一种片状纳米氧化镁粉体的制备方法与流程
本发明涉及纳米材料工艺的技术领域,尤其涉及一种片状纳米氧化镁粉体的制备方法。
背景技术:
纳米氧化镁是一种新型高功能精细无机材料。尤其是当mgo尺寸细化至纳米量级后,因纳米材料所特有的体积效应和表面效应,使得纳米mgo的低温烧结、微波吸收、催化性能等众多方面呈现出许多不同于本体材料的热光电和化学特性,因而引起了人们的广泛兴趣。在纺织领域,随着高性能阻燃纤维的需求越来越高,合成新型高性能阻燃剂就为发展功能面料提供了理想的材料,与传统的一些含磷或卤素有机阻燃剂相比,纳米氧化镁无毒、无味、添加量小,是开发阻燃纤维的理想添加剂。另外,纳米氧化镁可作为油漆、纸张及化妆品的填料、塑料和橡胶的添加剂和补强剂、脂肪的分解剂、医药品的擦光剂、化学吸附剂、以及各种电子材料、催化剂、超导体、耐火材料的辅助材料等,有着广阔的应用前景和巨大的经济潜力。
利用廉价的白云石制备有高经济价值的氧化镁产品是目前世界镁工业的一个主要发展方向之一,也提出了相当数量的制备工艺和研究成果,并大量转化为生产,实现了广泛的社会经济效益。目前,白云石制备轻质氧化镁极其副产品目前全世界主要采用碳化法、二次碳化法、碳氨双循环法等。
加压碳化法:白云石于950℃到1200℃煅烧成氧化钙和氧化镁后,用水消化成浆,通入已净化的二氧化碳气,待氢氧化钙转成碳酸钙后,再于常温下进行加压碳化,使镁转化为碳酸氢镁溶液。过滤出碳酸钙后的溶液,热解得到碱式碳酸镁产品[8]。工艺的优点是用自身产生的二氧化碳气体为钙、镁分离及沉淀的原料,而无需辅助材料。其不足之处有设备投资大,生产能力低,副产钙盐纯度不高。
氯化铵—二氧化碳法:用氯化铵和二氧化碳来处理用水消化好的白云石烧熟料,使料浆中的钙转成为碳酸钙沉淀,而镁以氯化镁溶液的形式与钙分离。该工艺无需加压,但白云石矿中的杂质和煅烧时引入的煤灰等与碳酸钙沉淀一道被滤出,使碳酸钙产品严重不纯。
硫酸铵一步浸出法:工艺利用硫酸钙与硫酸镁溶液解度上的差异,用硫酸铵作浸取剂,过程产生的氨回收用来沉淀镁,分离出来硫酸钙后的富镁液用回收的氨水将镁沉淀。反复洗涤氢氧化镁至无硫酸根后,灼烧制得优质氧化镁。该工艺的氧化镁产品中的硫酸根易超标,而且生产周期长,生产成本也比较高。
离子交换树脂法:用h型强酸性离子交换树脂来处理白云石烧熟料后的消化浆,然后将吸附钙后的交换树脂与氢氧化镁浆液分离,吸附钙后的树脂用盐酸将钙解脱下来,可制得氯化钙产品。解脱后的树脂可反复使用,氢氧化镁浆液经酸浸或铵浸后,过滤、沉淀、洗涤、烘干、灼烧的氧化镁。该工艺没有从根本上解决如何降低成本的问题,分离出的含钙树脂,还需要消耗与钙相当的盐酸。
技术实现要素:
本发明的目的是为了克服上述现有技术的缺点,提供一种片状纳米氧化镁粉体的制备方法,该片状纳米氧化镁粉体的制备方法所制得的纳米氧化镁粉体为片层纳米微片结构,排列规则有序,生长均匀,呈外延辐射状生长,微片彼此交叉生长,晶粒小,分散度高,活性高。
本发明解决其技术问题所采用的技术方案是:一种片状纳米氧化镁粉体的制备方法,包括如下步骤:
(1)将白云石矿粉碎后于900~1100℃下煅烧2~3h成白云石灰,加水配置成重量固液比为1:2~5的混合液,然后于50~80℃下消化20min~1h得到熟料,熟料为氢氧化钙和氢氧化镁的混合悬浊液;在熟料中缓慢加入10%~25%w/w浓度的盐酸溶液,混合悬浊液ph值由14突变为6~7时停止加入盐酸,然后过滤分离得到滤液和滤渣,滤渣洗涤数次后备用;
(2)将步骤(1)中的滤渣打散,机械搅拌下滴入10%~25%w/w浓度的硫酸溶液,当体系变成红灰色浑浊液及ph为5~6时停止加入硫酸溶液,然后过滤分离得到富镁滤液;
(3)于富镁滤液中加入5~20毫升peg或不加入peg,然后加入乙醇浓缩得到富镁液,富镁液的乙醇含量为25%,富镁液加热至60℃并搅拌,缓慢滴加15%浓度氨水在80℃下水热30min后真空抽滤得到氢氧化镁沉淀物滤渣和滤液,氢氧化镁沉淀物滤渣以蒸馏水冲洗后再用无水乙醇洗涤数次;
(4)所得氢氧化镁沉淀物滤渣于70~90℃干燥1~2h得到纳米氢氧化镁粉体;
(5)把纳米氢氧化镁粉体在400-750℃温度下煅烧2-3h得到纳米氧化镁粉体。
进一步的,所述的纳米氧化镁粉体的平均晶粒粒径为15nm。
进一步的,所述步骤(5)中的煅烧温度的升温速率为10℃/min。
综上所述,本发明的片状纳米氧化镁粉体的制备方法所制得的纳米氧化镁粉体为片层纳米微片结构,排列规则有序,生长均匀,呈外延辐射状生长,微片彼此交叉生长,晶粒小,分散度高,活性高。
附图说明
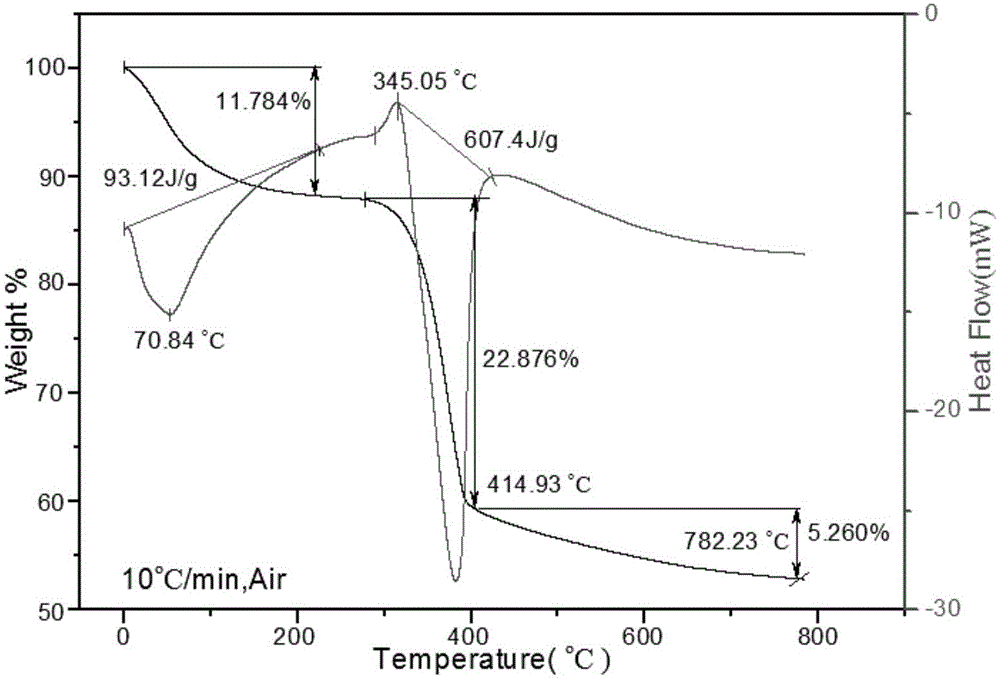
图1是前驱体mg(oh)2的dsc-tga曲线图;
图2是前驱体mg(oh)2在400℃煅烧1h后的sem图;
图3是前驱体mg(oh)2在550℃煅烧2h后的sem图;
图4是前驱体mg(oh)2在550℃煅烧不同时间后的红外光谱图;
图5是前驱体mg(oh)2在在不同温度下煅烧2h所得产物的x射线衍射曲线图;
图6是花瓣状氧化镁片的x射线衍射曲线图;
图7是花瓣状氧化镁片的的edx能谱图;
图8是前驱体mg(oh)2经650℃煅烧后mgo的红外吸收光谱图;
图9是15%(v/v)的氨水溶液沉淀含乙醇25%(v/v)的富镁液、650℃处理2h得到产物的sem图;
图10是25%(v/v)的氨水溶液沉淀含乙醇25%(v/v)的富镁液、650℃处理2h得到产物的sem图;
图11是25%(v/v)的氨水溶液沉淀含乙醇50%(v/v)的富镁液、650℃处理2h得到产物的sem图;
图12是15%(v/v)的氨水溶液沉淀含乙醇25%(v/v)的富镁液、750℃处理2h得到产物的sem图;
图13是35%(v/v)的氨水溶液沉淀含乙醇15%(v/v)的富镁液、不含peg、450℃处理3h得到产物的sem图;
图14是15%(v/v)的氨水溶液沉淀含乙醇25%(v/v)的富镁液、550℃处理2h得到产物的tem图;
图15是25%(v/v)的氨水溶液沉淀含乙醇25%(v/v)的富镁液、550℃处理2h得到产物的tem图;
图16是25%(v/v)的氨水溶液沉淀含乙醇50%(v/v)的富镁液、550℃处理2h得到产物的tem图。
具体实施方式
实施例1
本实施例1所描述的一种片状纳米氧化镁粉体的制备方法,包括如下步骤:
(1)将白云石矿粉碎后于900~1100℃下煅烧2~3h成白云石灰,加水配置成重量固液比为1:2~5的混合液,然后于50~80℃下消化20min~1h得到熟料,熟料为氢氧化钙和氢氧化镁的混合悬浊液;在熟料中缓慢加入10%~25%w/w浓度的盐酸溶液,混合悬浊液ph值由14突变为6~7时停止加入盐酸,然后过滤分离得到滤液和滤渣,滤渣洗涤数次后备用;
(2)将步骤(1)中的滤渣打散,机械搅拌下滴入10%~25%w/w浓度的硫酸溶液,当体系变成红灰色浑浊液及ph为5~6时停止加入硫酸溶液,然后过滤分离得到富镁滤液;
(3)于富镁滤液中加入5~20毫升peg或不加入peg,然后加入乙醇浓缩得到富镁液,富镁液的乙醇含量为25%,富镁液加热至60℃并搅拌,缓慢滴加15%浓度氨水在80℃下水热30min后真空抽滤得到氢氧化镁沉淀物滤渣和滤液,氢氧化镁沉淀物滤渣以蒸馏水冲洗后再用无水乙醇洗涤数次;
(4)所得氢氧化镁沉淀物滤渣于70~90℃干燥1~2h得到前驱体氢氧化镁粉体;
(5)把纳米氢氧化镁粉体在400-750℃温度下煅烧2-3h得到纳米氧化镁粉体。
在本实施例中,所述的纳米氧化镁粉体的平均晶粒粒径为15nm。
在本实施例中,所述步骤(5)中的煅烧温度的升温速率为10℃/min。
如图1所示,由tga曲线可知,mg(oh)2首先失去吸附水,随后表面吸附的有机分子在287.78℃左右开始分解,对应dsc曲线有个较小的放热峰,mg(oh)2在345.05℃开始剧烈分解,对应dsc曲线有最大吸收峰,414.93℃以后仍有失重,但失重量不大,约为11.78%。由此说明氢氧化镁的热分解过程分成两个阶段:
第一阶段为345.05℃~414.93℃,伴随着剧烈的吸热峰,反应速率常数较大,分解反应过程激烈,分解速度较快。对应的失重量约为22.87%;
第二阶段为400.89℃以后,分解过程缓慢,到782.23℃反应仍未完全,对应的失重量约为5.26%,说明反应速率常数较小。由此可选择前驱体mg(oh)2煅烧温度为450℃~700℃左右,煅烧时间为2~3h左右,且随着煅烧温度的减小,煅烧时间应相应延长。
如图2所示,清晰地反映了氧化镁的成核过程,其中原始氢氧化镁的微片状结构依然存在,微片面积基本保持不变,在每一个片状体的边缘可以看到十分细小的mgo晶核,其大小为单一片状体的0.1~0.2倍左右,从而证实了在氢氧化热分解的初期、中期的分解机理为成核控制机理。
如图3所示,氧化镁晶核已长大,但其颗粒大小远小于原始的氢氧化镁晶粒,其片层面积是原氢氧化镁片层面积的1.5倍。在氢氧化镁热分解的后期,随着热分解温度的上升,氢氧化镁表面形成的氧化镁晶核逐渐聚集并缓慢生长导致所形成的氧化镁膜厚度增,阻碍了分解产物水分子在其中的扩散,因而产物水在其中的扩散速度成为氢氧化镁热分解反应控制步骤,使得这一热分解过程变得缓慢。
如图4所示,图4曲线中a、b、c显示,随着煅烧时间的延长,氢氧化镁表面吸附的有机物随之分解,且其对称伸缩振动峰逐渐减弱,氧化镁的特征吸收峰逐渐形成并增强。图4中曲线d说明,550℃煅烧2h后,氢氧化镁特征峰已经完全消失,图4中a处634.8cm-1和b处418.7cm-1分别对应于氧化镁的伸缩及弯曲特征峰,说明氢氧化镁已经完全分解转变为氧化镁相,在400~800cm-1的波数范围内,mgo吸收谱窄而尖锐,说明经550℃煅烧2h后的mgo结晶较为完整。
如图5所示,当煅烧温度从400℃升高到650℃时,氢氧化镁的特征衍射峰减弱,而mgo的特征峰增强,550℃和650℃煅烧2h的产物,其氢氧化镁的特征峰已经完全消失,仅有氧化镁衍射峰,说明氢氧化镁已经完全分解转化为氧化镁相。
如图6所示,氧化镁主衍射峰尖锐且大大宽化,表明结晶良好,且晶粒尺寸很小,产物的各个衍射峰的位置和强度与面心立方晶相结构氧化镁的基本一致,表明产物为面心立方晶型的氧化镁,3个典型衍射峰分别为(200)、(220)和(222)晶面。图6上找不到杂峰,说明产物较纯。如图7所示,图7中的edx能谱显示,产物中仅含mg和o,其原子数目比例基本等于1:1,说明此产物杂质相含量很少,产品较纯净,基本仅由mg和o元素构成。采用scherrer公式计算得出mgo片层平均晶粒粒径约为15nm左右。
如图8所示,在400~800cm-1波数范围内,吸收谱窄而尖锐,说明经650℃煅烧处理后的mgo结晶较完整。mgo在3437.61cm-1处为水的吸收峰,1632.21cm-1及1128.80cm-1处为co2吸收峰。
氨水浓度的高低对mg(oh)2晶粒成长有较大的影响,前驱体形貌及晶粒大小的变化继而影响产物mgo的形貌和大小,较低浓度氨水沉淀的前驱体经煅烧后形成更复杂的菱片空间结构,并能形成类似花瓣状的片层结构,并且菱面微片空间取向性较好,但微片并不彼此交叉生长形成空间格子,而是由大量微片组成团簇空间,如图9、图10所示。mgso4溶液中乙醇含量的高低会影响最终产物氧化镁的微观结构,乙醇会在固液界面上形成一层致密的双电层保护膜,阻碍溶液中的mg2+和oh–在已经生成的氢氧化镁晶格表面结合,从而抑制了晶核的长大,促进了新晶核的形成,有利于小颗粒氢氧化镁的生长。mgso4溶液中乙醇含量较高时,反应后形成的菱形氧化镁片层厚度较小,表面积大,如图10、图11所示。显然低浓度氨水使得前驱体分子取向更加有序,进而生成有序性较高的氧化镁纳米片;煅烧温度对氧化镁形貌影响较大,合适的煅烧温度不仅能得到生长均匀、厚度较小的氧化镁,而且形成的菱形片取向性较好。
当煅烧温度升高到750℃时,有序生长的片层结构被破坏,氧化镁晶粒开始二次晶化,熔融、烧结形成微球,其表面张力的变化改变了片层结构的表面能分布,破坏了片层结构的连续性,这些熔融体微球彼此粘结,形成了珊瑚状的微枝结构,如图12所示。煅烧温度低于550℃时,分解不完全,煅烧温度过高,容易团聚,氧化镁粒径过大,550℃~650℃左右煅烧可形成厚度较小、面积较大、生长均匀的纳米微片,750℃左右煅烧可以形成珊瑚状纳米微枝结构氧化镁。
如图13所示,氧化镁呈板块状,长度大于5μm、厚度大于0.5μm,此氧化镁板块的侧表面具有的不同程度的纳米微片结构。显然peg对氧化镁微观形貌的影响较大。
综上所述,采用经添加edta和peg的含25%乙醇的mgso4溶液以15%浓度氨水沉淀得到的前驱体经650℃煅烧两小时可制得外延辐射状均匀生长的类似花瓣状的大面积纳米氧化镁微片;煅烧温度高于750℃时,可以得到珊瑚枝状纳米氧化镁;不添加peg时,可以得到板块状纳米氧化镁。
如图14所示,清晰的显示出,氧化镁为纳米微片结构,呈外延辐射状生长形成较大的团簇。
如图15所示,清晰的显示出,氧化镁为纳米微片结构,彼此交叉生长形成空间格子,其宽度约为200~600nm。
如图16所示,清晰的显示出,氧化镁为纳米微片结构,生长均匀,呈外延辐射状生长,微片彼此交叉生长,形成法向方向生长的空间团簇,其宽度为200~400nm左右。图16也清晰的显示出了氧化镁微片中具有大量的微观孔隙,尺寸很小,有利于比表面积的增加,这对氧化镁作为催化剂载体的应用具有重大意义。
以上所述,仅是本发明的较佳实施例而已,并非对本发明的技术方案作任何形式上的限制。凡是依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明的技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!