从约90摩尔%或更高以及在一些实施例中从约95摩尔%或更高。多种聚乳酸可以以任意百分比共混,其中每种具有不同的源自l-乳酸的单体单元与源自d-乳酸的单体单元之比。当然,聚乳酸也可以与其他类型的聚合物(例如,聚烯烃、聚酯等)共混。
在一个特定的实施例中,聚乳酸具有以下通式结构:
本发明中可使用的适合的聚乳酸聚合物的一个具体例子可从德国krailling的biomer,inc.以名称biomertml9000商购获得。其他适合的聚乳酸聚合物可从minnesota的minnetonka的natureworksllc
聚乳酸通常具有在从约40,000至约180,000克每摩尔,在一些实施例中从约50,000至约160,000克每摩尔以及在一些实施例中从约80,000至约120,000克每摩尔范围内的数均分子量(“mn”)。同样,聚合物还通常具有从约80,000至约250,000克每摩尔,在一些实施例中从约100,000至约200,000克每摩尔以及在一些实施例中从约110,000至约160,000克每摩尔范围内的重均分子量(“mw”)。重均分子量与数均分子量之比(“mw/mn”),即“多分散性指数”也相对较低。例如,多分散性指数通常在从约1.0至约3.0,在一些实施例中从约1.1至约2.0以及在一些实施例中从约1.2至约1.8的范围内。重均分子量和数均分子量可以通过本领域技术人员已知的方法测定。
一些类型的净聚酯(例如,聚乳酸)可以从周围环境中吸收水,而使其具有按起始聚乳酸的干重计约500至600份每一百万份(“ppm”),或甚至更高的含水量。含水量可以用本领域已知的多种方式测定,诸如根据astmd7191-05,诸如下文所述的。因为在熔融加工期间水的存在可以使聚酯水解降解并降低其分子量,所以有时期望在共混之前干燥聚酯。在大多数实施例中,例如,期望可再生的聚酯在与微米包合物添加剂共混之前具有约300份每一百万份("ppm")或更低,在一些实施例中约200ppm或更低,在一些实施例中从约1至约100ppm的含水量。聚酯的干燥可以例如在从约50℃至约100℃以及在一些实施例中从约70℃至约80℃的温度下发生。
不论采用的材料如何,均可以对热塑性组合物中的微米包合物添加剂的相对百分比进行选择,以获得期望的特性,而不显著影响所得的组合物。例如,按组合物中所用的聚烯烃基体的重量计,微米包合物添加剂通常的用量为热塑性组合物的从约1wt.%至约30wt.%,在一些实施例中从约2wt.%至约25wt.%以及在一些实施例中从约5wt.%至约20wt.%。微米包合物添加剂在整个热塑性组合物中的浓度可同样占从约0.1wt.%至约30wt.%,在一些实施例中从约0.5wt.%至约25wt.%以及在一些实施例中从约1wt.%至约20wt.%。
d.其他组分
出于多种不同的原因,可在组合物中采用多种不同的成分。例如,在一个特定的实施例中,可在热塑性组合物中采用相间改性剂以帮助降低纳米包合物和/或微米包合物添加剂与聚烯烃基体之间的摩擦和连通程度,并因此增强剥离的程度和均匀性。以此方式,孔可以按更加均匀的方式遍布在组合物中。改性剂可以在室温(例如25℃)下呈液体或半固体形式,以使得该改性剂具有相对低的粘度,从而允许其更容易地掺入热塑性组合物中并容易迁移至聚合物表面。通过降低聚烯烃基体与添加剂之间的界面处的物理力,据信,改性剂的低粘度、疏水性可帮助促进剥离。如本文所用,术语“疏水的”通常是指具有约40°或更大,且在一些情况下约60°或更大的空气中水的接触角的材料。相反,术语“亲水的”通常是指具有小于约40°的空气中水的接触角的材料。用于测量接触角的一种适合的测试是astmd5725-99(2008)。
虽然不是必需的,但是相间改性剂可尤其适于采用微米包合物添加剂且纳米包合物添加剂为固体(例如,聚合物材料)的实施例。适合的疏水性、低粘度相间改性剂可包括例如上文提及的液体和/或半固体。一种特别适合的相间改性剂是聚醚多元醇,诸如可以从basfcorp.以商品名
当采用时,按连续相聚烯烃基体的重量计,相间改性剂可占热塑性组合物的从约0.1wt.%至约20wt.%,在一些实施例中从约0.5wt.%至约15wt.%以及在一些实施例中从约1wt.%至约10wt.%。相间改性剂在整个热塑性组合物中的浓度可同样占从约0.05wt.%至约20wt.%,在一些实施例中从约0.1wt.%至约15wt.%以及在一些实施例中从约0.5wt.%至约10wt.%。按照上述量,相间改性剂具有这样的特性,该特性能够使其容易迁移至聚合物的界面表面并且在不破坏热塑性组合物的整体熔融特性的情况下促进剥离。例如,热塑性组合物的熔体流动速率也可以与聚烯烃基体的熔体流动速率相似。例如,在2160克的负荷和190℃的温度下根据astmd1238测定时,组合物的熔体流动速率(基于干燥状态)可以为从约0.1至约250克每10分钟,在一些实施例中从约0.5至约200克每10分钟以及在一些实施例中从约5至约150克每10分钟。
也可采用增容剂,增容剂改善结构域与基体之间的界面粘附并降低结构域与基体之间的界面张力,从而允许在混合过程中形成较小的结构域。适合的增容剂的例子可以包括例如用环氧部分或马来酸酐化学部分官能化的共聚物。马来酸酐增容剂的一个例子是丙烯-马来酸酐接枝共聚物,其可以从arkema以商品名orevactm18750和orevactmca100商购获得。当采用时,按连续相基体的重量计,增容剂可占热塑性组合物的从约0.05wt.%至约10wt.%,在一些实施例中从约0.1wt.%至约8wt.%以及在一些实施例中从约0.5wt.%至约5wt.%。
如果需要,也可采用可以充当聚烯烃基体的增塑剂的丁烯聚合物(均聚物或共聚物),从而改善其流动性和延展性,并继而改善其以相对高的速度加工而不在很大程度上断裂的能力。当采用时,按连续相(例如,一种或多种基体聚合物)的重量计,此类丁烯聚合物通常占从约0.01wt.%至约15wt.%,在一些实施例中从约0.1wt.%至约12wt.%以及在一些实施例中从约0.5wt.%至约10wt.%。按组合物的总重量计,丁烯聚合物还可以按从约0.01wt.%至约15wt.%,在一些实施例中从约0.1wt.%至约12wt.%以及在一些实施例中从约0.5wt.%至约10wt.%的量使用。
术语“丁烯聚合物”泛指具有四个碳原子的烯烃单体的均聚物或共聚物,所述烯烃单体包括1-丁烯(α-丁烯)、2-丁烯(顺式β-丁烯或反式β-丁烯)、2-甲基丙烯(异丁烯)、环丁烯以及它们的组合。丁烯聚合物也可以含有其他单体,诸如丙烯。在一个实施例中,例如,丁烯聚合物可以是1-丁烯的均聚物(称为“聚丁烯”)。其他合适的聚合物可包括异丁烯的均聚物(称为“聚异丁烯”)和1-丁烯、2-丁烯和/或异丁烯的共聚物(称为“聚丁烯”)。在一个特定的实施例中,例如,丁烯聚合物可以是聚丁烯(也称为聚-1-丁烯)。聚丁烯可通过1-丁烯的齐格勒-纳塔(ziegler-natta)低压聚合而制备,诸如通过用催化剂ticl3、ticl3alcl3和/或al(c2h5)2cl使1-丁烯聚合。虽然丁烯聚合物的分子量可基于聚合物的类型和聚合程度而变化,但是通常希望的是,丁烯聚合物具有相对低的分子量,诸如约10,000克/摩尔或更低、在一些实施例中约5,000克/摩尔或更低、在一些实施例中约2,000克/摩尔或更低以及在一些实施例中从约100至约1,000克/摩尔的数均分子量。当然,应当理解的是,也可以采用较高分子量的聚合物,诸如具有从约10,000至约250,000克/摩尔的数均分子量的那些聚合物。丁烯聚合物的密度也通常为约0.910克/立方厘米或更低、在一些实施例中约0.900克/立方厘米或更低以及在一些实施例中从约0.810至约0.890克/立方厘米,如根据astmd4052-11所测定的。丁烯聚合物的运动粘度可同样在从约50至约2,000厘沲("cs")、在一些实施例中从约100至约1,500cs以及在一些实施例中从约200至约1,000cs的范围内,如在40℃的温度下根据astmd445-14e2所测定的。
也可在热塑性组合物中采用其他适合的材料,诸如催化剂、抗氧化剂、稳定剂、表面活性剂、蜡、固溶剂、螯合剂、微粒、纳米填料以及被加入以增强热塑性组合物的可加工性和机械特性的其他材料。然而,本发明的一个有益方面是可在不需要各种传统添加剂诸如发泡剂(例如氯氟烃、氢氯氟烃、烃、二氧化碳、超临界二氧化碳、氮气等)和孔引发填料(例如,碳酸钙)。事实上,热塑性组合物通常可以不含发泡剂和/或孔引发填料。例如,这样的发泡剂和/或填料存在的量可以为热塑性组合物的不多于约1wt.%,在一些实施例中不多于约0.5wt.%以及在一些实施例中从约0.001wt.%至约0.2wt.%。另外,由于最终组合物的如在下文更详细描述的应力发白特性,所得的组合物可以在无需传统颜料诸如二氧化钛的情况下获得不透明颜色(例如白色)。在某些实施例中,例如,颜料可以按热塑性组合物的不多于约1wt.%,在一些实施例中不多于约0.5wt.%以及在一些实施例中从约0.001wt.%至约0.2wt.%的量存在。
ii.共混
为了形式热塑性组合物,通常采用多种已知技术中的任一种将组分共混在一起。在一个实施例中,例如,组分可以被单独提供或以组合方式提供。例如,组分可首先被干混在一起以形成基本均匀的干燥混合物,并且它们可同样地被同时或陆续供应至将材料分散共混的熔融加工装置内。可采用分批的和/或连续的熔融加工技术。例如,可利用混合机/捏合机、班布里混合机、法劳连续混合机、单螺杆挤出机、双螺杆挤出机、滚碎机等来共混和熔融加工这些材料。特别适合的熔融加工装置可以是共旋转双螺杆挤出机(例如可得自newjersey的ramsey的werner&pfleiderercorporation的zsk-30挤出机或可得自英国stone的thermoelectroncorp.的thermoprismtmusalab16挤出机)。这样的挤出机可包括进料口和通风口并提供高强度分布式和分散式混合。例如,组分可被进料至双螺杆挤出机的相同或不同的进料口并熔融共混以形成基本均匀的熔融混合物。如果需要,其他添加剂也可被注入到聚合物熔体中和/或在沿挤出机长度的不同位置处被分别进料至挤出机中。
无论所选的特定加工技术如何,所得的熔融共混组合物通常包含纳米包合物添加剂的纳米级结构域和任选地微米包合物添加剂的微米级结构域。可控制剪切/压力程度和热量来确保充分分散,但不会高到不利地降低结构域尺寸以使得它们不能实现期望特性的程度。例如,共混通常在从约180℃至约300℃,在一些实施例中从约185℃至约250℃,在一些实施例中从约190℃至约240℃的温度下进行。同样,在熔体加工过程中的表观剪切速率可以在从约10-1至约3000s-1,在一些实施例中从约50s-1至约2000s-1以及在一些实施例中从约100s-1至约1200s-1的范围内。表观剪切速率可以等于4q/□r3,其中q是聚合物熔体的体积流动速率(“m3/s”),而r是熔融聚合物所流动经过的毛细管(例如,挤出机模头)的半径(“m”)。当然,也可控制其他变量,诸如与生产率成反比的在熔融加工过程中的停留时间,以实现期望程度的均质性。
为了获得期望的剪切条件(例如速率、停留时间、剪切速率、熔融加工温度等),一个或多个挤出机螺杆的速率可在一定范围内选择。通常,随着螺杆速度的增加,由于额外的机械能输入系统中,可观察到产品温度的升高。例如,螺杆速度可以在从约50至约600转每分钟(“rpm”),在一些实施例中从约70至约500rpm以及在一些实施例中从约100至约300rpm的范围内。这可在不负面影响所产生结构域的尺寸的情况下产生用于分散纳米包合物添加剂的足够高的温度。熔体剪切速率以及还有添加剂被分散的程度也可通过在挤出机的混合部段中使用一个或多个分布式和/或分散式混合元件来增加。用于单螺杆挤出机的适合的分布式混合机可包括例如saxon混合机、dulmage混合机、cavitytransfer混合机等。同样,适合的分散式混合机可包括blisterring混合机、leroy/maddock混合机、crd混合机等。如在本领域中熟知的那样,可通过在圆桶中使用导致聚合物熔体折叠和再取向的销钉来进一步改善混合,诸如在busskneader挤出机、cavitytransfer混合机和vortexintermeshingpin(vip)混合机中所使用的那些。
iii.纤维形成
如本文所用,术语“纤维”通常是指通过使聚合物穿过成型孔口诸如模头而形成的细长挤出物。除非另外指明,否则术语“纤维”既包括具有一定长度的不连续纤维也包括基本上连续的长丝。基本上连续的长丝可例如具有远大于其直径的长度,诸如大于约15,000至1以及在一些情况下大于约50,000至1的长度与直径比(“长宽比”)。如果需要,纤维可以是“中空的”达到其包括沿着纤维的至少一部分在纵向方向上延伸的中空腔体的程度。在一些情况下,腔体可沿着纤维的整个长度延伸。
由热塑性组合物形成的纤维通常可以具有任何所需的构型,包括单组分和多组分(例如,皮芯构型、并排构型、分割饼式构型、海中岛构型等)。也可以采用中空纤维(单组分和/或多组分),诸如在授予carter等人的美国专利no.6,642,429中所述。在一些实施例中,纤维可包含一种或多种另外的聚合物作为其组分(例如,双组分)或成分(例如,双成分)以进一步增强强度、可加工性和/或其他性质。例如,热塑性组合物可形成皮/芯双组分纤维的芯组分,而另外的聚合物可形成皮组分,或反之亦然。该另外的聚合物可以是任何所需的聚合物,诸如聚酯(例如,聚乳酸、聚对苯二甲酸乙二醇酯等);聚烯烃(例如,聚乙烯、聚丙烯、聚丁烯等);聚四氟乙烯;聚乙酸乙烯酯;聚氯乙烯-乙酸乙烯酯;聚乙烯醇缩丁醛;丙烯酸树脂(例如,聚丙烯酸酯、聚丙烯酸甲酯、聚甲基丙烯酸甲酯等);聚酰胺(例如,尼龙);聚氯乙烯;聚偏二氯乙烯;聚苯乙烯;聚乙烯醇;和聚氨酯。
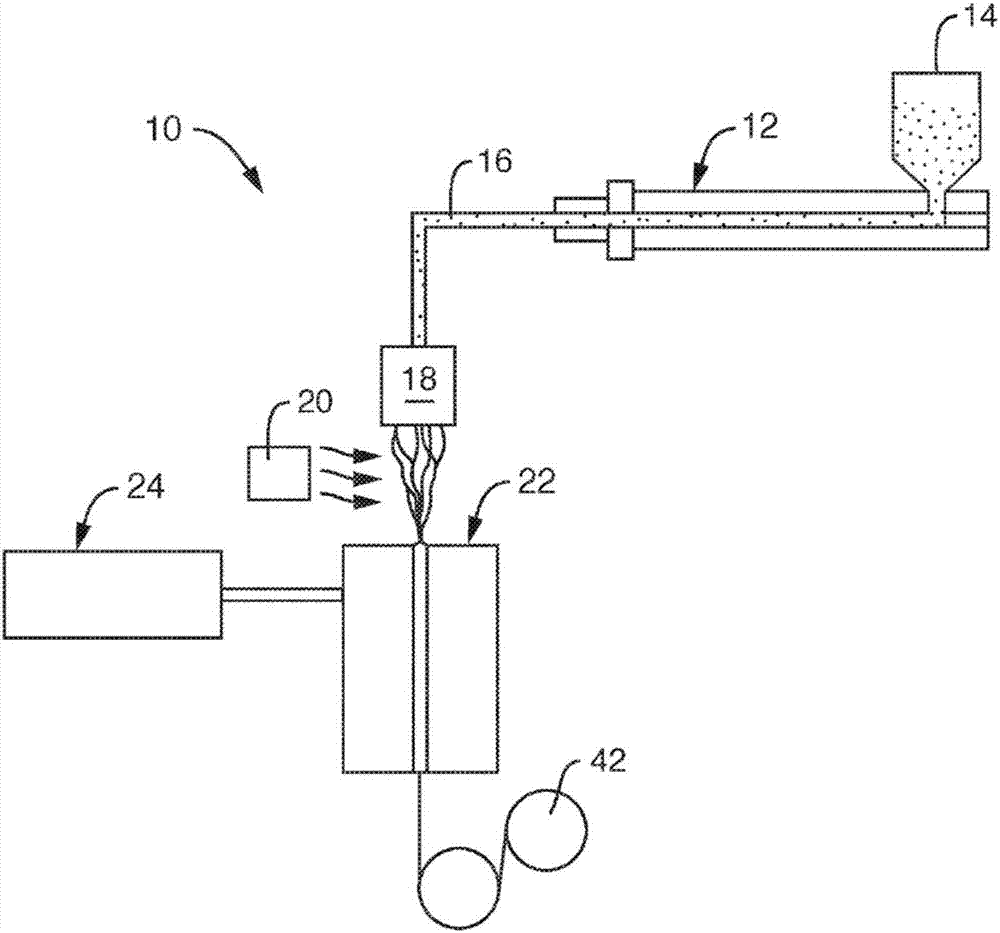
无论其特定配置如何,多种方法中的任一种均可用于形成本发明的纤维。例如,可使用这样的方法形成纤维,其中将热塑性组合物通过模头系统(或喷丝头)挤出,该系统可包括容纳纺丝组的外壳,纺丝组具有多个彼此堆叠的板并具有被布置成形成流路以引导热塑性组合物的毛细管图案。参见图2,例如,更详细地示出了用于形成纤维的方法的一个实施例。在该特定实施例中,本发明的热塑性组合物可从料斗14进料到挤出机12中。可使用任何常规技术向料斗14提供共混物。将挤出机12加热到足以挤出熔融聚合物的温度。通常将组合物在从约180℃至约300℃,在一些实施例中从约200℃至约260℃以及在一些实施例中从约210℃至约250℃的温度下熔纺。
然后使挤出的组合物穿过聚合物导管16通向喷丝头18,该喷丝头具有以一行或多行布置的开口。这些开口在当聚合物被从中挤出时形成向下挤出的长丝帘。方法10还采用紧邻从喷丝头18延伸的纤维帘定位的骤冷鼓风机20。来自骤冷鼓风机20的空气使从喷丝头18延伸的纤维骤冷。骤冷空气可从纤维帘的一侧导入(如图2所示)或从纤维帘的两侧导入。为了形成具有所需长度的纤维,通常将骤冷后的纤维熔融牵伸,诸如使用如图2所示的纤维牵伸单元22。用于熔纺聚合物的纤维牵伸单元或抽吸器是本领域熟知的。适用于本发明的方法的纤维牵伸单元包括美国专利号3,802,817和3,423,255中所示类型的线性纤维抽吸器。纤维牵伸单元22通常包括细长的竖直通道,通过抽吸空气从通道侧面进入并穿过通道向下流动而从通道中牵伸纤维。加热器或鼓风机24为纤维抽吸单元22供应抽吸空气。抽吸空气熔融牵伸纤维和环境空气通过纤维牵伸单元22。气体的流动导致纤维熔融牵伸或缩减,这增加形成纤维的聚合物的分子取向或结晶度。当采用纤维牵伸单元时,可选择“拉伸”比以有助于实现所需的纤维长度。“拉伸”比是纤维被牵伸后的线性速度(例如,导丝辊42或多孔表面(未示出)的线性速度)除以纤维被挤出后的线性速度。例如,在熔融牵伸过程中的拉伸比可如下计算:
拉伸比=a/b
其中,
a是纤维被熔融牵伸后的线性速度(例如,导丝速度)且直接测量;而
b是挤出后的纤维的线性速度且可如下计算:
挤出机线性纤维速度=c/(25*π*d*e2)
其中,
c是通过单个孔的通量(克每分钟);
d是聚合物的熔体密度(克每立方厘米);以及
e是纤维被挤出通过的孔口的直径(单位厘米)。在某些实施例中,拉伸比可为从约5:1至约4000:1,在一些实施例中从约10:1至约2000:1,在一些实施例中从约15:1至约1000:1以及在一些实施例中从约20:1至约800:1。
一旦形成后,纤维便可通过纤维牵伸单元22的出口沉积到导丝辊42上。如果需要,可使收集在导丝辊42上的纤维任选地接受另外的在线加工和/或转化步骤(未示出),如本领域的技术人员将理解的那样。例如,可收集纤维,之后卷曲、纹理化和/或切成从约3至约80毫米,在一些实施例中从约4至约65毫米以及在一些实施例中从约5至约50毫米范围内的平均纤维长度。
iv.牵伸
一旦形成后,便将多根纤维扭转在一起形成束。束中的纤维的至少一部分(即使不是全部)由本发明的热塑性组合物形成。多种不同技术中的任一种通常都可以用于扭转纤维。在一个实施例中,例如,扭转可通过将纤维从线筒(bobbin)越过末端(over-end)退绕而形成,如本领域已知的那样。此外,例如,也可以采用本领域已知用于扭转复丝纱的其他技术,诸如加捻、交织等。
无论所用的技术如何,均将所得的扭转纤维束牵伸以形成所需的多孔网络。束通常以“固态”牵伸,因为纤维的热塑性组合物保持在低于聚烯烃基体聚合物的熔融温度的温度下。这尤其有助于确保聚合物链不被改变至使多孔网络变得不稳定的程度。例如,可将束在从约-50℃至约150℃,在一些实施例中从约-40℃至约100℃,在一些实施例中从约-20℃至约50℃以及在一些实施例中从约20℃至约50℃的温度下牵伸。该温度可任选地比具有最高玻璃化转变温度的组分(例如,微米包合物添加剂)的玻璃化转变温度低至少约10℃,在一些实施例中至少约20℃以及在一些实施例中至少约30℃。牵伸温度可以用多种方式实现,诸如通过使纤维束与所需温度下的空气、水(例如,水浴)等接触。
可采用多种牵伸技术,诸如抽吸(例如,纤维牵伸单元)、拉伸架牵伸、双轴牵伸、多轴牵伸、型材牵伸(profiledrawing)、s型卷绕牵伸(s-wrapdrawing)、真空牵伸等。在一个实施例中,例如,可使束横穿一系列辊,这些辊可以s型构型(例如,s型卷绕)牵伸束。无论所采用的技术如何,纤维束通常被牵伸(例如沿机器方向)至使得纤维被拉伸到从约1.1至约25,在一些实施例中从约1.5至约15以及在一些实施例中从约2至约10的牵伸比。牵伸比可以通过将纤维束的长度除以其在牵伸前的长度而确定。牵伸率也可以变化,以帮助实现所期望的特性,诸如在从约5%至约1500%每分钟变形,在一些实施例中从约20%至约1000%每分钟变形以及在一些实施例中从约25%至约850%每分钟变形的范围内。虽然组合物通常在不施加外部热量(例如,加热的辊)的情况下牵伸,但是可任选地采用这样的热以改善可加工性、降低牵伸力、增加牵伸速率并改善纤维均匀性。
以上述方式牵伸可导致形成稳定的多孔网络,而不在纤维中存在很大程度的断裂。网络中的孔可具有“纳米级”尺寸(“纳米孔”),诸如约800纳米或更小,在一些实施例中从约5至约700纳米以及在一些实施例中从约10至约500纳米的平均横截面尺寸。纳米孔还可例如具有从约100至约5000纳米,在一些实施例中从约50至约2000纳米以及在一些实施例中从约100至约1000纳米的平均轴向尺寸(例如,长度)。在牵伸过程中也可以形成微米孔,该微米孔具有约0.2或更大,在一些实施例中约0.5微米或更大以及在一些实施例中从约0.5微米至约5微米的平均横截面尺寸。在某些情况下,微米孔和/或纳米孔的轴向尺寸可以大于横截面尺寸,以使得长宽比(纵向尺寸与横截面尺寸之比)为从约1至约30,在一些实施例中从约1.1至约15以及在一些实施例中从约1.2至约5。例如,微米孔的轴向尺寸可以为1微米或更大,在一些实施例中约1.5微米或更大以及在一些实施例中从约2至约30微米。
无论该具体大小如何,本发明人已发现,孔(例如,纳米孔、微米孔或二者)可以按基本均匀的方式分布在整个组合物中。例如,孔可以分布成列,这些列沿着与应力施加方向大致垂直的方向取向。这些列可以跨过组合物的宽度大致彼此平行。无意受理论的限制,据信,这样的均匀分布的多孔网络的存在可导致良好的机械特性(例如,负载下的能量耗散,和冲击强度)。这与涉及使用发泡剂的形成孔的常规技术形成鲜明对比,发泡剂往往会导致不可控的孔分布和不良的机械特性。
除了形成多孔网络外,牵伸也可明显增大某些离散结构域的轴向尺寸以使得它们具有大致线性的、伸长的形状。例如,伸长的微米级结构域可具有比牵伸前的结构域的轴向尺寸大大约10%或更多,在一些实施例中从约20%至约500%以及在一些实施例中从约50%至约250%的平均轴向尺寸。牵伸后的轴向尺寸(例如,长度)可在从约1μm至约400μm,在一些实施例中从约5μm至约200μm以及在一些实施例中从约10μm至约150μm的范围内。微米级结构域也可以相对薄并因此具有小横截面尺寸,诸如从约0.02至约20微米,在一些实施例中从约0.1至约10微米以及在一些实施例中从0.4至约5微米。这可导致从约2至约150,在一些实施例中从约3至约100以及在一些实施例中从约4至约50的结构域长宽比(轴向尺寸与正交于轴向尺寸的尺寸之比)。由于它们的小尺寸,纳米级结构域通常不以与微米级结构域相同的方式伸长。因此,纳米级结构域可以保持从约1至约1000纳米,在一些实施例中从约5至约800纳米,在一些实施例中从约10至约500纳米以及在一些实施例中从约20至约200纳米的平均轴向尺寸(例如,长度)。
牵伸还可沿着纤维的纵向方向形成一个或多个局部颈缩区,这些颈缩区在未颈缩区之间间隔开。颈缩纤维还可具有沿着其纵向方向的非均匀的横截面直径,这可提供多种不同的益处,诸如增加的表面积等。颈缩区的数量通常可以变化,并可基于所选的拉伸比进行控制。然而,通常的是,颈缩区的数量可在从约1至约400个颈缩区每厘米,在一些实施例中从约2至约200个颈缩区每厘米以及在一些实施例中从约5至约50个颈缩区每厘米的范围内。颈缩区的数量可由以下等式确定:
n=(1-l2)/(l1+l2)
其中n为颈缩区的数量,l1为颈缩区的平均长度,并且l2为未颈缩区(包括从颈缩区到未颈缩区的过渡)的平均长度。
甚至在由本发明实现的极低密度下,所得的纤维也不是脆的并因此可在施加应变时变形而不是碎裂。纤维因此可继续充当承重构件,甚至在纤维已表现出实质性伸长后。就这一点而言,本发明的纤维能够表现出改善的“峰值伸长特性”,即,纤维在其峰值负荷下的伸长百分比。例如,本发明的纤维可表现出约50%或更多,在一些实施例中约100%或更多,在一些实施例中从约200%至约1500%以及在一些实施例中从约400%至约800%的峰值伸长率,诸如根据astmd638-10在23℃下测定时。具有多种多样平均直径的纤维均可实现这样的伸长,诸如直径在从约0.1至约50微米,在一些实施例中从约1至约40微米,在一些实施例中从约2至约25微米以及在一些实施例中从约5至约15微米范围内的那些纤维。
虽然具有在应变下延伸的能力,但是本发明的纤维也可保持相对强韧。例如,纤维可表现出从约25至约600兆帕(“mpa”),在一些实施例中从约50至约450mpa以及在一些实施例中从约60至约350mpa的峰值拉伸应力,诸如根据astmd638-10在23℃下测定时。指示本发明的纤维的相对强度的另一个参数是“韧性”,其指示纤维的拉伸强度,以每单位线密度的力表示。例如,本发明的纤维可具有从约0.75至约7.0克力(“gf”)每旦(纤度单位,denier),在一些实施例中从约1.0至约6.0gf每旦以及在一些实施例中从约1.5至约5.0gf每旦的韧性。纤维的纤度可根据所需的应用而变化。通常,形成具有小于约15,在一些实施例中小于约12以及在一些实施例中从约0.5至约6的单丝纤度(即,线性密度单位,其等于每9000米纤维的质量克数)。
v.纤维材料
牵伸后,可解开纤维束以使得可将单根纤维用于各种应用。或者,可将纤维束自身用于所需的应用。该纤维和/或纤维束可以单独使用或并入纤维材料中,诸如织造织物、针织织物、非织造网等。例如,来源于牵伸后的纤维束的单根纤维可通过将纤维随机沉积在成型表面上(任选地借助真空)然后使用任何已知的技术粘结所得的网而形成连贯的非织造网结构。
可使用任何常规的技术粘结非织造网,诸如通过粘合剂或以自发方式(例如,在不用施加外部粘合剂的情况下,纤维发生融合和/或自粘)。自发粘结例如可通过使纤维在它们为半熔融或发粘状态时接触,或只是通过将增粘树脂和/或溶剂与用于形成纤维的聚合物共混而实现。适合的自发粘结技术可包括超声粘结、热粘结、空气穿透粘结、热轧粘结等等。例如,网可通过热机械过程进一步粘结或压花上图案,在该过程中使网在加热的平滑砧辊与加热的图案辊之间通过。图案辊可具有提供所需的网性质或外观的任何升高的图案。有利地,图案辊限定升高的图案,该图案限定多个粘结位置,这些位置限定占辊总面积的约2%与30%之间的粘结面积。示例性粘结图案包括例如在授予hansen等人的美国专利3,855,046、授予levy等人的美国专利号5,620,779、授予haynes等人的美国专利号5,962,112、授予sayovitz等人的美国专利6,093,665以及授予romano等人的美国设计专利号428,267、授予brown的美国设计专利号390,708、授予zander等人的美国设计专利号418,305、授予zander等人的美国设计专利号384,508、授予zander等人的美国设计专利号384,819、授予zander等人的美国设计专利号358,035和授予blenke等人的美国设计专利号315,990中描述的那些。辊之间的压力可为从约5至约2000磅每线性英寸。使辊之间的压力和辊的温度得以平衡以得到所需的网性质或外观同时维持像布一样的性质。如本领域技术人员熟知的是,所需的温度和压力可根据许多因素而变化,这些因素包括但不限于图案粘结面积、聚合物性质、纤维性质和非织造物性质。
除了纺粘网外,多种其他非织造网也可由根据本发明的热塑性组合物形成,诸如熔喷网、粘合梳理网、湿法网、气流法网、共成形网、水力缠结网。例如,可将热塑性组合物挤出通过多个细模头毛细管进入会聚的高速气体(例如,空气)流,该气体流将纤维缩减以减小其直径。之后,熔融的纤维由高速气体流携带并沉积在收集表面上以形成随机分散的熔融纤维的网。作为另外一种选择,可通过将成捆的由热塑性组合物形成的纤维置于将纤维分开的清棉机中而将聚合物形成梳理网。接着,将纤维送往精梳或梳理单元,该单元将纤维进一步分开并沿机器方向对齐,从而形成沿机器方向取向的纤维非织造网。一旦形成后,通常将非织造网通过一种或多种如上所述的已知粘结技术稳定化,以形成粘合梳理网。
如果需要,非织造网还可以为包含本发明的纤维与其他类型的纤维(例如,短纤维、长丝等)的组合的复合物。例如,可以使用另外的合成纤维,诸如由以下材料形成的那些纤维:聚烯烃,例如聚乙烯、聚丙烯、聚丁烯等;聚四氟乙烯;聚酯,例如聚对苯二甲酸乙二醇酯等;聚乙酸乙烯酯;聚氯乙烯-乙酸乙烯酯;聚乙烯醇缩丁醛;丙烯酸树脂,例如聚丙烯酸酯、聚丙烯酸甲酯、聚甲基丙烯酸甲酯等;聚酰胺,例如尼龙;聚氯乙烯;聚偏二氯乙烯;聚苯乙烯;聚乙烯醇;聚氨酯;聚乳酸等。如果需要,也可采用可再生的聚合物。已知的合成纤维的一些例子包括可以名称t-255和t-256(两者均使用聚烯烃皮)或t-254(其具有低熔点共聚酯皮)得自charlotte,northcarolina的kosainc.的皮芯双组分纤维。可以使用的另外其他已知的双组分纤维包括可得自moriyama,japan的chissocorporation或可得自wilmington,delaware的fibervisionsllc那些。也可采用聚乳酸短纤维,诸如可从台湾fareasterntextile,ltd.商购获得的那些。
复合物也可包含纸浆纤维,诸如高平均纤维长度的纸浆、低平均纤维长度的纸浆或它们的混合物。适合的高平均长度的绒毛浆纤维的一个例子包括软木牛皮纸浆纤维。软木牛皮纸浆纤维来源于针叶树并包括诸如但不限于以下浆纤维:北方、西方和南方软木树种,包括红杉、红刺柏、铁杉、花旗松、真枞、松树(例如,南方松)、云杉(例如,黑云杉)、竹子、它们的组合等等。北方软木牛皮纸浆纤维可用于本发明。适用于本发明的可商购获得的南方软木牛皮纸浆纤维的例子包括可以商品名“nf-405”得自在federalway,washington具有办事处的weyerhaeusercompany的那些。另一种适用于本发明的纸浆是可以商品名coosabsorbs纸浆得自在greenville,southcarolina具有办事处的bowatercorp.的经漂白的硫酸盐木浆,其主要包含软木纤维。低平均长度的纤维也可用于本发明。适合的低平均长度的浆纤维的例子为硬木牛皮纸浆纤维。硬木牛皮纸浆纤维来源于阔叶树并包括诸如但不限于桉树、枫树、桦树、山杨等浆纤维。桉树牛皮纸浆纤维对于增加柔软度、增强亮度、增加不透明度并改变片材的孔结构以增强其芯吸能力可能是尤其理想的。也可采用竹纤维或棉纤维。
非织造复合物可使用多种已知的技术形成。例如,非织造复合物可以是包含热塑性组合物纤维和吸收材料的混合物或稳定化基体的“共成形材料”。例如,共成形材料可通过这样的过程制成,其中将至少一个熔喷模头布置在斜槽附近,通过该斜槽在形成网的同时向网添加吸收材料。这样的吸收材料可包括但不限于纸浆纤维、超吸收性颗粒、无机和/或有机吸收材料、经处理的聚合物短纤维等。吸收材料的相对百分比可在宽范围内变化,具体取决于非织造复合物的所需特性。例如,非织造复合物可包含从约1wt.%至约60wt.%,在一些实施例中从约5wt.%至约50wt.%以及在一些实施例中从约10wt.%至约40wt.%的热塑性组合物纤维。非织造复合物可同样包含从约40wt.%至约99wt.%,在一些实施例中从50wt.%至约95wt.%以及在一些实施例中从约60wt.%至约90wt.%的吸收材料。这样的共成形材料的一些例子在授予anderson等人的美国专利号4,100,324、授予everhart等人的美国专利号5,284,703和授予georger等人的美国专利号5,350,624中有所公开。
在本发明中也可以形成非织造层合物,其中一个或多个层由热塑性组合物形成。例如,一个层的非织造网可以是含有热塑性组合物的纺粘网,而另一层的非织造网包含热塑性组合物、其他可再生的聚合物和/或任何其他聚合物(例如,聚烯烃)。在一个实施例中,非织造层合物包含定位在两个纺粘层之间的熔喷层以形成纺粘/熔喷/纺粘(“sms”)层合物。如果需要,纺粘层可由热塑性组合物形成。熔喷层可由热塑性组合物、其他可再生的聚合物和/或任何其他聚合物(例如,聚烯烃)形成。形成sms层合物的各种技术在授予brock等人的美国专利号4,041,203、授予timmons等人的美国专利号5,213,881、授予timmons等人的美国专利号5,464,688、授予bornslaeger的美国专利号4,374,888、授予collier等人的美国专利号5,169,706和授予brock等人的美国专利号4,766,029以及授予fitting等人的美国专利申请公布号2004/0002273中有所描述。当然,非织造层合物可具有其他构型并具有任何所需数量的熔喷和纺粘层,诸如纺粘/熔喷/熔喷/纺粘层合物(“smms”)、纺粘/熔喷层合物(“sm”)等。虽然非织造层合物的基重可针对所需的应用而定制,但是其通常在从约10至约300克每平方米(“gsm”),在一些实施例中从约25至约200gsm以及在一些实施例中从约40至约150gsm的范围内。
也可形成非织造膜层合物。在这样的实施例中,膜通常为液体不可渗透的和蒸气可渗透的或蒸气不可渗透的。液体不可渗透的和蒸气可渗透的膜通常称为“可呼吸的”并且它们通常具有约100克/平方米/24小时(g/m2/24h)或更大,在一些实施例中从约500至约20,000g/m2/24h以及在一些实施例中从约1,000至约15,000g/m2/24h的水蒸气透过率(“wvtr”)。可呼吸膜可以为微孔的或一体的膜。微孔膜通常通过将填料(例如,碳酸钙)掺入聚合物基体中之后将膜拉伸形成孔而形成。这样的膜的例子例如在授予mccormack的美国专利号5,843,057、授予mccormack的美国专利号5,855,999、授予morman等人的美国专利号5,932,497、授予mccormack等人的美国专利号5,997,981、授予kobylivker等人的美国专利号6,002,064、授予mccormack等人的美国专利号6,015,764、授予mathis等人的美国专利号6,037,281、授予mccormack等人的美国专利号6,111,163和授予taylor等人的美国专利号6,461,457中有所描述。
如果需要,可对纤维、非织造网等进行退火以有助于确保它们保持所需的形状。退火通常在从约40℃至约120℃,在一些实施例中从约50℃至约110℃以及在一些实施例中从约80℃至约100℃的温度下进行。也可使用多种已知技术中的任一种对纤维进行表面处理以改善其性质。例如,可以使用高能束(例如,等离子体、x射线、电子束等)移除或减少在纤维上形成的任何表皮层,以改变表面极性、使表面层变脆等。如果需要,可在形成网之前和/或之后以及在冷牵伸纤维之前和/或之后进行这样的表面处理。
vi.制品
本发明的纤维可用于多种多样的不同制品。在一个实施例中,例如,该纤维可用于吸收制品。吸收制品能够吸收水或其他流体。一些吸收制品的例子包括但不限于个人护理吸收制品,诸如尿布、训练裤、吸收性内裤、成人失禁用制品、女性卫生产品(例如,卫生巾)、泳衣、婴儿湿巾、手套式擦拭布(mittwipe)等等;医疗用吸收制品,诸如衣物、开窗术材料、垫料、绷带、吸收性布单(absorbentdrape)和医用擦拭布;食品服务纸巾;服装制品等等。无论预期应用如何,吸收制品通常包括定位在底片与顶片之间的吸收构件(例如,芯层、涌流层、转移延迟层、包裹片材、通气层等)。值得注意的是,吸收构件、底片和/或顶片以及吸收制品的一个或多个其他部件(例如,耳部、防漏翼片、侧片、腰带或腿带等)可包括本发明的纤维,该纤维为单独的形式或包含此类纤维的非织造网的形式。
就这一点而言,将描述吸收制品的多种示例性实施例。参见图1,例如,吸收制品201的一个特定实施例以尿布的形式示出。然而,如上所述,本发明可体现为其他类型的吸收制品,诸如失禁用制品、卫生巾、尿裤、卫生护垫、训练裤等等。在所示的实施例中,吸收制品201示为在未紧固构型中具有沙漏形。然而,当然也可以利用其他形状,诸如大致矩形、t形或i形。如图所示,吸收制品201包括由各种部件形成的底座202,这些部件包括底片217、顶片205以及包括吸收芯层203和涌流层207的吸收构件。然而,应当理解的是,其他层也可用于本发明。同样,图1中提及的一个或多个层在本发明的某些实施例中也可以取消。
如上所指出的那样,底片217可包括本发明的纤维,任选地以非织造网的形式。例如,非织造网可以设置为使得其限定吸收制品201的面向衣物的表面333。吸收制品201还包括顶片205。顶片205通常被设计成接触使用者的身体并且为液体可渗透的。例如,顶片205可限定面向身体的表面218,该表面通常为顺应性的、柔软感的且对穿戴者皮肤无刺激的。如果需要,顶片205可包括本发明的纤维,任选地以非织造网的形式。例如,非织造网可被定位成使得其限定面向身体的表面218(如果需要这样的话)。顶片可围绕吸收芯层203,以使得其完全包裹吸收制品。作为另外一种选择,顶片205和底片217可延伸到吸收构件之外,并在周边使用已知的技术(诸如通过粘合剂粘结、超声粘结等)完全或部分地接合在一起。如上所指出的那样,顶片205可包括根据本发明形成的非织造网。顶片205还可包括常规非织造网(例如,纺粘网、熔喷网或粘合梳理网)。包括非织造网的其他示例性顶片构造在美国专利号5,192,606、5,702,377、5,931,823、6,060,638和6,150,002以及美国专利申请公开号2004/0102750、2005/0054255和2005/0059941中有所描述。顶片205也可包括多个贯穿形成的开孔以允许体液更容易地进入吸收芯层203。这些开孔可以随机或均匀布置在整个顶片205中,或者它们可仅位于沿着吸收制品的纵向轴线布置的窄纵向带或条中。这些开孔允许体液快速向下渗入吸收构件中。开孔的大小、形状、直径和数量可以变化以适应某人的特定需求。
吸收制品还包括定位在顶片与底片之间的吸收构件。吸收构件可以由单个吸收层或包括单独和不同的吸收层的复合物形成。然而,应当理解的是,许多吸收层均可用于本发明。在图1中,例如,吸收构件包括吸收芯层203和涌流层207,该涌流层有助于减速并扩散可快速引入吸收芯层203中的涌流或迸发液体。有利地,涌流层207在将液体释放进吸收芯层203的储存或保持部分前快速接纳并暂时保留液体。在所示实施例中,例如,涌流层207插在顶片205的面向内的表面216与吸收芯层203之间。作为另外一种选择,涌流层207可位于顶片205面向外的表面218上。涌流层207通常由液体可高度渗透的材料构造而成。适合的材料可包括多孔织造材料、多孔非织造材料和开孔膜。在一个实施例中,涌流层207可包括本发明的纤维,任选地以非织造网的形式。适合的涌流层的其他例子在授予ellis等人的美国专利号5,486,166和授予ellis等人的美国专利号5,490,846中有所描述。
如果需要,吸收构件也可包括垂直定位在涌流层下方的转移延迟层。转移延迟层可包含亲水性低于其他吸收层的材料,并且通常可被表征为基本上疏水性的。例如,转移延迟层可以是由本发明的纤维形成的非织造网(例如,纺粘网)。纤维的横截面形状可以是圆形的、三叶形的或多叶形的,并且其结构可以是中空的或实心的。通常,诸如通过在约3%至约30%的网面积上热粘结而使网粘结。可用于转移延迟层的适合材料的其他例子在授予meyer等人的美国专利号4,798,603和授予serbiak等人的美国专利号5,248,309中有所描述。为了调节本发明的性能,转移延迟层也可以用选定量的表面活性剂处理以增加其初始可润湿性。
转移延迟层通常可以具有任何大小,诸如约150mm至约300mm的长度。通常,转移延迟层的长度约等于吸收制品的长度。转移延迟层在宽度上也可等于涌流层,但通常更宽。例如,转移延迟层的宽度可介于约50mm至约75mm之间,更具体地讲约48mm。转移延迟层通常具有比其他吸收构件低的基重。例如,转移延迟层的基重通常小于约150克每平方米(gsm)以及在一些实施例中介于约10gsm至约100gsm之间。如果需要,转移延迟层可包括本发明的纤维,任选地以非织造网的形式。
除了上述部件以外,吸收制品201还可以包括如本领域已知的各种其他部件。例如,吸收制品201还可以包括基本上亲水性的包裹片材(未示出),该片材有助于维持吸收芯层203的纤维结构的完整性。包裹片材通常围绕吸收芯层203在其至少两个主表面上放置,并由吸收性纤维素材料构成,诸如皱纹包装纸或高湿强度薄纸(highwet-strengthtissue)。包裹片材可被构造成提供芯吸层,该层有助于在吸收芯层203的吸收纤维团块上快速分布液体。在吸收芯纤维团块的一侧上的包裹片材材料可粘结到位于纤维团块相对侧上的包裹片材以有效地夹带吸收芯层203。此外,吸收制品201还可以包括定位在吸收芯层203与底片217之间的通气层(未示出)。当利用时,通气层可有助于将底片217与吸收芯层203隔绝,从而减少底片217中的湿气。这样的通气层的例子可包括层合到可呼吸膜的非织造网,诸如在授予blaney等人的美国专利号6,663,611中所述。如果需要,包裹片材和/或通气层可包括本发明的纤维,任选地以非织造网的形式。
在一些实施例中,吸收制品201也可以包括一对耳部(未示出),该耳部从吸收制品201的侧边缘232延伸到腰部区之一中。耳部可与所选的尿布部件整体形成。例如,耳部可与底片217整体形成或由用于提供顶部表面的材料形成,该材料可包括本发明的纤维,任选地以非织造网的形式。在可供选择的构型中,耳部可由连接并组装到底片217、顶部表面,在底片217与顶部表面之间的或处于各种其他构型的构件提供。如上所述,耳部可包括本发明的纤维,任选地以非织造网的形式。
如在图1中代表性示出,吸收制品201还可以包括一对防漏翼片212,该翼片被构造成提供屏障并容纳侧向流动的身体排出物。防漏翼片212可沿着与吸收芯层203的侧边缘相邻的顶片205的侧向相对的侧边缘232定位。防漏翼片212可沿着吸收芯层203的整个长度延伸,或可以仅沿着吸收芯层203的长度部分延伸。当防漏翼片212在长度上比吸收芯层203更短时,它们可选择性定位在裆区210中沿着吸收制品201的侧边缘232的任何位置。在一个实施例中,防漏翼片212沿着吸收芯层203的整个长度延伸以更好地容纳身体排出物。这样的防漏翼片212通常是本领域的技术人员熟知的。例如,防漏翼片212的适合构造和布置在授予enloe的美国专利号4,704,116中有所描述。如果需要,防漏翼片可包括本发明的纤维,任选地以非织造网的形式。
吸收制品201可包含各种弹性的或可拉伸的材料,诸如一对腿部弹性构件206,它们附连到侧边缘232以进一步防止身体排出物的渗漏并支持吸收芯层203。此外,一对腰部弹性构件208可附连到吸收制品201的纵向相对的腰部边缘215。腿部弹性构件206和腰部弹性构件208通常适于在使用中围绕穿戴者的腿部和腰部紧密配合,以维持与穿戴者的积极接触关系,并有效地减少或消除从吸收制品201的身体排出物渗漏。吸收制品201还可以包括一个或多个紧固件230。例如,两个柔性紧固件130在图1中示出,它们位于腰部区的相对侧边缘上以形成腰部开口和一对围绕穿戴者的腿部开口。紧固件230的形状通常可以变化,但可包括例如大致矩形、方形、圆形、三角形、卵形、线性形状等等。紧固件可包括例如扣件材料。在一个特定的实施例中,每个紧固件230包括附连到柔性背衬的内表面的扣件材料的单独件。弹性构件(例如,腿、腰等)和/或紧固件可在需要时包括本发明的纤维,任选地以非织造网的形式。
吸收制品201的各个区和/或部件可使用任何已知的附连机构诸如粘合剂、超声、热粘结等组装在一起。适合的粘合剂可包括例如热熔粘合剂、压敏粘合剂等。当利用时,粘合剂可以作为均匀的层、图案化层、喷雾图案或任何单独的线、漩涡或点而施加。在所示实施例中,例如,底片217和顶片205用粘合剂组装到彼此并组装到吸收芯层203。作为另外一种选择,吸收芯层203可用常规的紧固件诸如纽扣、钩环型紧固件、胶带紧固件等连接到底片217。类似地,诸如腿部弹性构件206、腰部弹性构件208和紧固件230的其他尿布部件也可用任何附连机构组装成吸收制品201。
虽然上文已经描述了尿布的各种构型,但是应当理解的是,其他尿布和吸收制品构型也包括在本发明的范围内。此外,本发明绝不限于尿布。事实上,可根据本发明形成任何其他吸收制品,包括但不限于其他个人护理吸收制品,诸如训练裤、吸收性内裤、成人失禁用产品、女性卫生产品(例如,卫生巾)、泳衣、婴儿湿巾等;医疗用吸收性制品,诸如衣物、开窗术材料、垫料、绷带、吸收性布单和医用擦拭布;食品服务纸巾;服装制品等等。
通过参照以下实例可以更好地理解本发明。
测试方法
熔体流动速率:
熔体流动速率("mfr")是指,当通常在190℃、210℃或230℃下经受10分钟的2160克的负荷时,聚合物被迫使通过挤出流变仪孔口(直径为0.0825英寸)的重量(以克计)。除非另外指明,否则熔体流动速率用tiniusolsen挤压式塑性计根据astm测试方法d1238测量。
热特性:
玻璃化转变温度(tg)可以根据astme1640-09通过动态力学分析(dma)测定。可以使用得自tainstruments的q800仪器。实验运行可以在3℃/min的加热速率下用从-120℃至150℃范围内的温度扫描模式以张力/张力几何图形来实施。应变幅度频率在测试期间可以保持恒定(2hz)。可以测试三(3)个独立的样品,以获得用tanδ曲线的峰值来定义的平均玻璃化转变温度,其中tanδ被定义为损耗模量与储能模量之比(tanδ=e”/e’)。
熔融温度可以通过差示扫描量热法(dsc)来测定。差示扫描量热仪可以是dscq100差示扫描量热仪,其配备有液氮冷却附件和universalanalysis2000(4.6.6版)分析软件程序,二者均可以得自delaware的newcastle的t.a.instrumentsinc.。为了避免直接操作样品,使用镊子或其他工具。将样品放入铝盘并在分析天平上称重至0.01毫克的精确度。在材料样品上方,轧盖至所述盘上。通常,将树脂粒料直接放置在称量盘上。
如在差示扫描量热仪的操作手册中所述,差示扫描量热仪采用铟金属标准品来校准,并且执行基线校正。将材料样品放入差示扫描量热仪的测试室中来测试,并且使用空盘作为参比物。所有测试均在测试室上以每分钟55立方厘米的氮气(工业级)吹扫下运行。对于树脂粒料样品而言,加热和冷却程序为2个循环的测试,该测试首先是平衡所述室至-30℃,随后是以每分钟10℃的加热速率加热至200℃的温度的第一加热段,然后将样品在200℃下平衡3分钟,接着是以每分钟10℃的冷却速率冷却到-30℃的温度的第一冷却段,随后将样品在-30℃下平衡3分钟,再是以每分钟10℃的加热速率加热至200℃的温度的第二加热段。所有测试均在测试室上以每分钟55立方厘米的氮气(工业级)吹扫下运行。
使用universalanalysis2000分析软件程序来评价结果,该软件程序识别并量化拐点(inflection)的玻璃化转变温度(tg)、吸热峰和放热峰,以及dsc图上的峰下面积。玻璃化转变温度被确定为图线上斜率发生明显变化的区域,而熔融温度则使用自动拐点计算来确定。
拉伸特性:
拉伸特性可根据astm638-10在23℃下测定。例如,最初可将各个纤维试件的长度缩短(例如,用剪刀剪短)至38毫米,然后单独地置于黑色天鹅绒布上。可以此方式收集10至15个纤维试件。然后可将纤维试件以基本上竖直的状态安装在外部尺寸为51毫米×51毫米且内部尺寸为25毫米×25毫米的矩形纸框上。可通过用胶带小心地将纤维末端固定到纸框的侧面而将每个纤维试件的末端可操作地附接到纸框。可采用常规实验室显微镜测量每个纤维试件的外部、相对更短的纤维横截面尺寸,显微镜可适当地校准并设为40倍放大率。该纤维横截面尺寸可记录为各个纤维试件的直径。纸框有助于将样本纤维试件的末端以避免对纤维试件造成过度损坏的方式固定在恒定速率的延伸式拉伸试验机的上下夹具中。
恒定速率的延伸式拉伸试验机和适当的测力传感器可用于该试验。测力传感器可被选择(例如,10n)成使得试验值落在全量程负荷的10-90%内。拉伸试验机(即,mtssynergy200)和测量传感器可得自edenprairie,michigan的mtssystemscorporation。然后可将纸框组件中的纤维试件安装在拉伸试验机的夹具之间以使得纤维的末端可操作地被拉伸试验机的夹具保持。然后,可将纸框平行于纤维长度延伸的侧切掉或以其他方式分开,以使得拉伸试验机仅对纤维施加试验力。纤维可以12英寸每分钟的拉伸速率和夹具速度接受拉伸试验。所得的数据可使用得自mtscorporation的testworks4软件程序通过以下试验设置而分析:
韧性值以克力每旦表示。也可测量峰值伸长率(断裂应变%)和峰值应力。
网的峰值负荷可使用沿着长度(md)和宽度方向(cd)切割的2英寸×6英寸的条带测定。试验可在配有两个1英寸×3英寸的涂有橡胶的夹具的通用拉伸试验机中进行。标距长度可以为76±1mm(3±0.04英寸)。
密度和空隙体积百分比:
为测定密度和空隙体积百分比,在牵伸前,可初步测量试件的宽度(wi)和厚度(ti)。牵伸前的长度(li)也可以通过测量试件表面上的两个标记之间的距离来测定。之后,可将试样牵伸,以引发孔的形成。然后,可利用digimatic卡尺(mitutoyocorporation)测量试件的宽度(wf)、厚度(tf)和长度(lf),精确至0.01mm。牵伸前的体积(vi)可通过wixtixli=vi计算。牵伸后的体积(vf)也可通过wfxtfxlf=vf计算。密度(ρf)可通过ρf=ρi/φ计算,其中ρi为前体材料的密度,而空隙体积百分比(%vv)如下计算:%vv=(1–1/φ)x100。
流体静压试验(“静压头”):
流体静压试验是材料在静压力下抵抗液态水渗透的量度,并根据aatcc试验方法127-2008进行。可对每个试件的结果取平均值并以厘米(cm)记录。值越高表示对水渗透的抵抗力越高。
水蒸气透过率(“wvtr”):
用于测定材料的wvtr的试验可基于材料的性质而变化。一种测量wvtr值的技术是astme96/96m-12程序b。另一种方法涉及使用inda试验程序ist-70.4(01)。inda试验程序归纳如下。通过固定式警戒膜和将测试的样品材料从已知温度和湿度的湿室中分离出干燥室。警戒膜的目的是限定明确的空气间隙并在表征该空气间隙时用于使该空气间隙中的空气安静或静止。干燥室、警戒膜和湿室构成扩散单元,将测试膜密封在其中。样品架是由mocon/modemcontrols,inc.,minneapolis,minnesota制造的permatran-wmodel100k。首先测试警戒膜以及产生100%相对湿度的蒸发器组件之间的空气间隙的wvtr。水蒸气扩散通过空气间隙和警戒膜然后和与水蒸气浓度成比例的干燥气流混合。电信号被输入计算机进行处理。计算机计算空气间隙和警戒膜的透过率并储存该值以供进一步使用。
警戒膜和空气间隙的透过率作为caic存储在计算机中。然后将样品材料密封在测试单元中。再一次,水蒸气通过空气间隙向警戒膜和测试材料扩散然后与吹扫测试材料的干燥气流混合。同样,再一次,将该混合物传送至蒸气传感器。然后计算机计算空气间隙、警戒膜和测试材料的组合的透过率。接着根据以下公式将该信息用于计算水分通过测试材料传输的透过率:
tr-1测试材料=tr-1测试材料、警戒膜、空气间隙–tr-1警戒膜、空气间隙
然后如下计算水蒸气透过率("wvtr"):
其中,
f=以cm3/分钟表示的水蒸气流量;
ρsat(t)=在温度t下饱和空气中水的密度;
rh=在所述单元中的具体位置的相对湿度;
a=所述单元的横截面积;以及
psat(t)=在温度t下水蒸气的饱和蒸气压。
弗雷泽孔隙度(frazierporosity):
通过以下方式在
实例1
形成包含芯和皮的双组分纤维,芯由93wt.%的全同立构的聚丙烯(m3661,230℃下的熔体流动速率为14g/10min,且熔融温度为150℃,totalpetrochemicals)和7wt.%的
实例2
如实例1中所述牵伸纤维束,但使用不提供扭转的退绕纱架(unwindcreel)使线筒退绕。收起五重辊速度为4米每分钟,并且最后两套五重辊的速度为24米每秒以得到6的牵伸比。水浴设为44℃。
实例3
如实例1中所述牵伸纤维束,但通过基于导丝辊的牵伸单元牵伸纤维束。将纤维束越过末端退绕,以向纤维提供轻微扭转,如实例1中所述。进料辊速度为48米每分钟,第一牵伸辊速度为50米每分钟,第二牵伸辊速度为100米每分钟,第三和第四牵伸辊速度为250米每分钟,而松弛辊速度为245米每分钟。所得的牵伸比为5.2。第一、第二、第三和第四牵伸辊的温度设为40℃。
虽然本发明已经就其具体实施例进行了详细描述,但是将领会的是,本领域技术人员在获得前述内容的理解后可以容易地设想出这些实施例的替代形式、变型形式和等同方案。因此,本发明的范围应被评估为所附权利要求及其任何等同方案的范围。
权利要求书(按照条约第19条的修改)
1.一种包含围绕纵向轴线扭转的多根纤维的纤维束,其中所述纤维的至少一部分由热塑性组合物形成,所述热塑性组合物包含含有聚烯烃基体聚合物的连续相和以离散结构域的形式分散在所述连续相中的纳米包合物添加剂,其中在所述组合物中限定包括多个纳米孔的多孔网络。
2.根据权利要求1所述的纤维束,其中所述束包含5根或更多根纤维。
3.根据权利要求1所述的纤维束,其中所述纤维螺旋扭转。
4.根据权利要求3所述的纤维束,其中所述纤维以从0.1°至20°的螺旋角和/或从1至300转每米的间距扭转。
5.根据权利要求1所述的纤维束,其中所述纤维束具有从1至30千克每9000米的总纤度。
6.根据权利要求1所述的纤维束,其中所述纳米孔具有800纳米或更小的平均横截面尺寸。
7.根据权利要求1所述的纤维束,其中所述聚烯烃基体聚合物在2160克的负荷和230℃下根据astmd1238测定时具有从0.5至80克/10分钟的熔体流动速率。
8.根据权利要求1所述的纤维束,其中所述聚烯烃基体聚合物为包含至少90重量%的丙烯的基本上全同立构的聚丙烯均聚物或共聚物。
9.根据权利要求1所述的纤维束,其中按所述连续相的重量计,所述连续相占所述热塑性组合物的从60wt.%至99wt.%并且所述纳米包合物添加剂占所述组合物的从0.05wt.%至20wt.%。
10.根据权利要求1所述的纤维束,其中所述纳米包合物添加剂包括官能化聚烯烃。
11.根据权利要求10所述的纤维束,其中所述官能化聚烯烃为聚环氧化物。
12.根据权利要求1所述的纤维束,其中所述纳米包合物添加剂在2160克的负荷和比所述熔融温度高至少40℃的温度下根据astmd1238测定时具有从0.1至100克/10分钟的熔体流动速率。
13.根据权利要求1所述的纤维束,其中所述组合物还包含以离散结构域的形式分散在所述连续相中的微米包合物添加剂。
14.根据权利要求13所述的纤维束,其中所述微米包合物添加剂为聚乳酸。
15.根据权利要求13所述的纤维束,其中所述微米包合物添加剂具有0℃或更高的玻璃化转变温度。
16.根据权利要求1所述的纤维束,其中所述热塑性组合物还包含相间改性剂。
17.根据权利要求1所述的纤维束,其中所述多孔网络还包括微米孔。
18.一种形成多孔纤维的方法,所述方法包括:
在低于所述基体聚合物的熔融温度的温度下牵伸根据前述权利要求中任一项所述的纤维束,从而形成包括多个纳米孔的多孔网络。
19.根据权利要求18所述的方法,其中将所述纤维束拉伸到从1.1至25的牵伸比。
20.根据权利要求18所述的方法,其中所述纤维束在从-50℃至150℃的温度下牵伸。
21.根据权利要求18所述的方法,还包括解开所述束以形成至少一根多孔纤维。
22.一种非织造网,所述非织造网包含根据权利要求21所述的多孔纤维。
- 还没有人留言评论。精彩留言会获得点赞!