一种掺杂多主元贮氢合金及其制备方法与流程
1.本发明涉及多主元贮氢合金领域,具体涉及一种掺杂多主元贮氢合金及其制备方法。
背景技术:
2.开发清洁高效的绿色能源将是解决能源危机与全球变暖的重要手段。尽管氢气是一种清洁高效的绿色能源,是化石燃料重要的替代品。但氢能的实际应用仍然面临这许多困难。而制约氢能利用最关键的技术瓶颈之一,就是氢气的储存与运输。相比于高压气态储氢、液态储氢,固态贮氢同时兼具了高贮氢密度、高安全性以及价格相对低廉的优势,是一种十分有前景的技术。尽管如此,传统的贮氢合金也存在一些不足之处,使得贮氢合金的大规模运用仍然难以实现。如mg及其合金,其放氢动力学性能缓慢,放氢温度较高。而zrco合金存在歧化效应,这将导致zrco合金性能的衰减。tife虽然成本低廉,但其储氢量不高,且活化困难。lani5虽然易于活化,且动力学性能较好,但储氢量仍然达不到使用需求。因此,发展具有优异储氢性能的新型贮氢合金是实现氢能利用的关键基础。
3.多主元合金一般具有四种及以上金属组元,其多元素种类及成分比例的可调节性,使得其在新型贮氢合金的开发上有更多的可能性。多主元合金具有一些独特晶格特征,如严重的晶格畸变使得其有可能具有与传统合金完全不同的性能。对tivnb,tivzrnb,以及tivzrnbhf多主元合金及其氘化物的研究表明多主元合金的氘原子占位与其品格畸变有着非常重要的关系。tizrhfvnb高熵合金中的品格畸变提供了更多包容氢的间隙位置,贮氢容量得到提高。虽然多主元合金具有优异的氢化性能以及易于调控的优异特性,但目前多主元贮氢合金中存在的如贮氢量较低、活化性能较差等问题成为了限制其发展的关键因素之一。因此急需发展一种针对多主元合金的贮氢性能提升技术。
4.为了解决上述问题,研究人员采用了元素成分比例调节、复合材料、添加催化剂、元素掺杂等多种技术。其中元素掺杂是提升储氢合金的储氢量、活化性能、动力学性能的重要手段。研究表明la、ce、ho掺杂的ti
1.02
cr
1.1
mn
0.3
fe
0.6
合金相比掺杂前品格常数更大,储氢量提高约10%;同时,掺杂也给氢原子的扩散提供了更多的通道,使得其活化性能也大大提升。另外,关于nd掺杂的nd
4.3
mg
87.0
ni
8.7
合金的研究发现合金在氢化过程中原位形成了ndh2‑
mg
‑
mg2ni复合结构,大量的晶界给氢原子扩散提供了通道,从而提升了其动力学性能。对于贮氢合金而言,元素掺杂能够提升其动力学性能,降低氢化物的热稳定性,但是对储氢量的影响却极其有限。最近,研究发现用pd对zrco合金进行表面修饰可以大大降低其吸氢活化能,从而提升其动力学性能,pd在其中起到了催化h2解离的作用。但是pd修饰的zrco合金的储氢量却没有明显提升。
5.此外,大多数元素掺杂技术的掺杂元素含量在(1%~10%),需要消耗更多的原料,且难以实现极其微量掺杂元素在合金中的均匀分布。对于co掺杂tife
0.8
mn
0.2
的研究发现5%的co(tife
0.8
mn
0.2
co
0.05
)元素掺杂有效吸氢量仅提升了6.51%,但是动力学性能以及最大吸氢量均有所降低。另外,掺杂5%的y后tiv
1.1
mn
09
的最大吸氢量提升了18%。在上述案
例中,虽然掺杂对贮氢合金的储氢量有了明显改善,但掺杂元素的含量均较高。由于活性金属如pd、pt、au等价格昂贵,为了降低贮氢合金的制造成本就需要减少掺杂元素的浓度。因此,降低掺杂元素的含量,提高掺杂元素的均匀分布,同时大大提高贮氢合金的最大吸氢量及活化性能是急需解决的问题。
技术实现要素:
6.本发明的目的在于,提供一种掺杂多主元贮氢合金及其制备方法,以达到降低掺杂元素含量,提高掺杂元素分布均匀性,同时提高其储氢量以及活化性能的目的。
7.本发明采用的技术方案如下:
8.一种掺杂多主元贮氢合金,所述合金成分为(ti
a
zr
b
hf
c
)
x
(a
d
b
e
)
y
m,其中元素m为au、pd、pt中任意一种,(ti
a
zr
b
hf
c
)
x
(a
d
b
e
)
y
与m的质量份数比为1∶0.0005~1∶0.0015;其中,元素a、b分别为w、mo、nb、al、sc中的任意一种,各元素原子比例范围分别为:0.2≤a≤0.4,0.2≤b≤0.4,0.2≤c≤0.4,a+b+c=1;0.3≤d≤0.7,0.3≤e≤0.7,d+e=1;0.5≤x≤0.7,0.3≤y≤0.5,x+y=1。
9.一种基于前述的掺杂多主元贮氢合金的制备方法,所述制备方法按以下步骤完成:
10.(1)制备由活性金属元素m制成的活性金属元素m金属丝,所述活性金属元素m金属丝的直径小于0.05mm,所述m为au、pd、pt中任意一种。
11.(2)根据各元素的摩尔质量,按照一定原子比称取ti金属颗粒、zr金属颗粒、hf金属颗粒、a金属颗粒、b金属颗粒;按照一定质量份数比称取m金属丝;
12.(3)为了使元素分布均匀,将所述活性金属元素m金属丝缠绕在金属颗粒上;
13.(4)将所述金属颗粒和金属丝放入真空电弧熔炼炉的样品腔室的铜坩埚中,然后用真空泵对样品腔室抽真空至真空度为30~50pa;
14.(5)对所述金属颗粒和金属丝进行熔炼,直至所述金属颗粒和金属丝溶解均匀,得到掺杂多主元贮氢合金;
15.(6)将所制备的微量掺杂多主元贮氢合金退火处理。
16.进一步,步骤(6)具体为:将掺杂多主元贮氢合金切割成若干小方片,将小方片放入吸氢测试系统的样品室中,依次采用机械泵、分子泵、离子泵对样品室抽真空,当吸氢测试系统的样品室真空度低于真空度阈值时,将吸氢测试系统的样品室温度缓慢升温至400℃~600℃,并保温4小时以上,得到单相掺杂多主元贮氢合金。
17.进一步,步骤(2)中,所述ti、zr、hf、a、b金属颗粒纯度均在99%以上。
18.进一步,步骤(1)中,所述m金属丝原料纯度在99.9%以上。
19.进一步,步骤(4)中,所述真空度阈值为5
×
10
‑3pa。
20.本发明的优点与积极效果如下:
21.本发明应用一种微量元素掺杂技术,解决了微量元素在合金中分布均匀的瓶颈难题,实现在降低掺杂元素含量的同时极大提升多主元贮氢合金的储氢量,同时降低了其活化时间。本发明在ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元贮氢合金中进行微量pd、pt、au活性金属元素掺杂,通过热处理工艺,得到单相掺杂多主元贮氢合金,有效提高了合金的贮氢密度、活化性能。
22.本发明对多主元贮氢合金进行微量的活性金属掺杂,所得的合金储氢量高,动力学以及活化性能均得到了有效改善。相比未掺入活性金属的多主元贮氢合金,本发明使用了微量元素掺杂技术,使得多主元贮氢合金在极微量(≤0.0015)的活性金属元素掺杂条件下,其储氢量提高高达54.7%,且活化性能得到明显提升。
附图说明
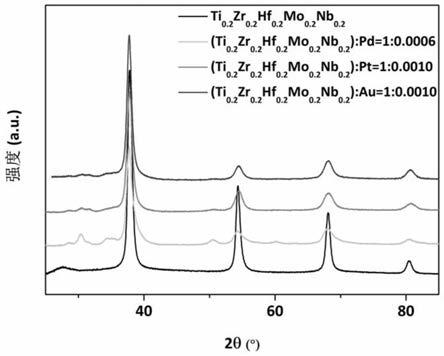
23.图1为本发明中铸态ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010掺杂多主元贮氢合金的xrd谱;
24.图2为本发明中ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010掺杂多主元贮氢合金经过热处理后的xrd谱;
25.图3为本发明中ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010掺杂多主元贮氢合金吸氢后的xrd谱;
26.图4为本发明中ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010掺杂多主元贮氢合金的活化曲线;
27.图5为本发明中ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010掺杂多主元贮氢合金的动力学曲线。
具体实施方式
28.以下结合说明书附图和具体优选的实施例对本发明作进一步描述,但并不因此而限制本发明的保护范围。本领域技术人员可由本说明书所揭示的内容对这些技术方案做进一步改良而不违背本发明的精神和范围。
29.本发明提出的元素掺杂改性多主元贮氢合金的实施例详细说明如下:
30.实施例1
31.本实施例制备掺杂多主元贮氢合金(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006,具体实施方式如下:
32.步骤1:制备活性金属元素pd的金属丝,pd金属丝的直径为0.05mm;
33.步骤2:根据金属元素的摩尔质量,按照原子比ti∶zr∶hf∶mo∶nb=0.2∶0.2∶0.2∶0.2∶0.2计算并称量出总质量约为15g的高纯纯金属颗粒,按照质量份数比(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006称取pd活性金属丝;
34.步骤3:为了使元素分布均匀,将pd金属丝缠绕在ti、zr、hf、mo和nd金属颗粒上;
35.步骤4:按照金属元素熔点从低到高的顺序,依次将缠绕pd金属丝的ti、zr、hf、mo和nd金属颗粒放入真空电弧熔炼炉的样品腔室的铜坩埚中,然后用真空泵对样品腔室抽真空至真空度为30~50pa;
36.步骤5:对缠绕pd金属丝的ti、zr、hf、mo和nd金属颗粒进行熔炼,起弧电流为300a,
缓慢将电流升高至600a,并用电极杆缓慢搅拌;当上层金属熔化后将电流升高至800a,待金属熔化后继续保持搅拌3~5min;然后缓慢将电流降低到300a并关闭熔炼炉电流;待合金冷却后将其翻转,反复熔炼合金5次以上,得到掺杂多主元贮氢合金。
37.步骤6:将掺杂多主元贮氢合金切成1em
×
1cm
×
0.1cm的小方片,将若干小方片放入吸氢测试系统的样品室中,依次采用机械泵、分子泵、离子泵对吸氢测试系统的样品室抽真空,当样品室真空度优于5
×
10
‑3pa后缓慢升温,升温速率为10℃/min,当样品室温度达到600℃后,保温4h,然后随炉降温。最后将一部分退火后的小方片破碎并用研钵研磨成粉末,其他试样小方片用作物相分析与元素成分分布分析。
38.物相分析与元素成分分布:将退火后的试样小方片,用220、500、800、1200、2400目的sic砂纸依次打磨,并用去离子水、丙酮、酒精依次超声清洗样品至清洗液体澄清。随后将样品用真空干燥箱烘干后用xrd谱仪检测样品物相结构。由图1可知,相比于未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金,除bcc主相外还形成了其他杂相;而经过高温真空退火后,其物相如图2所示,为单相bcc结构,高温退火后杂相消失,形成了均匀的单相掺杂多主元贮氢合金,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006吸氢后结构如图3所示,为单相fcc结构,单相掺杂多主元贮氢合金在吸氢过程中经过了bcc到fcc相的转变。
39.吸氢性能测试:
40.(1)样品除气:称取步骤6中研磨后的掺杂多主元贮氢合金(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006粉末约200mg装入吸氢测试系统的样品室,用机械泵、分子泵、离子泵依次对吸氢测试系统的样品室抽真空,当真空度至少为5.0
×
10
‑3pa时对样品进行加热除气;保持真空度低于5.0
×
10
‑3pa对样品进行缓慢升温至600℃并保温1h。
41.(2)活化性能测试:除气完成后,降温至250℃并保温30min;随后在吸氢测试系统的缓冲室中加入25bar氢气,打开吸氢测试系统的缓冲室阀门,测试样品的活化性能。
42.(3)动力学性能测试:样品活化后,关闭吸氢测试系统的加热模块,待吸氢测试系统的样品室降温至室温后,用机械泵、分子泵、离子泵依次对吸氢测试系统的样品室抽真空,当真空度至少为5.0
×
10
‑3pa时对样品进行加热排除氢气;保持真空度低于5.0
×
10
‑3pa对样品进行缓慢升温至600℃并保温1h排除样品中残余的氢气。待氢气排干净后降温至250℃并保温30min;随后在吸氢测试系统的缓冲室中加入5bar氢气,打开吸氢测试系统的缓冲室阀门,测试样品的动力学性能。
43.1)吸氢性能测试:图4给出了样品的活化曲线,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006掺杂多主元贮氢合金需要的孕育时间相较于未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金有所减少,其最大储氢量从1.1wt.%快速提升至1.66wt.%。
44.2)活化完成后,测试了样品的动力学性能,动力学曲线如图5所示,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pd=1∶0.0006与未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金相比仍然具有快速的吸氢动力学性能,且储氢量相比于ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金有显著提升,最大储氢量从1.038%提升至1.607%,最大储氢量提升约54.7%。
45.实施例2
46.本实施例制备掺杂多主元贮氢合金(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010,具体实施步骤与实例1相同,区别在于:掺杂元素从pd元素换成了pt元素,质量份数比为(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010。
47.物相分析与元素成分分布:用xrd谱仪检测样品物相结构。由图1可知,相比于未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金,除bcc主相外还形成了其他杂相;经过高温真空退火后,其物相如图2所示,为单相bcc结构,高温退火后杂相消失,形成了均匀的单相掺杂多主元贮氢合金,而(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010吸氢后结构如图3所示,为单相fcc结构,单相掺杂多主元贮氢合金在吸氢过程中经过了bcc到fcc相的转变。
48.1)吸氢性能测试:图4给出了样品的活化曲线,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010掺杂多主元贮氢合金需要的孕育时间相较于未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金大大减少,只需30s就能快速吸氢,其最大储氢量从1.1wt.%快速提升至1.66wt.%。
49.2)活化完成后,测试了样品的动力学性能,动力学曲线如图5所示,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):pt=1∶0.0010与未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金相比仍然具有快速的吸氢动力学性能,且储氢量相比于ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金有较大提升,最大储氢量从1.038%提升至1.425%,最大储氢量提升约37.2%。
50.实施例3
51.本实施例制备掺杂多主元贮氢合金(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010,具体实施步骤与实例1相同,区别在于:掺杂元素从pd元素换成了au元素,质量份数比为(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010。
52.物相分析与元素成分分布:用xrd谱仪检测样品物相结构。由图1可知,相比于未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金,除bcc主相外还形成了其他杂相;经过高温真空退火后,其物相如图2所示,为单相bcc结构,为单相bcc结构,高温退火后杂相消失,形成了均匀的单相掺杂多主元贮氢合金,而(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010吸氢后结构如图3所示,为单相fcc结构,单相掺杂多主元贮氢合金在吸氢过程中经过了bcc到fcc相的转变。
53.1)吸氢性能测试:图4给出了样品的活化曲线,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010掺杂多主元贮氢合金需要的孕育时间相较于未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金大大减少,无需经过孕育就能快速吸氢,其最大储氢量从1.1wt.%快速提升至1.73wt.%。
54.2)活化完成后,测试了样品的动力学性能,动力学曲线如图5所示,(ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
):au=1∶0.0010与未掺杂的ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金相比仍然具有快速的吸氢动力学性能,且储氢量相比于ti
0.2
zr
0.2
hf
0.2
mo
0.2
nb
0.2
多主元合金有较大提升,最大储氢量从1.038%提升至1.586%,最大储氢量提升约52.8%。
55.综上所述,我们对高熵合金进行了极微量的活性金属掺杂改性(质量份数比0.06%
‑
0.10%),不仅提升了其活化性能,同时大大提高了其储氢量,最大储氢量相比于未掺杂前样品提升了37
‑
55%,使得其氢化性能有了很大改善。退火工艺消除了掺杂过程中形成的杂相,使得掺杂多主元贮氢合金吸氢前后物相与未掺杂的多主元贮氢合金相同,避免了掺杂引入杂相造成的吸氢量的损失。
56.上述实例仅为发明的具体实施方式。本发明的保护范围并不限于此,任何熟悉本专业领域的技术人员在本发明揭露的技术范围内,可以对本发明所作的任何修改、调整,都应当涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1