制备用于增材制造的可光固化配制剂的制作方法
最近经常遇到的任务是快速制造原型和/或小型系列。增材制造方法是用于制造的逐层增加方法,其设计用来转移已有的三维计算机辅助设计数据(cad),理想地不需要直接和快速地手动转换成工件。
对于增材制造,已知有不同的方法,这可细分为基于光照射的方法和不使用光照射的方法。
利用基于光照射的方法将可固化液体组合物逐层固化,这优选使用uv发光激光器、uv发光二极管(led)或uv灯进行,其或者通过uv照射点的快速扫描或通过使用微镜阵列的大面积uv光投影进行。作为可固化液体组合物,例如使用单体或单体混合物。借助uv照射诱导所述单体的聚合。
其他增材制造方法例如为光聚合物喷射方法(ppj)、选择性激光烧结方法(sls)和熔融长丝制造方法(fff)(也称为熔融沉积成型(fdm))。
ep2151214公开了用于立体光刻制备牙科陶瓷的光固化浆料。所述浆料包含5-65重量%的可聚合粘合剂、0.001-1重量%的光引发剂和35-90重量%的表面改性的陶瓷和/或玻璃陶瓷颗粒。陶瓷或玻璃陶瓷颗粒优选具有10-200纳米的粒度。此外,所述浆料可包含一种或多种分散剂和颜料。
ep2404590也公开了一种用于立体光刻制备高强度陶瓷的光固化陶瓷浆料。该浆料包含1-30重量%的至少一种酸性单体、0-50重量%的至少一种非酸性的可自由基聚合单体、0.001-2重量%的光引发剂和30-90重量%的陶瓷和/或玻璃陶瓷颗粒。陶瓷或玻璃陶瓷颗粒的粒径粒度为10-200纳米。此外,所述浆料可包含分散剂和颜料。
us2010/0003619公开了用于制造三维物体的系统和方法。us2010/0003619的示例性可uv固化树脂包含76重量%的平均粒度为7微米的二氧化硅粉末、19.17重量%的sr238单体、2.34重量%的sr494单体、1.58重量%的variquatc55分散剂和0.86重量%的irgacure819。
现有技术中描述的配制剂高度填充有陶瓷粉末。因此,这些配制剂的陶瓷组分倾向于随时间沉淀,从而在输送或打印设备的容器底部形成沉积物。该沉积层通常很难再分散。这使得几乎不可能在增材制造方法中使用陶瓷组分会沉淀的配制剂。如果陶瓷颗粒很大(微米尺寸,这是用打印模具实施的某些金属铸造方法所需的),则所述的沉淀特别强烈。
因此,本发明的技术问题是提供一种制备可光固化配制剂的改进方法,其不具有上述现有技术的缺点或者仅以显著降低的程度具有这些缺点。此外,所述方法应以简单、安全和成本有效的方式进行。
该目的通过一种制备用于增材制造方法的可光固化配制剂(f)的方法来实现,所述方法包括如下步骤:
a)提供包含如下组分的陶瓷分散体(cd):
(a)至少一种d50值为至少2μm的陶瓷材料,
(b1)至少一种第一丙烯酸化物,
(c)至少一种分散剂,
b)提供包含如下组分的溶液(s):
(b2)至少一种第二丙烯酸化物,
(d)至少一种光引发剂,
c)将步骤a)中提供的陶瓷分散体(cd)与步骤b)中提供的溶液(s)混合,从而获得可光固化配制剂(f)。
可光固化配制剂(f)优选通过暴露于uv光源而可光固化。因此,本发明的另一目的为一种制备可光固化配制剂(f)的方法,其中可光固化配制剂(f)通过暴露于uv光源而可光固化。
陶瓷分散体(cd)和溶液(s)可在步骤c)中通过常规混合设备混合。这使得本发明的方法非常简单、稳健且成本有效。
此外,令人惊讶地发现陶瓷分散体(cd)即使在较长时间内也不会塌陷,尽管陶瓷材料具有至少2μm的d50值。
获得的可光固化配制剂(f)具有足以用于增材制造方法的低粘度。
此外,由本发明方法获得的可光固化配制剂(f)可直接用于增材制造方法中,而不需要添加其他组分并且在该方法中在至少12小时内抗沉积稳定。
在增材制造方法中由可光固化配制剂(f)获得的三维零件具有高精度。此外,未固化的可光固化配制剂(f)可容易地从增材制造方法中获得的模制品中移除。此外,通过增材制造技术获得的可光固化配制剂(f)的模制品是机械稳定的,从而使得它们可容易地进一步加工,例如容易地移除粘合剂和烧结。
陶瓷分散体(cd)和溶液(s)在输送期间是稳定的,并且可储存至少12周。
已发现如果陶瓷分散体(cd)的粘度较高(例如用布鲁克菲尔德粘度计(转子64,6rpm)在25℃的温度下测得为15-50pa·s),则其是特别稳定的。
下文将更详细地描述本发明的方法。
步骤a)
在步骤a)中,提供陶瓷分散体(cd)。该陶瓷分散体(cd)包含至少一种d50值为至少2μm的陶瓷材料作为组分(a)、至少一种第一丙烯酸化物作为组分(b1)和至少一种分散剂作为组分(c)。
就本发明而言,术语“至少一种陶瓷材料”和“组分(a)”是同义的,并且在本发明通篇中可互换使用。
这同样适用于术语“至少一种第一丙烯酸化物”和“组分(b1)”,以及术语“至少一种分散剂”和“组分(c)”。就本发明而言,术语“至少一种第一丙烯酸化物”和“组分(b1)”是同义的,并且在本发明通篇中可互换使用。就本发明而言,术语“至少一种分散剂”和“组分(c)”也是同义的,并且在本发明通篇中可互换使用。
下文将更详细地讨论组分(a)、(b1)和(c)。
陶瓷分散体(cd)可以以任意比例包含组分(a)、(b1)和(c)。优选地,本发明方法的步骤a)中提供的陶瓷分散体(cd)包含74.9-95重量%的组分(a)、0.1-25重量%的组分(b1)和0.1-15重量%的组分(c),基于组分(a)、(b1)和(c)的重量百分比之和,优选基于陶瓷分散体(cd)的总重量。
特别优选地,步骤a)中提供的陶瓷分散体(cd)包含78-92重量%的组分(a)、3-20重量%的组分(b1)和0.4-11重量%的组分(c),基于组分(a)、(b1)和(c)的重量百分比之和,优选基于陶瓷分散体(cd)的总重量。
更优选地,步骤a)中提供的陶瓷分散体(cd)包含81-89重量%的组分(a)、6-15重量%的组分(b1)和0.7-7重量%的组分(c),基于组分(a)、(b1)和(c)的重量百分比之和,优选基于陶瓷分散体(cd)的总重量。
在优选实施方案中,本发明方法的步骤a)中提供的陶瓷分散体(cd)优选包含57-90体积%的组分(a)、10-42体积%的组分(b1)和0.1-15体积%的组分(c),基于组分(a)、(b1)和(c)的体积百分比之和,优选基于陶瓷分散体(cd)的总体积。
特别优选地,步骤a)中提供的陶瓷分散体(cd)包含62.1-84.1体积%的组分(a)、10-37.2体积%的组分(b1)和0.7-15%体积的组分(c),基于组分(a)、(b1)和(c)的体积百分比之和,优选基于陶瓷分散体(cd)的总体积。
更优选地,步骤a)中提供的陶瓷分散体(cd)包含66.3-78.9体积%的组分(a)、11.3-28.1%体积的组分(b1)和1.3-13.2体积%的组分(c),基于组分(a)、(b1)和(c)的体积百分比之和,优选基于陶瓷分散体(cd)的总体积。
因此,本发明的另一目的为一种方法,其中步骤a)中提供的陶瓷分散体(cd)包含74.9-95重量%的组分(a)、0.1-25重量%的组分(b1)和0.1-15重量%的组分(c),基于组分(a)、(b1)和(c)的重量百分比之和。
陶瓷分散体(cd)中所含的组分(a)、(b1)和(c)的重量百分比通常加和为100重量%。此外,陶瓷分散体(cd)中所含的组分(a)、(b1)和(c)的体积百分比通常加和为100体积%。
在优选实施方案中,陶瓷分散体(cd)不含光引发剂,特别是不含组分(d)。
因此,本发明的另一目的也为一种方法,其中陶瓷分散体(cd)不含光引发剂。
在步骤a)中提供陶瓷分散体(cd)可通过本领域技术人员所已知的任何方法进行。通常,通过混合组分(a)、(b1)和(c)来提供陶瓷分散体(cd)。优选的是高剪切混合设备,优选剪切速率为20-100s-1。合适的混合设备是本领域技术人员所已知的。合适混合设备的实例包括捏合机、行星式混合机和立式混合机(例如eirich强力混合机)。
优选地,通过首先混合组分(b1)和(c)来提供陶瓷分散体(cd)。然后逐步加入组分(a)并混合。混合期间的温度优选为至多60℃,特别优选为5-60℃。
陶瓷分散体(cd)通常包含分散在组分(b1)和(c)中的组分(a)。
因此,组分(b1)和(c)为连续相,组分(a)为陶瓷分散体(cd)中的分散相。
陶瓷分散体(cd)可在低于组分(b1)和组分(c)的分解温度的任何温度下提供。
优选地,陶瓷分散体(cd)在步骤a)中在5-40℃,更优选10-35℃,最优选15-30℃的温度下提供。
陶瓷分散体(cd)的粘度优选为15-50pa·s,特别优选为18-40pa·s,更优选为20-30pa·s,用布鲁克菲尔德粘度计(转子类型64,6rpm)在25℃温度下测量。
优选地,陶瓷分散体(cd)在-20℃至80℃的温度下储存至少12周内抗沉积稳定。
步骤b)
在步骤b)中,提供溶液(s),其包含至少一种第二丙烯酸化物作为组分(b2)和至少一种光引发剂作为组分(d)。
就本发明而言,术语“至少一种第二丙烯酸化物”和“组分(b2)”是同义的,并且在本发明通篇中可互换使用。
这同样适用于术语“至少一种光引发剂”和“组分(d)”。就本发明而言,术语“至少一种光引发剂”和“组分(d)”也是同义的,并且在本发明通篇中可互换使用。
下文将更详细地描述组分(b2)和组分(d)。
例如,步骤b)中提供的溶液(s)包含75-99.9重量%的组分(b2)和0.1-25重量%的组分(d),基于组分(b2)和(d)的重量百分比之和,优选基于溶液(s)的总重量。
优选地,步骤b)中提供的溶液(s)包含80-99.9重量%的组分(b2)和0.1-20重量%的组分(d),基于组分(b2)和(d)的重量百分比之和,优选基于溶液(s)的总重量。
特别优选地,步骤b)中提供的溶液(s)包含84-97重量%的组分(b2)和3-16重量%的组分(d),基于组分(b2)和(d)的重量百分比之和,优选基于溶液(s)的总重量。
因此,本发明的另一目的还为一种方法,其中步骤b)中提供的溶液(s)包含75-99.9重量%的组分(b2)和0.1-25重量%的组分(d),基于组分(b2)和(d)的重量百分比之和。
步骤b)中提供的溶液(s)优选包含组分(c2)—至少一种第二分散剂。
术语“组分(c2)”和“至少一种第二分散剂”是同义的,并且在本发明通篇中可互换使用。
例如,溶液(s)包含0.01-50重量%,优选0.01-30重量%,特别优选0.01-15重量%的组分(c2),基于溶液的总重量(s)。
对于组分(c2),使用下文对组分(c)给出的实施方案和优选方案。
因此,本发明的另一目的还为一种方法,其中步骤b)中提供的溶液(s)进一步包含组分(c2)—至少一种分散剂。
此外,步骤b)中提供的溶液(s)可包含至少一种添加剂。合适的至少一种添加剂是本领域技术人员所已知的,并且优选选自uv吸收剂。
因此,本发明的另一目的还为一种方法,其中步骤b)中提供的溶液(s)进一步包含至少一种选自uv吸收剂的添加剂。
合适的uv吸收剂是本领域技术人员所已知的。优选的uv吸收剂选自2-羟基苯基二苯甲酮类、2-(2-羟基苯基)苯并三唑类和2-羟基苯基-s-三嗪类。这些uv吸收剂是本领域技术人员所已知的,并且可由basfse以商品名
如果溶液(s)包含至少一种添加剂,则溶液(s)优选以0.1-3.7重量%,更优选0.1-2.5重量%,特别优选0.1-1.3重量%的量包含所述添加剂,基于溶液(s)的总重量。
溶液(s)中所含的组分(b2)、组分(d)和任选的组分(c2)和所述至少一种添加剂的重量百分比通常加和为100重量%。
在优选实施方案中,溶液(s)不含陶瓷材料,特别是不含组分(a)。
因此,本发明的另一目的还为一种方法,其中步骤b)中提供的溶液(s)不含陶瓷材料。
溶液(s)通常包含溶解在组分(b2)中的组分(d)。
溶液(s)的提供可通过本领域技术人员已知的任何方法进行。
优选地,通过混合组分(b2)和(d)以及任选的组分(c2)和至少一种添加剂来提供溶液(s)。混合可通过本领域技术人员已知的任何方法进行,例如用螺旋桨式搅拌器、桨式混合机或分散盘。将组分以任意顺序加在一起,然后混合1-2小时,或者直至均匀。
溶液(s)的提供可在低于组分(b2)和(d)的分解温度的任何温度下进行。
优选地,溶液(s)在步骤b)中在5-40℃,特别优选10-35℃,更优选15-30℃的温度下提供。
组分(a)
组分(a)为至少一种d50值为至少2μm的陶瓷材料。
在本发明的上下文中,“至少一种陶瓷材料”意指正好一种陶瓷材料以及两种或更多种陶瓷材料的混合物。
所述至少一种陶瓷材料具有至少2μm的d50值,优选d50值为2-10μm。
在另一优选实施方案中,组分(a)具有:
0.01-5μm的d10值,
2-10μm的d50值,和
20-35μm的d90值。
特别优选地,组分(a)具有:
0.1-2μm的d10值,
4-10μm的d50值,
25-35μm的d90值。
在本发明的上下文中,“d10值”是指基于颗粒总体积的10体积%颗粒小于或等于d10值且基于颗粒总体积的90体积%颗粒大于d10值时的粒度。在本发明的上下文中,“d50值”是指基于颗粒总体积的50体积%颗粒小于或等于d50值且基于颗粒总体积的50体积%颗粒大于d50值时的粒度。在本发明的上下文中,“d90值”是指基于颗粒总体积的90体积%颗粒小于或等于d90值且基于颗粒总体积的10体积%颗粒大于d90值时的粒度。
d10、d50和d60值使用malvernmastersizer2000粒度分析仪通过激光衍射测量。在即将测量之前,通过搅拌和超声处理将陶瓷材料在去离子水中分散10分钟。
组分(a)为至少一种陶瓷材料。在本发明的上下文中,陶瓷材料意指金属或第一准金属和非金属或第二准金属的非金属化合物。
“金属”意指正好一种金属以及两种或更多种金属的混合物。这同样适用于“非金属”、“第一准金属”和“第二准金属”。“非金属”意指正好一种非金属以及两种或更多种非金属的混合物。“第一准金属”意指正好一种第一准金属以及两种或更多种第一准金属的混合物。“第二准金属”意指正好一种第二准金属以及两种或更多种第二准金属的混合物。
合适的金属是本领域技术人员所已知的。优选地,金属选自锆、铝、锌、铁、钛和钇。
合适的非金属是本领域技术人员所公知的。本发明的非金属可选自周期表的任何非金属,优选非金属选自碳、氮、氧、磷和硫,特别优选非金属为氧。
准金属是本领域技术人员所公知的。第一准金属和第二准金属可选自周期表的任何准金属。优选地,第一准金属和/或第二准金属选自硼和硅。应知晓的是,第一准金属和第二准金属彼此不同。例如,如果第一准金属为硼,则第二准金属选自除硼之外的元素周期表的任何其他准金属。
优选地,组分(a)选自氧化物、碳化物、硼化物、氮化物和硅化物,特别优选组分(a)选自氧化物。
在优选实施方案中,组分(a)选自sio2、zro2、al2o3、zno、fe2o3、fe3o4、y2o3、tio2、sic、si3n4、tib和aln。
特别优选地,组分(a)选自sio2、zro2、al2o3、zno、fe2o3、fe3o4、y2o3和tio2。更特别优选的组分(a)选自sio2和zro2。
因此,本发明的另一目的还为一种方法,其中组分(a)选自sio2、zro2、al2o3、zno、fe2o3、fe3o4、y2o3、tio2、sic、si3n4、tib和aln。
组分(b1)和(b2)
组分(b1)为至少一种第一丙烯酸化物。
在本发明的上下文中,“至少一种第一丙烯酸化物”意指正好一种第一丙烯酸化物以及两种或更多种第一丙烯酸化物的混合物。
组分(b2)为至少一种第二丙烯酸化物。
在本发明的上下文中,“至少一种第二丙烯酸化物”意指正好一种第二丙烯酸化物以及两种或更多种第二丙烯酸化物的混合物。优选两种或更多种第二丙烯酸化物的混合物。
组分(b1)和组分(b2)可彼此相同或不同。
优选地,组分(b1)和组分(b2)彼此独立地选自丙烯酸化物和甲基丙烯酸化物。
因此,本发明的另一目的为一种方法,其中组分(b1)和组分(b2)彼此独立地选自丙烯酸化物和甲基丙烯酸化物。
合适的丙烯酸化物和甲基丙烯酸化物是本领域技术人员所已知的。
在下文中,将丙烯酸化物和甲基丙烯酸化物概括为(甲基)丙烯酸化物。
合适的(甲基)丙烯酸化物为单(甲基)丙烯酸化物以及多官能(甲基)丙烯酸化物。
因此,本发明的另一目的还为一种方法,其中组分(b1)和组分(b2)彼此独立地选自单(甲基)丙烯酸化物和多官能(甲基)丙烯酸化物。
合适的单(甲基)丙烯酸化物是本领域技术人员所已知的。合适的单(甲基)丙烯酸化物优选选自(甲基)丙烯酸异冰片基酯、(甲基)丙烯酸冰片基酯、(甲基)丙烯酸三环癸基酯、(甲基)丙烯酸二环戊基酯、(甲基)丙烯酸二环戊烯基酯、(甲基)丙烯酸环己基酯、(甲基)丙烯酸苄基酯、(甲基)丙烯酸-4-丁基环己基酯、丙烯酰吗啉、(甲基)丙烯酸、(甲基)丙烯酸2-羟乙酯、(甲基)丙烯酸2-羟丙酯、(甲基)丙烯酸2-羟丁酯、(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丙酯、(甲基)丙烯酸异丙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸戊酯、(甲基)丙烯酸异丁酯、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸戊酯、己内酯丙烯酸酯、(甲基)丙烯酸异戊酯、(甲基)丙烯酸己酯、(甲基)丙烯酸庚酯、(甲基)丙烯酸辛酯、(甲基)丙烯酸异辛酯、(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸壬酯、(甲基)丙烯酸癸酯、(甲基)丙烯酸异癸酯、(甲基)丙烯酸十三烷基酯、(甲基)丙烯酸十一烷基酯、(甲基)丙烯酸月桂基酯、(甲基)丙烯酸硬脂基酯、(甲基)丙烯酸异硬脂基酯、(甲基)丙烯酸四氢糠基酯、(甲基)丙烯酸丁氧基乙酯、乙氧基二甘醇(甲基)丙烯酸酯、(甲基)丙烯酸苄基酯、(甲基)丙烯酸苯氧基乙酯、聚乙二醇单(甲基)丙烯酸酯、聚丙二醇单(甲基)丙烯酸酯、甲氧基乙二醇(甲基)丙烯酸酯、(甲基)丙烯酸乙氧基乙酯、甲氧基聚乙二醇(甲基)丙烯酸酯、甲氧基聚丙二醇(甲基)丙烯酸酯、双丙酮(甲基)丙烯酰胺、(甲基)丙烯酸β-羧乙酯、邻苯二甲酸(甲基)丙烯酸酯、(甲基)丙烯酸二甲基氨基乙酯、(甲基)丙烯酸二乙基氨基乙酯、(甲基)丙烯酸丁基氨基甲酰基乙酯、正-异丙基(甲基)丙烯酰胺、氟代(甲基)丙烯酸酯和(甲基)丙烯酸7-氨基-3,7-二甲基辛酯。
合适的多官能(甲基)丙烯酸化物是本领域技术人员所公知的,并且例如选自三羟甲基丙烷三(甲基)丙烯酸酯、季戊四醇(甲基)丙烯酸酯、乙二醇二(甲基)丙烯酸酯、双酚a二缩水甘油醚二(甲基)丙烯酸酯、二环戊二烯二甲醇二(甲基)丙烯酸酯、[2-[1,1-二甲基-2-[(1-氧代烯丙基)氧基]乙基]-5-乙基-1,3-二
特别优选的组分(b1)和组分(b2)彼此独立地选自双酚a二缩水甘油醚二(甲基)丙烯酸酯、乙氧基化或丙氧基化双酚a或双酚f二(甲基)丙烯酸酯、二环戊二烯二甲醇二(甲基)丙烯酸酯、[2-[1,1-二甲基-2-[(1-氧代烯丙基)氧基]乙基]-5-乙基-1,3-二
最优选地,组分(b1)和组分(b2)彼此独立地选自乙氧基化三羟甲基丙烷三(甲基)丙烯酸酯和1,6-己二醇二(甲基)丙烯酸酯。
特别优选地,组分(b1)为1,6-己二醇二(甲基)丙烯酸酯,组分(b2)为1,6-己二醇二(甲基)丙烯酸酯与乙氧基化三羟甲基丙烷三(甲基)丙烯酸酯的混合物。
因此,本发明的另一目的为一种方法,其中组分(b1)和组分(b2)彼此独立地选自双酚a二缩水甘油醚二(甲基)丙烯酸酯、乙氧基化或丙氧基化双酚a或双酚f二(甲基)丙烯酸酯、二环戊二烯二甲醇二(甲基)丙烯酸酯、[2-[1,1-二甲基-2-[(1-氧代烯丙基)氧基]乙基]-5-乙基-1,3-二
优选地,组分(b1)和组分(b2)彼此独立地具有≥1至6,更优选1-4,特别优选2-3的c-c双键官能度。
因此,本发明的另一目的为一种方法,其中组分(b1)和组分(b2)彼此独立地具有≥1至6的c-c双键官能度。
本发明上下文中,c-c双键官能度意指每分子中的丙烯酰基和甲基丙烯酰基单元的数量。该c-c双键官能度的测量方法是本领域技术人员所已知的。
如果组分(b1)为两种或更多种第一丙烯酸化物的混合物和/或组分(b2)为两种或更多种第二丙烯酸化物的混合物,则c-c双键官能度是每分子的所有丙烯酰基和甲基丙烯酰基单元的平均值。
组分(c)
组分(c)为至少一种分散剂。
在本发明的上下文中,“至少一种分散剂”意指正好一种分散剂以及两种或更多种分散剂的混合物。
合适的分散剂是本领域技术人员所已知的。优选地,组分(c)选自乙氧基化脂肪醇、聚氧亚丙基/亚乙基嵌段共聚物、乙氧基化壬基酚、聚乙二醇对辛基苯基醚、烷氧基化二胺、月桂基硫酸钠和阳离子分散剂。阳离子分散剂是特别优选的。
因此,本发明的另一目的还为一种方法,其中组分(c)选自乙氧基化脂肪醇、聚氧亚丙基/亚乙基嵌段共聚物、乙氧基化壬基酚、聚乙二醇对辛基苯基醚、烷氧基化二胺、月桂基硫酸钠和阳离子分散剂。
合适的阳离子分散剂是本领域技术人员所已知的。优选烷氧基化季铵盐,尤其是聚丙氧基季铵氯化物。因此,组分(c)优选选自聚丙氧基季铵氯化物。聚丙氧基季铵氯化物以evonik的商品名
对任选包含在溶液(s)中的组分(c2)而言,适用上述实施方案和优选方案。
组分(d)
组分(d)为至少一种光引发剂。
在本发明的上下文中,“至少一种光引发剂”意指正好一种光引发剂以及两种或更多种光引发剂的混合物。
在本发明的上下文中,光引发剂为暴露于uv照射时产生反应性物种的分子。
作为组分(d),在暴露于uv照射时产生反应性物种的任何光引发剂都是合适的。
优选地,组分(d)选自二苯甲酮、烷基二苯甲酮、卤代甲基化二苯甲酮、米蚩酮、苯偶姻、苯偶姻醚、苄基缩酮(benzylketal)、苯乙酮衍生物、苯基乙醛酸、蒽醌、甲基蒽醌、酰基氧化膦和双酰基氧化膦。特别优选为苯乙酮衍生物。
因此,本发明的另一目的为一种方法,其中组分(d)选自二苯甲酮、烷基二苯甲酮、卤代甲基化二苯甲酮、米蚩酮、苯偶姻、苯偶姻醚、苄基缩酮、苯乙酮衍生物、苯基乙醛酸、蒽醌、甲基蒽醌、酰基氧化膦和双酰基氧化膦。
优选的组分(d)为α-羟基酮,单-和双-酰基氧化膦。
上述优选组分(d)是本领域技术人员所已知的。米蚩酮也称为4,4'-双(n,n-二甲基氨基)二苯甲酮。
优选的酰基氧化膦为2,4,6-三甲基苯甲酰基二苯基氧化膦和2,4,6-三甲基苯甲酰基苯基次膦酸乙酯。双酰基氧化膦的实例为双(2,4,6-三甲基苯甲酰基)苯基氧化膦。
苯乙酮衍生物的实例为羟基-2-甲基-1-苯基丙-1-酮和羟基环己基苯基酮。
对组分(d),作为实例可提及二苯甲酮、苯乙酮、乙酰萘醌、甲基乙基酮、苯戊酮、苯己酮、α-苯基苯丁酮、对吗啉代苯丙酮、二苯并环庚酮、4-吗啉代二苯甲酮、4-吗啉代脱氧苯偶姻、对二乙酰苯、4-氨基二苯甲酮、4'-甲氧基苯乙酮、β-甲基蒽醌、叔丁基蒽醌、蒽醌羧酸酯、苯甲醛、α-四氢萘酮、9-乙酰基菲、2-乙酰基菲、10-噻吨酮、3-乙酰基菲、3-乙酰基吲哚、9-芴酮、2,3-二氢1-茚酮、1,3,4-三乙酰基苯、噻吨-9-酮、呫吨-9-酮、2,4-二甲基噻吨酮、2,4-二乙基噻吨酮、2,4-二异丙基噻吨酮、2,4-二氯噻吨酮、羟基环己基苯基酮、苯偶姻、苯偶姻异丁醚、氯呫吨酮、苯偶姻四氢吡喃醚、苯偶姻甲醚、苯偶姻乙醚、苯偶姻丁醚、苯偶姻异丙醚、7h-苯偶姻甲醚、苯并[de]蒽-7-酮、1-萘甲醛、4,4'-双(二甲氨基)二苯甲酮、4-苯基二苯甲酮、4-氯二苯甲酮、米蚩酮、1-乙酰萘、2-乙酰萘、1-苯甲酰基环己-1-醇、2-羟基-2,2-二甲基苯乙酮、2,2-二甲氧基-2-苯基苯乙酮、2,2-二乙氧基-2-苯基苯乙酮、1,1-二氯苯乙酮、1-羟基苯乙酮、苯乙酮二甲基缩酮、邻甲氧基二苯甲酮、三苯基膦、三邻甲苯基膦、苯并[a]蒽-7,12-二酮、2,2-二乙氧基苯乙酮,苯偶酰缩酮如苯偶酰二甲基缩酮,2-甲基-1-[4-(甲硫基)苯基]-2-吗啉代丙-1-酮,蒽醌如2-甲基蒽醌、2-乙基蒽醌、2-叔丁基蒽醌、1-氯蒽醌、2-戊基蒽醌和2,3-丁二酮。
特别优选地,组分(d)为羟基环己基苯基酮。羟基环己基苯基酮由basfse以商品名
步骤c)
在步骤c)中,将步骤a)中提供的陶瓷分散体(cd)和步骤b)中提供的溶液(s)混合,从而获得可光固化配制剂(f)。
陶瓷分散体(cd)和溶液(s)可通过本领域技术人员所已知的任何方法混合。优选的是通常已知的混合设备,例如行星式混合机、螺旋桨式搅拌器和分散盘。将陶瓷分散体(cd)和溶液(s)以任意顺序加入混合容器中,并根据混合设备混合至少30分钟至2小时。高剪切速率和延长的混合时间可促进可光固化配制剂(f)的稳定性。
陶瓷分散体(cd)和溶液(s)的混合可在优选低于60℃的任何温度下进行。优选地,混合在15-50℃,特别优选17-35℃,最优选20-30℃的温度下进行。
因此,本发明的另一目的为一种方法,其中将步骤a)中提供的陶瓷分散体(cd)与步骤b)中提供的溶液(s)混合以获得可光固化配制剂(f)的步骤c)在15-40℃的温度下进行。
陶瓷分散体(cd)和溶液(s)可在步骤c)中以任意比例混合。优选地,在步骤c)中,将75-99重量%的陶瓷分散体(cd)与1-25重量%的溶液(s)混合,从而获得可光固化配制剂(f),基于陶瓷分散体(cd)和溶液(s)的重量百分比之和。
特别优选地,在步骤c)中,将85-95重量%的陶瓷分散体(cd)与5-15重量%的溶液(s)混合,从而获得可光固化配制剂(f),基于陶瓷分散体(cd)和溶液(s)的重量百分比之和。
因此,本发明的另一目的为一种方法,其中在步骤c)中,将75-99重量%的陶瓷分散体(cd)与1-25重量%的溶液(s)混合,从而获得可光固化配制剂(f),基于陶瓷分散体(cd)和溶液(s)的重量百分比之和。
可光固化配制剂(f)
在步骤c)中,获得了可光固化配制剂(f)。
可光固化配制剂(f)包含陶瓷分散体(cd)和溶液(s)中所含的组分。可光固化配制剂(f)可包含反应和/或未反应形式的组分。优选地,可光固化配制剂(f)包含未反应形式的组分。
如果陶瓷分散体(cd)包含其他添加剂和/或溶液(s)包含至少一种添加剂和/或组分(c2),则步骤c)中获得的可光固化配制剂(f)通常还包含其他添加剂和/或组分(c2)和/或所述至少一种添加剂。
例如,可光固化配制剂(f)包含72-83重量%的组分(a)、14-25重量%的组分(b1)和(b2)、0.8-6.0重量%的组分(c)和0.1-2.0重量%的组分(d),基于组分(a)、(b1)、(b2)、(c)和(d)的重量百分比之和,优选基于可光固化配制剂(f)的总重量。
所述配制剂的粘度优选为0.1-3pa·s,特别优选为0.15-2.8pa·s,最优选为0.2-2.5pa·s,使用布鲁克菲尔德粘度计(转子类型62,12rpm)在25℃温度下测量。
可光固化配制剂(f)优选在增材制造方法中在20-30℃的温度下在至少12小时内抗沉积稳定。
因此,本发明的另一目的为一种可通过本发明方法获得的可光固化配制剂(f)。
增材制造方法
本发明的另一目的为一种在增材制造方法中制备模制品的方法,包括如下步骤:
i)提供根据权利要求11的可光固化配制剂(f),
ii)形成步骤i)中提供的可光固化配制剂(f)的第一部分的层,并使用uv光源固化形成的层的至少一部分以获得模制品。
增材制造方法是本领域技术人员所已知的,例如描述于us4,575,330中。
在步骤i)中,提供本发明的可光固化配制剂(f)。可光固化配制剂(f)可通过本领域技术人员已知的任何方法在步骤i)中提供。优选地,可光固化配制剂(f)以液态提供。特别优选地,可光固化配制剂(f)以液态提供在容器中。合适的容器是本领域技术人员所已知的。容器优选包括工作表面。
在步骤ii)中,形成可光固化配制剂(f)的第一部分的层,并使用uv光源固化该层的至少一部分。可光固化配制剂(f)的第一部分的层优选通过使用工作表面形成。该层的厚度优选为50-150μm,特别优选为70-130μm,最优选为80-120μm。
然后,通过暴露于uv光源使可光固化配制剂的第一部分的层固化。
优选的uv光源为uv激光器、uv灯和uv发光二极管。
在固化期间,形成所述至少一种光引发剂(组分(d))的自由基并引发所述至少一种第一丙烯酸化物和所述至少一种第二丙烯酸化物的聚合。由于组分(b1)和(b2)的聚合,这导致可光固化配制剂(f)硬化。
优选地,在第二步骤中,形成可光固化配制剂(f)的第二部分的第二层。该第二层通常通过将工作表面向下移动而形成,从而使得包含在容器中的液体可光固化配制剂(f)流到工作表面上并且流到可光固化配制剂(f)的固化部分上。通常使用再涂刮刀使该层变平。
因此,优选地,步骤ii)包括如下步骤:
ii-1)形成步骤i)中提供的可光固化配制剂(f)的第一部分的层,
ii-2)使用uv光源固化步骤ii-1)中形成的可光固化配制剂(f)的第一部分的
层的至少一部分,从而获得固化层,
ii-3)在步骤ii-2)中获得的固化层上形成步骤i)中提供的可光固化配制剂(f)
的第二部分的第二层,
ii-4)使用uv光源固化步骤ii-3)中形成的可光固化配制剂(f)的第二部分的
第二层的至少一部分,从而获得模制品。
因此,本发明的另一目的为一种制备模制品的方法,其中步骤ii)包括如下步骤:
ii-1)形成步骤i)中提供的可光固化配制剂(f)的第一部分的层,
ii-2)使用uv光源固化步骤ii-1)中形成的可光固化配制剂(f)的第一部分的
层的至少一部分,从而获得固化层,
ii-3)在步骤ii-2)中获得的固化层上形成步骤i)中提供的可光固化配制剂(f)
的第二部分的第二层,
ii-4)使用uv光源固化步骤ii-3)中形成的可光固化配制剂(f)的第二部分的
第二层的至少一部分,从而获得模制品。
优选地,步骤ii-1)和ii-2)重复至少一次。
因此,本发明的另一目的还为一种方法,其中步骤ii-1)和ii-2)重复至少一次。
步骤ii)中获得的模制品通常在其表面上包含未固化的可光固化配制剂(f)。该未固化的可光固化配制剂(f)可通过用水溶性有机溶剂洗涤所得模制品而移除。合适的水溶性有机溶剂是本领域技术人员所已知的。优选的水溶性有机溶剂为二醇及其衍生物。特别优选三丙二醇甲醚。在用水溶性有机溶剂洗涤后,优选用水洗涤模制品,然后干燥。
因此,本发明的另一目的为可光固化配制剂(f)在增材制造方法中的用途。可光固化配制剂(f)优选通过暴露于uv光源而可光固化。
下文参考实施例说明本发明,但不限于此。
实施例
陶瓷分散体(cd)
陶瓷分散体通过使用作为高剪切分散设备的双螺旋混合机(螺旋间距1:1,搅拌器直径12.5英寸)在至多100rpm的搅拌器速度下将作为组分(a)的86.7重量%d50值为9μm的二氧化硅(sio2)粉末与作为组分(c)的1.73重量%聚丙氧基季铵氯化物(商品名:variquatccns42,evonik)在作为组分(b1)的己二醇二丙烯酸酯(商品名:laromerhdda,basfse)中混合来制备。
混合程序在10加仑反应器中进行。将组分(b1)和组分(c)倾入容器中并以最小速度(15-20rpm)混合。然后逐步加入组分(a),同时以最低速度混合。使用温度控制来将批料温度保持为低于35℃。在加入组分(a)后,将混合物以基于扭矩确定的速度搅拌2小时。将扭矩保持为低于90ft-lb是合乎需要的。
获得的陶瓷分散体(cd)的粘度为21pa·s,使用布鲁克菲尔德粘度计(转子类型64,6rpm)在25℃的温度下测量。
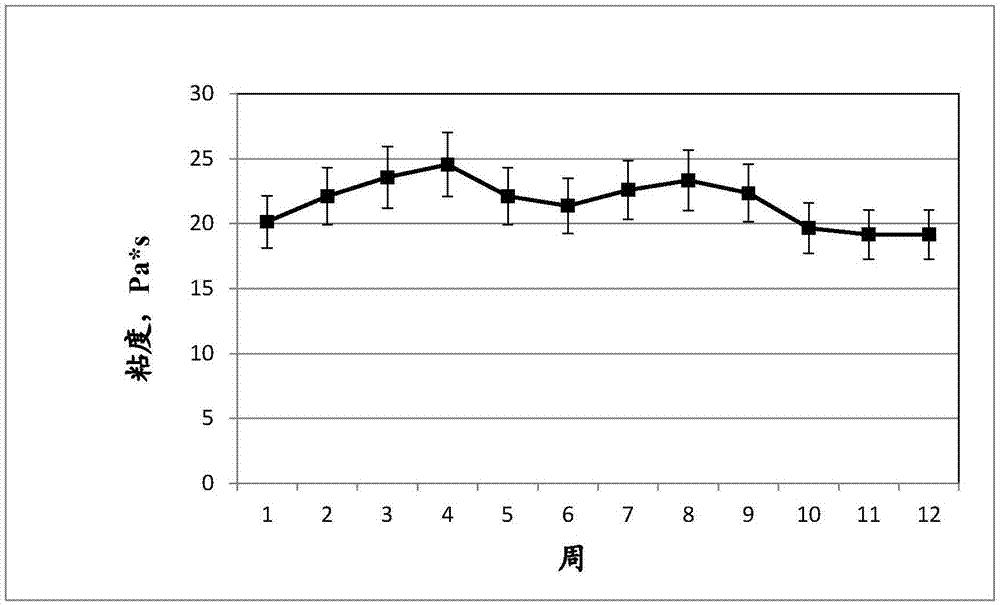
在填充至10cm高度的容纳罐底部没有获得沉积物形成,在下拉材料的光学显微镜观察中没有看到附聚物,并且没有获得粘度随时间的显著变化。陶瓷分散体(cd)在经历循环温度变化(循环:在-20℃下保持6小时,经6小时升温至50℃,在50℃下保持6小时,经6小时降至-20℃)下稳定至少12周。
图1显示了所得陶瓷分散体(cd)在12周内的粘度。可以看出,在标准偏差内,粘度随时间的变化是恒定的。
可光固化配制剂(f)
将获得的陶瓷分散体(cd)与包含作为组分(b2)的己二醇二丙烯酸酯(商品名:
获得的可光固化配制剂(f)的粘度为2.1pa·s,使用布鲁克菲尔德粘度计(转子类型62,12rpm)在25℃下测量。
可光固化配制剂的(f)稳定性定义为在室温(20℃)下形成沉积的起始时间。沉积形成通过用刮刀刮擦填充有10cm高度的可光固化配制剂(f)的容纳罐的底部来测试。可光固化配制剂(f)稳定12小时。
将可光固化配制剂(f)用于立体光刻装置(raplasrps450)中以获得模制品。然后,通过用三丙二醇甲醚(tpm)漂洗和温和刷洗,然后用水漂洗来清除模制品中的未反应材料。获得的模制品显示出高精度和低卷曲变形。
- 还没有人留言评论。精彩留言会获得点赞!