一种氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料及其制备方法与应用与流程
本发明涉及水污染处理技术领域,更具体地,涉及一种氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料及其制备方法与应用。
背景技术:
随着城市化工行业等不断繁荣发展,工业废水污染逐渐成为一个全球化环境问题。工业废水成分复杂,其中难降解的有机物其分子结构复杂,化学稳定性强,可造成持久性污染,传统城市污水生化处理系统不能达到完全净化的效果。因此,探寻更有效的水处理方法对缓解当前的水环境污染问题具有深刻意义。在众多新兴的水处理工艺中,(类)芬顿技术在处理水中复杂有机物时表现出广泛的应用前景。
(类)芬顿高级氧化技术包括均相体系和非均相体系两大类。与均相(类)芬顿法相比,非均相法具有如下优点:(1)反应后非均相催化剂,尤其是铁磁性材料易于从体系中分离回收;(2)大部分非均相催化剂化学稳定性强,可循环使用,甚至还可以进行再生,材料利用率高;(3)非均相体系中的金属溶出较少,一般不会对水环境造成影响;(4)非均相催化剂种类繁多、形貌结构可控、制备方法多样,丰富拓宽了(类)芬顿高级氧化技术领域的发展和应用。因此,非均相(类)芬顿法具有极大的实际推广潜能,越来越引起人们的密切关注和研究。
目前,非均相催化剂的研究主要集中在纳米零价铁、铁氧体、金属以及金属氧化物等方面,而关于金属磷化物基材料在高级氧化中的研究鲜有报道。近年研究发现,金属磷化物在(类)芬顿高级氧化中可表现出优越的性能,是一类非常有潜力催化剂。然而,目前金属磷化物材料仍然面临着催化活性不高、化学稳定性不佳、颗粒易团聚、金属离子易流失等问题,极大阻碍了其推广实际应用。另一方面,材料的微观物理结构以及表面化学结构等对其性能具有决定性影响,故通过对金属磷化物材料的形貌、组成、结构等的理性设计和调控可进一步改善其性能,此方面的工作亦亟待开展。
技术实现要素:
本发明的目的在于针对现有技术中,金属磷化物作为(类)芬顿高级氧化非均相催化剂的应用存在的缺点和不足,提供一种氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料。
本发明的另一目的在于提供所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料的制备方法。
本发明的再一目的在于提供所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料的应用。
本发明的上述目的是通过以下方案予以实现的:
一种氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料的制备方法,包括如下步骤制备得到:
s1.将金属盐、碳源化合物及膨化剂均匀混合后,在惰性气体氛围下热解,得到m-g-c3n4;
s2.将所述m-g-c3n4分散于溶剂中,得到悬浮液a,然后将溶有六氯环三磷腈和4,4-二羟基二苯砜的混合溶液b滴加至悬浮液a中,混合反应;然后再滴加碱性辅剂,混匀反应,待反应结束后,分离得到m-g-c3n4@pzs;
s3.将所述m-g-c3n4@pzs在气体氛围下,高温热解,得到所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料mpx-nps-c。
本发明通过将高分散的超细金属磷化物纳米粒子负载在多掺杂碳多孔纳米片上构筑纳米复合材料,得到具有高催化活性和稳定性的纳米复合材料,其作为催化剂应用时,具有如下优势:1)碳材料的高比表面积和多孔结构,以及金属磷化物纳米粒子的超细尺寸,使复合催化剂材料能够充分暴露反应活性位点;2)多孔超薄纳米片结构有利于传质扩散;3)多掺杂碳多孔纳米片能够高度分散金属磷化物,防止金属流失和纳米粒子的团聚,提高材料的稳定性,方便回收再利用;4)杂原子共掺杂可为材料提供更多的活性位点,提高催化活性;5)碳材料绿色无污染,同时自身也具有一定的催化性能。
优选地,步骤s1中所述金属盐为金属乙酸盐、金属硝酸盐、金属硫酸盐、金属碳酸盐或金属氯化物中的至少一种。
优选地,步骤s1中所述金属盐中的金属为(过渡金属)mn、fe、co、cu、ni、ce、cr、zn、v、ti、sc、mo、w、cd、zr、nb、tc、bh,(稀土金属)lu、ce、pr、tb、y、sc,(贵金属)pd、ru、rh、pt、ir、os、au或ag中的至少一种。
优选地,所述碳源化合物为三聚氰胺、二氰二胺、单氰胺、尿素、葡萄糖、麦芽糖、糖醇、蔗糖、淀粉、纤维素、木质素、柠檬酸、环氧树脂、酚醛树脂、聚乙烯醇、聚乙二醇、聚丙烯腈或炭黑中的至少一种。
所述膨化机为氯化铵、硫酸铵、碳酸铵、碳酸氢铵或碳酸氢钠中的至少一种。
优选地,步骤s1中,所述热解的升温速率为1~50℃/min;优选地为1~30℃/min;更优选地为2℃/min、5℃/min、10℃/min、15℃/min、20℃/min、2-5℃/min、5~10℃/min、5~15℃/min、10~15℃/min、10~20℃/min、5~20℃/min、15~20℃/min、2~10℃/min、2~15℃/min或2~20℃/min。
优选地,步骤s1中,所述热解的温度为400~650℃;优选地为450~600℃;更优选地为480℃、500℃、550℃、580℃、480~500℃、500~550℃、550~580℃、480~550℃、480~580℃或500~580℃。该温度范围内所用碳源可分解转化为g-c3n4,所用金属盐转化为金属基纳米粒子负载在上面,生成m-g-c3n4材料。
优选地,步骤s1中,所述热解的时间为1~20h;优选地为2~16h;更优选地为2h、5h、10h、15h、2~5h、2~10h、3~4h、2~15h、5~10h、5~15h或10~15h。足够的热解时间保证金属盐及碳源的充分分解,完全转化为m-g-c3n4。但热解时间太长可能导致金属基纳米粒子过量团聚问题。因此需要适宜的热解时间范围。优选地,步骤s1中,所述金属盐、碳源化合物及膨化剂先混溶于水中后,再加热去除水分,得到混合均匀的混合粉末;或,所述金属盐、碳源化合物及膨化剂经研磨成粉末后再混合均匀,得到混合粉末。
优选地,步骤s1中,所述金属盐、碳源化合物及膨化剂的物质的量之比可为1︰(10~100)︰(0~500);更优选地,为1︰24︰(100~200)、1︰30︰(130~240)、1︰40︰(180~310)或1︰60︰(270~470)。
优选地,步骤s1中,热解后的降温方式为自然降温或程序强制降温。
优选地,步骤s1中,所述惰性气体为氮气(n2)、氩气(ar)或氦气(he)中的至少一种。
优选地,步骤s2中,所述碱性辅剂为甲胺、二甲胺、三甲胺、乙胺、二乙胺、三乙胺、苯胺、对甲苯胺、对硝基苯胺、二苯胺、苄胺、氢氧化钠、氢氧化钾、氢氧化镁、氢氧化铝、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、氨水、吡啶,二甲基咪唑、苯并咪唑、2-羟基苯并咪唑、1-正丁基咪唑或4-硝基咪唑中的至少一种。
优选地,步骤s2中,所述溶剂可为甲醇、乙醇、丙醇、丁醇、异丙醇、乙二醇、丙二醇、1,4-丁二醇、1,2,4-丁三醇、1,6-己二醇、戊二醇、丙三醇、苯甲醇、环乙醇、丙酮、二甘醇、三甘醇、乙腈、乙二醇单甲醚、乙二醇单乙醚、乙二醇单正丁醚、乙酸甲酯、乙酸乙酯、二甲基亚砜、二甲基甲酰胺或去离子水中的至少一种。
优选地,步骤s2中,所述混合溶液b中的溶剂与悬浮液a中的溶剂种类相同。
优选地,步骤s2中,所述悬浮液a中m-g-c3n4与溶剂的质量体积比为(0.01~30)︰1000,具体为(0.1~25)︰1000;更具体为(0.1~5)︰1000、(5~10)︰1000、(0.1~10)︰1000、(0.1~20)︰1000、(10~20)︰1000或(5~20)︰1000。
优选地,步骤s2中,混合溶液b与悬浮液a混合后,溶液中的m-g-c3n4与hccp的物质的量的比为1︰(0~40),具体可为1︰(0~10)、1︰(0~20)、1︰(0~30)、1︰(10~20)、1︰(20~30)或1︰(10~30),其中hccp用量不为0。
优选地,步骤s2中,混合溶液b中,所述hccp、bps及溶剂的质量体积比为0.2︰(0.1~10)︰(10~10000),更具体为0.2︰(0.3~8)︰(50~8000)。
优选地,步骤s2中,混合溶液b的滴加速度为20~100ml/h。滴加速度不可过快,以形成均匀的体系,有助于后续pzs的均匀包覆。
优选地,步骤s2中,混合溶液b和悬浮液a的混合时间为10~60min;更优选地为15~30min,更优选地为15min、18min、20min、24min、15~18min、15~20min、15~24min、18~24min、18~20min、20~24min。
优选地,步骤s2中,所述hccp与碱性辅助剂的质量体积比为(0.01~2):1;优选地为(0.05~1)︰1;更优选地为(0.1~0.3)︰1、(0.1~0.5)︰1、(0.1~0.8)︰1、(0.3~0.5)︰1、(0.3~0.8)︰1或(0.5~0.8)︰1。
优选地,步骤s2中,所述碱性辅剂的滴加速度为5~50ml/h。该速度足够缓慢以保证pzs在m-g-c3n4材料表面的均匀形成。
优选地,步骤s2中,滴加碱性辅剂后反应的时间为5~48h;优选地为6~36h,更优选地为6h、8h、10h、12h、24h、6~16h、6~8h、8~10h、6~10h、10~12h、8~12h、6~12h、12~24h、10~24h、8~24h或6~24h。
优选地,步骤s2中反应结束后的分离过程为:将反应液经自然沉降、抽滤或离心,得到固体物质,然后进行洗涤,烘干,得到m-g-c3n4@pzs。
更优选地,所述洗涤采用甲醇、乙醇或乙腈中的至少一种溶剂进行洗涤。
更优选地,所述干燥条件可设置为30~150℃条件下干燥3~48h。
优选地,步骤s3中,所述热解过程的升温速率为1~50℃/min;优选地为1~30℃/min;更优选地为2℃/min、5℃/min、10℃/min、30℃/min、2~5℃/min、2~10℃/min、2~30℃/min、5~30℃/min、5~10℃/min或10~30℃/min。
优选地,步骤s3中,所述热解过程的温度为700~1200℃;优选地为800~1100℃;更优选地为800℃、850℃、900℃、1000℃、800~850℃、850~900℃、800~900℃、900~1000℃、850~1000℃或800~1000℃。在该温度范围内,能够使pzs发生剧烈分解,一方面与高温碳化的m-g-c3n4共同生成氮磷硫共掺杂的多孔碳,另一方面聚合物分解的p与m-g-c3n4中的金属结合形成金属磷化物负载于多孔碳上。
优选地,步骤s3中,所述热解时间为1~20h;优选地为1~15h;更;优选地为1h、5h、8h、15h、1~5h、1~8h、1~15h、5~8h、5~15h或8~15h。
优选地,步骤s3中,热解反应结束后的降温方式为自然降温或程序强制降温。
优选地,步骤s3中,热解过程中的气体可为惰性气体,或惰性气体与反应气体的混合气体;优选地,所述惰性可为氮气(n2)、氩气(ar)或氦气(he)中的至少一种;优选地,反应气体可为氨气(nh3)、硫化氢(h2s)或磷化氢(ph3)中的至少一种。
本发明同时还保护由上述方法制备得到的氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料。
优选地,所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料的表观物理形态为多孔纳米片状。
优选地,所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料的直径为0.5~10μm,厚度为2~30nm,料比表面积为500~1400m2g-1,孔容为0.45~1.50cm3g-1。
所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料作为电化学催化、光化学催化、光催化水裂解或(类)芬顿高级氧化催化剂的应用同样在本发明的保护范围之类。
优选地,所述电化学催化包括电催化析氢反应或电催化水裂解反应。
优选地,所述光化学催化包括光催化析氢反应或光催化水裂解反应。
优选地,所述(类)芬顿高级氧化包括活化过氧化氢的芬顿技术、活化过氧化单硫酸盐(pms)或过硫酸盐(ps)。
优选地,所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料作为光电协同催化析氢反应、光电协同催化水裂解反应或光催化活化pms协同的高级氧化反应催化剂中的应用。
优选地,所述氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料作为生物传感器材料的应用。
与现有技术相比,本发明具有以下有益效果:
本发明制备的氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料中,具有如下优势:1)金属磷化物纳米粒子有利于反应活性位点的充分暴露;2)以碳材料作为载体使极大地提高了材料地比表面积和隙率,有利于反应物的吸附以及与活性位点的接触;3)多孔碳材料能够较好地分散金属磷化物纳米颗粒,防止纳米粒子团聚,同时抑制金属离子溶出,提高了材料的稳定性;4)pzs包覆为原位生成金属磷化物提供磷源,无需外加其他磷源,同时其也是n、p、s杂原子共掺杂的来源,可以为纳米复合材料提供更多的活性位点;
同时,本发明提供的合成方法具有操作简单、反应迅速、条件灵活、普遍适用性高的优势;且制备的氮磷硫共掺杂多孔碳负载的金属磷化物纳米复合材料在催化活化h2o2、pms、ps降解复杂有机化合物中表现出优异的性能,拓宽了金属磷化物材料在高级氧化水处理中的应用。
附图说明
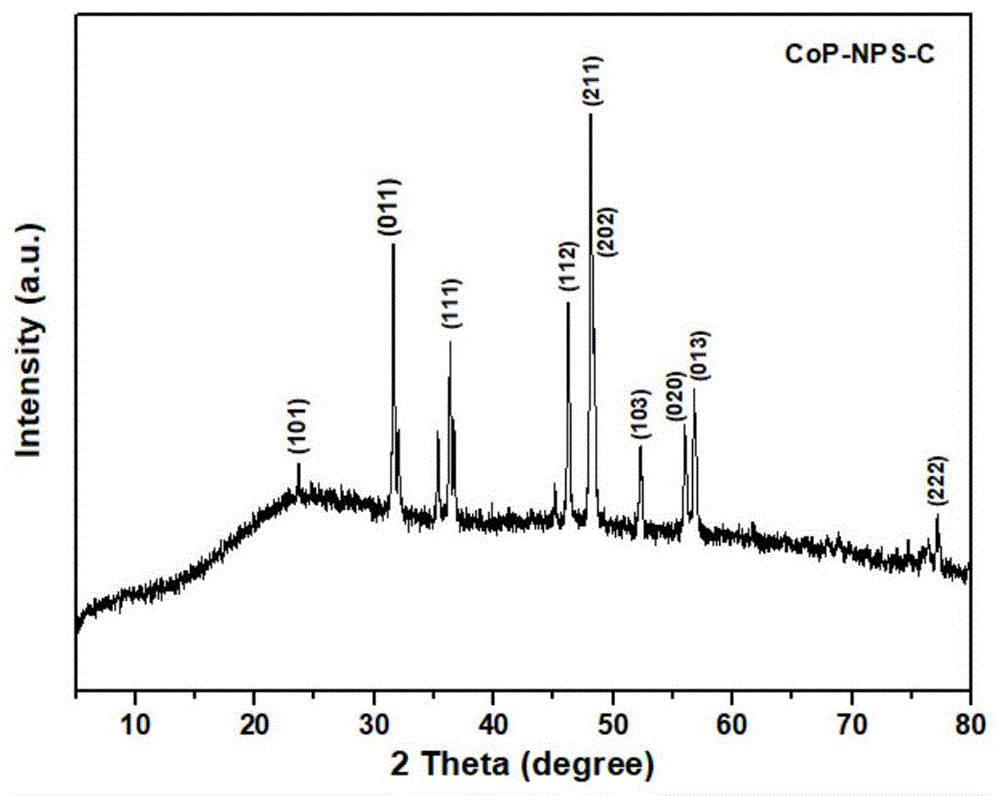
图1为实施例1所制得的氮磷硫共掺杂多孔碳负载的磷化钴纳米复合材料x射线粉末衍射图。
图2为实施例2所制得的氮磷硫共掺杂多孔碳负载的磷化钛纳米复合材料sem图。
图3为实施例3所制得的氮磷硫共掺杂多孔碳负载的磷化锰纳米复合材料sem图。
图4为实施例1所制得的氮磷硫共掺杂多孔碳负载的磷化钴纳米复合材料活化pms降解对氯苯酚性能图。
图5为实施例3所制得的氮磷硫共掺杂多孔碳负载的磷化锰纳米复合材料活化pms降解亚甲基蓝性能图。
图6为实施例6所制得的氮磷硫共掺杂多孔碳负载的钴镍双金属磷化物纳米复合材料sem图。
图7为实施例6所制得的氮磷硫共掺杂多孔碳负载的钴镍双金属磷化物纳米复合材料tem图。
图8为实施例7所制得的氮磷硫共掺杂多孔碳负载的锰钒双金属磷化物纳米复合材料sem图。
图9为实施例6所制得的氮磷硫共掺杂多孔碳负载的钴镍磷化物纳米复合材料活化ps降解罗丹明b性能图。
图10为实施例7所制得的氮磷硫共掺杂多孔碳负载的锰钒磷化物纳米复合材料活化pms降解四环素性能图。
图11为实施例10所制得的氮磷硫共掺杂多孔碳负载的铂铁铜三金属磷化物纳米复合材料tem图。
图12为实施例11所制得的氮磷硫共掺杂多孔碳负载的磷化镍纳米复合材料x射线粉末衍射图。
图13为实施例11所制得的氮磷硫共掺杂多孔碳负载的磷化镍纳米复合材料活化pms降解阿昔洛韦性能图。
图14为实施例12所制得的氮磷硫共掺杂多孔碳负载的铁钼双金属磷化物纳米复合材料sem。
图15为实施例12所制得的氮磷硫共掺杂多孔碳负载的铁钼双金属磷化物纳米复合材料tem。
图16为实施例12所制得的氮磷硫共掺杂多孔碳负载的铁钼双金属磷化物纳米复合材料活化h2o2降解双酚a性能图。
具体实施方式
下面结合具体实施例对本发明做出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的试验方法如无特殊说明,均为常规方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
实施例1、氮磷硫共掺杂多孔碳负载的磷化钴纳米复合材料的制备
具体制备过程如下:
将0.0747g(0.3mmol)的乙酸钴[co(ch3coo)2·4h2o]、3g二氰二胺、15g氯化铵在30ml去离子水中以80℃加热回流4h,得到溶解完全的混合溶液,再升温至100℃蒸干去离子水,在100℃烘箱中完全干燥后,得到三者混合均匀的粉末;将混合粉末在管式炉中以2℃/min的升温速率由室温升高至550℃,在550℃下热解2h,管式炉中通入n2,流速控制在20-30ml/min,自然降温后即得到co-g-c3n4材料;
将200mgco-g-c3n4材料在50ml的甲醇中超声30min,得到分散良好的悬浮液a;将溶解有150mghccp和350mgbps的甲醇溶液15ml,缓慢滴加至悬浮液a中;混合15-20min后,继续缓慢滴加500μl三乙胺和500μl甲醇的混合液,持续反应12h后进行分离操作。上述过程中悬浮液始终保持搅拌状态,转速维持在650rpm。反应后的材料经孔径为0.45μm的有机混合尼龙新亚过滤膜抽滤,用乙醇洗涤3次后,在60℃的烘箱中干燥6h,得到co-g-c3n4@pzs材料;
取400mgco-g-c3n4@pzs材料于管式炉中进行热解,以2℃/min的升温速率,由室温升至800℃,在800℃下停留1.5h,在管式炉中通入n2,流速控制在20-30ml/min,自然降温后即得到氮磷硫共掺杂多孔碳负载的磷化钴纳米复合材料(cop-nps-c)。
材料的表征:
产物经x射线粉末衍射仪鉴定其晶相为磷化钴(如图1所示);
实施例2、氮磷硫共掺杂多孔碳负载的磷化钛纳米复合材料的制备
具体制备过程如下:
将24g(100mmol)的硫酸钛[ti(so4)2]、800g尿素、2kg碳酸铵研磨混合,得到三者混合均匀的粉末;将混合粉末在管式炉中以5℃/min的升温速率由室温升高至580℃,在580℃下热解4h,管式炉中通入he,流速控制在50ml/min左右,自然降温后即得到ti-g-c3n4材料;
将80gti-g-c3n4材料在4l的乙醇中超声45min,得到分散良好的悬浮液a;将溶解有80ghccp和200gbps的乙醇溶液1l,滴加至悬浮液a中;混合40min后,继续缓慢滴加400ml的二甲胺,持续反应30h后进行分离操作。上述过程中悬浮液始终保持搅拌状态,转速维持在1000rpm。反应后的材料先经过自然沉降除去上清液后,再以10000rpm,5min的离心操作进一步分离固液两相,并且用甲醇洗涤1次,在80℃的烘箱中干燥3h,得到ti-g-c3n4@pzs材料;
取30gti-g-c3n4@pzs材料于管式炉中进行热解,以10℃/min的升温速率,由室温升至900℃,在900℃下停留6h,在管式炉中通入he,流速控制在40ml/min左右,自然降温后即得到氮磷硫共掺杂多孔碳负载的磷化铬纳米复合材料(tip-nps-c)。
材料的表征:
用sem(如图2所示)对其形貌进行表征,可以看出其为多孔片状堆叠的结构,其片状直径约为1-4μm;
实施例3、氮磷硫共掺杂多孔碳负载的磷化锰纳米复合材料的制备
具体制备过程如下:
将12.25g(50mmol)乙酸锰[mn(ch3coo)2·4h2o]、100g三聚氰胺、100g硫酸铵在1l去离子水中以60℃加热回流12h,得到溶解完全的混合溶液,再升温至90℃蒸干去离子水,在100℃烘箱中完全干燥后,研磨得到三者混合均匀的粉末;将混合粉末在管式炉中以20℃/min的升温速率由室温升高至650℃,在650℃下热解2h,管式炉中通入n2,流速控制在30-40ml/min,以10℃/min速率降至室温后即得到mn-g-c3n4材料;
将5gmn-g-c3n4材料在1l的乙腈中超声60min,得到分散良好的悬浮液a;将溶解有3ghccp和10gbps的乙腈溶液200ml加至悬浮液a中;混合40min后,继续缓慢滴加40ml苯胺和60ml乙醇的混合液,持续反应24h后进行分离操作。上述过程中悬浮液始终保持搅拌状态,转速维持在900rpm。反应后的材料经孔径为0.45μm的有机混合尼龙新亚过滤膜抽滤,并用乙醇洗涤5次后,在80℃的烘箱中干燥1h,得到mn-g-c3n4@pzs材料;
取10gmn-g-c3n4@pzs材料于管式炉中进行热解,以24℃/min的升温速率,由室温升至1000℃,在1000℃下停留4h,在管式炉中通入n2,流速控制在30-40ml/min左右,以20℃/min速率降至室温后即得到氮磷硫共掺杂多孔碳负载的磷化钒纳米复合材料(mnp-nps-c)。
材料的表征:
用sem(如图3所示)对其形貌进行表征,可以看出其表现出疏松多孔的片状堆叠结构,其片状直径约为1-3μm;
实施例4、氮磷硫共掺杂多孔碳负载的磷化钴纳米复合材料在活化过氧化单硫酸盐高级氧化中的性能测试
以对氯苯酚为底物,测试实施例1制备的材料活化pms降解对氯苯酚溶液的催化性能。体系中对氯苯酚初始浓度为50mg/l,催化剂初始浓度为50mg/l,pms初始浓度为500mg/l。具体步骤如下:
称取实施例1制备的氮磷硫共掺杂多孔碳负载的磷化钴纳米复合材料(cop-nps-c)50mg,超声分散在1l的50mg/l的对氯苯酚(4-cp)水溶液中,搅拌30min,取样待测。接着,再往体系中加入500mgpms,每隔一段时间取样待测。取样的操作为:取1ml样液,用1ml无水乙醇进行猝灭,再经0.22μm的滤膜过滤,后取1ml滤液于液相瓶中待测。将待测液通过高效液相色谱(hplc)检测,分析溶液中的对氯苯酚的浓度。另外按照上述方法,不加入上述材料,以探究pms单独氧化降解4-cp的能力,作为对照。
测试结果表明(如图4所示),当反应体系中只有4-cp和催化剂时,30min后4-cp浓度仅仅下降了0.75%,可能是4-cp吸附在材料的表面而使浓度下降,且该吸附作用可以忽略;当继续往体系中投加pms后,5min时4-cp降解率达到了90.01%,20min时达到了98.84%,4-cp几乎完全降解。另外地,由图可以得知,当单独采用pms氧化降解4-cp时,20min后4-cp的浓度降低了不到5%,说明pms自身对4-cp的氧化降解能力很差。由此看出,4-cp的降解主要是依靠催化剂活化pms的高级氧化反应实现的。本发明制备的氮磷硫共掺杂多孔碳负载的磷化钴纳米复合材料在活化pms降解4-cp中具有优异的催化活性。
实施例5、氮磷硫共掺杂多孔碳负载的磷化锰纳米复合材料在活化过氧化单硫酸盐高级氧化中的性能测试
以亚甲基蓝(mb)为底物,测试实施例3制备的材料活化pms降解亚甲基蓝溶液的催化性能。体系中亚甲基蓝初始浓度为150mg/l,催化剂初始浓度为100mg/l,pms初始浓度为800mg/l。具体步骤如下:
称取实施例3制备的氮磷硫共掺杂多孔碳负载的磷化锰纳米复合材料(mnp-nps-c)50mg,超声分散在500ml的150mg/l的亚甲基蓝(mb)水溶液中,搅拌30min,取样待测。接着,再往体系中加入400mgpms,每隔一段时间取样待测。取样的操作为:取2ml样液,用2ml无水乙醇进行猝灭,再经0.22μm的滤膜过滤,后对滤液进行适度稀释,待测。将待测液通过紫外分光光度计检测,分析溶液中的mb的浓度变化。另外按照上述方法,不加入上述材料,来探究pms单独氧化降解mb的能力,形成对照。
由测试结果可知(如图5所示),当反应体系中只有mb和催化剂时,30min后溶液中mb的浓度仅仅下降了0.14%,这可能是因为材料的表面会吸附少量的mb,该吸附作用较弱,可以忽略;当继续往体系中投加pms后,10min时mb降解了达到了27.25%,60min时mb降解率为43.03%。并且,当体系中不加入催化剂时,60min时溶液中mb只降解了10.34%,说明pms自身氧化降解mb能力较差。由此看出,mb的降解主要是依靠材料催化活化pms的高级氧化反应实现的。本发明制备的氮磷硫共掺杂多孔碳负载的磷化锰纳米复合材料在活化pms降解mb中具有良好的催化活性。实施例6、氮磷硫共掺杂多孔碳负载的钴镍双金属磷化物纳米复合材料的制备
具体制备过程如下:
将32.5g(150mmol)的乙酸钴[co(ch3coo)2·4h2o]、12.45g(50mol)的乙酸镍[ni(ch3coo)2·4h2o]、200g蔗糖、1.2kg碳酸氢钠直接研磨至完全混合;将混合粉末在管式炉中以5℃/min的升温速率由室温升高至500℃,在500℃下热解8h,管式炉中通入ar,流速控制在50mlmin左右,以5℃/min速率降至室温后即得到coni-g-c3n4材料;
将200gconi-g-c3n4材料在16l的苯甲醇中超声50min,得到分散良好的悬浮液a;将溶解有150ghccp和500gbps的苯甲醇溶液3l加至悬浮液a中;混合20min后,继续缓慢滴加1.6l三乙胺,持续反应10h后进行分离操作。上述过程中悬浮液始终保持搅拌状态,转速维持在700rpm。反应后的材料经孔径为0.45μm的有机混合尼龙新亚过滤膜抽滤,并用甲醇洗涤2次后,在60℃的烘箱中干燥12h,得到coni-g-c3n4@pzs材料;
取120gconi-g-c3n4@pzs材料于管式炉中进行热解,以20℃/min的升温速率,由室温升至800℃,在800℃下停留7h,在管式炉中通入ar和ph3的混合气体,流速控制在10ml/min左右,以40℃/min速率降至200℃后自然降温,冷却后即得到氮磷硫共掺杂多孔碳负载的钴铁双金属磷化物纳米复合材料(conip-nps-c)。
材料的表征:
用sem(如图6所示)和tem(如图7所示)对其形貌表征,可以看出材料具有多孔的纳米薄片形貌,片状直径约为2-6μm,金属磷化物高度均匀分散在纳米片中,其尺寸为5-10nm。
实施例7、氮磷硫共掺杂多孔碳负载的锰钒双金属磷化物纳米复合材料的制备
具体制备过程如下:
将49g(200mmol)的乙酸锰[mn(ch3coo)2·4h2o]、3.7752g(30mmol)的氯化钒(vcl3)、250g单氰胺、700g氯化铵在去离子水中以80℃加热回流4h,再升温至120℃至体系中的液体蒸干,于120℃烘箱中进一步干燥12h,研磨得到完全混合的粉末;将混合粉末在管式炉中以40℃/min的升温速率由室温升高至520℃,在520℃下热解12h,管式炉中通入he,流速控制在30mlmin左右,自然降温后即得到mnv-g-c3n4材料;
将260gmnv-g-c3n4材料在10l的乙醇中超声25min,得到分散良好的悬浮液a;将溶解有150ghccp和400gbps的乙醇溶液1l加至悬浮液a中;混合20min后,继续缓慢滴加0.3m氢氧化钠溶液150ml,持续反应32h后进行分离操作。上述过程中悬浮液始终保持搅拌状态,转速维持在950rpm。反应后的材料经孔径为0.45μm的有机混合尼龙新亚过滤膜抽滤,并用乙醇洗涤4次后,在80℃的烘箱中干燥16h,得到mnv-g-c3n4@pzs材料;
取300gmnv-g-c3n4@pzs材料于管式炉中进行热解,以15℃/min的升温速率,由室温升至860℃,在860℃下停留15h,在管式炉中通入he和h2s的混合气体,流速控制在35ml/min左右,以15℃/min速率降至100℃后自然降温,冷却后即得到氮磷硫共掺杂多孔碳负载的锰钒双金属磷化物纳米复合材料(mnvp-nps-c)。
材料的表征:
用sem(如图8所示)对其形貌表征,可以看出材料为纳米薄片的形貌,且表面疏松多孔,片状直径约为1-2μm。
实施例8、氮磷硫共掺杂多孔碳负载的钴镍磷化物纳米复合材料在活化过硫酸盐高级氧化中的性能测试
以罗丹明b(rhb)为底物,测试材料活化ps降解rhb溶液的催化性能。体系中rhb初始浓度为200mg/l,催化剂初始浓度为80mg/l,ps初始浓度为600mg/l。
具体步骤如下:
取实施例6制备的氮磷硫共掺杂多孔碳负载的钴镍磷化物纳米复合材料160mg,超声分散在2l的200mg/l的rhb溶液中,搅拌30min,取样待测。接着,再往体系中加入900mgps,每隔一段时间取样待测。取样的操作为:取2ml样液,用2ml无水乙醇进行猝灭,再经0.22μm的滤膜过滤,后对滤液进行适度稀释,待测。将待测液通过紫外分光光度计检测,分析溶液中的rhb的浓变化。另外按照上述方法,不加催化剂,探究ps单独氧化降解rhb的能力,作为对照。
测试结果表明(如图9所示),当反应体系中只有rhb和催化剂时,30min后rhb减少了2.05%,该阶段发生的是rhb在催化剂表面的吸附,且吸附量较少。当继续往体系中投加ps后,2min时rhb降解率达到了68.85%,5min降解率为83.21%,20min降解率已经高达93.79%,60min时降解率为97.33%,rhb几乎完全降解。而当单独采用ps氧化降解rhb时,60min后4-cp的浓度只下降了8.5%,说明仅仅依靠ps自身氧化降解rhb无法达到较好的去除效果。由此看出,rhb的降解依靠的是材料活化ps产生活性自由基来实现的。本发明制备的氮磷硫共掺杂多孔碳负载的钴镍双金属纳米复合材料在活化ps降解rhb中具有优异的催化活性。
实施例9、氮磷硫共掺杂多孔碳负载的锰钒磷化物纳米复合材料在活化过氧化单硫酸盐高级氧化中的性能测试
以四环素(tc)为底物,测试材料活化pms降解tc溶液的催化性能。体系中四环素初始浓度为120mg/l,催化剂初始浓度为200mg/l,pms初始浓度为1g/l。
具体步骤如下:
取实施例7制备的氮磷硫共掺杂多孔碳负载的锰钒磷化物纳米复合材料800mg,超声分散在4l的120mg/l的tc溶液中,搅拌30min,取样待测。接着,再往体系中加入3.2gpms,每隔一段时间取样待测。取样的操作为:取2ml样液,用2ml无水甲醇进行猝灭,再经0.22μm的滤膜过滤,后对滤液进行适度稀释,待测。将待测液通过紫外分光光度计检测,分析溶液中的tc的浓变化。
测试结果表明(如图10所示),当反应体系中只有tc和催化剂时,30min后tc减少了3.87%,该阶段发生的是tc在材料表面的吸附,且吸附量较少。当继续往体系中投加pms后,10min时tc降解了56.28%,40min时降解率达到了79.19%tc大部分被降解完成。当体系中不加入上述材料时,单单依靠pms自身的氧化作用,60min时tc的的去除率只有6%,降解效果差。因此,tc的降解主要依靠材料活化pms来实现。本发明制备的氮磷硫共掺杂多孔碳负载的锰钒双金属纳米复合材料在活化pms降解四环素中具有良好的催化活性。
实施例10、氮磷硫共掺杂多孔碳负载的铂铁铜三金属磷化物纳米复合材料的制备
具体制备过程如下:
将2.8992g(12mmol)的硝酸铜[cu(no3)2·3h2o]、2.7030g(10mmol)氯化铁(fecl3·6h2o)、1.0358g(2mmol)氯铂酸(h2ptcl6·6h2o)、28g柠檬酸、135g氯化铵在去离子水中以70℃加热回流10h,再升温至110℃至体系中的液体蒸干,于110℃烘箱中进一步干燥36h,再研磨得到完全混合的粉末;将混合粉末在管式炉中以40℃/min的升温速率由室温升高至530℃,在530℃下热解8h,管式炉中通入he和ar的混合气体,流速控制在35-40ml/min,以15℃/min速率进行程序降温,即得到ptfecu-g-c3n4材料;
将15gptfecu-g-c3n4材料在1l的乙腈中超声30min,得到分散良好的悬浮液a;将溶解有40ghccp和100gbps的乙腈溶液500ml加至悬浮液a中;混合40min后,继续缓慢滴加150ml二乙胺和100ml乙腈的混合液,持续反应15h后进行分离操作。反应后的材料经孔径为0.45μm的有机混合尼龙新亚过滤膜抽滤,并用乙腈洗涤2次后,在90℃的烘箱中干燥7h,得到ptfecu-g-c3n4@pzs材料;
取20gptfecu-g-c3n4@pzs材料于管式炉中进行热解,以45℃/min的升温速率,由室温升至1150℃,在1150℃下停留8h,在管式炉中通入he、nh3和ph3的混合气体,流速控制在10-20ml/min,自然降温冷却后即得到氮磷硫共掺杂多孔碳负载的铂钛铁三金属磷化物纳米复合材料(ptfecu-nps-c)。
材料的表征:
用tem(如图11所示)对其形貌表征,可以看出材料为纳米薄片的形貌,呈疏松状,金属磷化物均匀地分散在纳米片上,其尺寸约为5-15nm。
实施例11、氮磷硫共掺杂多孔碳负载的磷化镍纳米复合材料的制备及其在活化过氧化单硫酸盐高级氧化中的性能测试
具体制备过程如下:
将249g(1mol)的乙酸镍[ni(ch3coo)2·4h2o]、2kg淀粉、6kg氯化铵在4l去离子水中以80℃加热回流4h,得到溶解完全的混合溶液,再升高温度至150℃蒸干去离子水,在150℃烘箱中完全干燥后,得到三者混合均匀的粉末;将混合粉末在管式炉中由室温升高至550℃,在550℃下热解15h,升温速率为10℃/min,在管式炉中通入ar,流速控制在20-30ml/min,以20℃/min速率降至200℃后自然降温后即得ni-g-c3n4材料;
将500gni-g-c3n4材料在30l的丙醇中超声25min,得到分散良好的悬浮液a;将溶解有1kghccp和2.4kgbps的丙醇溶液4l加至悬浮液a中;混合20min后,继续缓慢滴加0.5m氨水1l,持续反应48h后进行分离操作。反应后的体系先通过静置使材料自然沉降,除去上清液后再经孔径为0.45μm的有机混合尼龙新亚过滤膜抽滤除去剩余液体成分,再用甲醇洗涤8次,在100℃的烘箱中干燥24h,得到ni-g-c3n4@pzs材料;
取1kgni-g-c3n4@pzs材料于管式炉中进行热解,以6℃/min的升温速率,由室温升至1200℃,在1200℃下停留18h,在管式炉中通入ar、n2、he、h2s和ph3的混合气体,流速控制在10-15ml/min,以20℃/min速率降温即得到氮磷硫共掺杂多孔碳负载的磷化镍纳米复合材料(nip-nps-c)。
材料的表征:
产物经x射线粉末衍射仪鉴定其晶相为磷化镍(如图12所示);
材料的催化性能测试:
以阿昔洛韦(acv)为底物,测试材料活化pms降解acv溶液的催化性能。体系中acv的初始浓度为25mg/l,催化剂初始浓度为60mg/l,pms初始浓度为800mg/l。具体步骤如下:
取上述制备的氮磷硫共掺杂多孔碳负载的磷化镍纳米复合材料600mg,超声分散在10l的25mg/l的acv溶液中,搅拌30min,取样待测。接着,再往体系中加入8gpms,每隔一段时间取样待测。取样的操作为:取1ml样液,用1ml无水乙醇进行猝灭,再经0.22μm的滤膜过滤,后取1ml滤液于液相瓶中待测。将待测液通过高效液相色谱(hplc)检测,分析溶液中的acv的浓度。
测试结果表明(如图13所示),当反应体系中只有acv和催化剂时,30min时acv的浓度降低了0.61%,表明材料本身对acv吸附量较少。当继续往体系中投加pms后,5min时acv降解了40.27%,40min时总共降解率了68.57%。另外地,由图得知,当pms单独氧化降解acv时,60min后仅有5.67%的acv被去除,说明pms自身对avc的氧化降解能力很弱。由此看出,材料能够活化pms在降解acv中起到主要作用。本发明制备的氮磷硫共掺杂多孔碳负载的磷化镍纳米复合材料在活化pms降解acv中具有良好的催化活性。
实施例12、氮磷硫共掺杂多孔碳负载的铁钼双金属磷化物纳米复合材料的制备及其在活化过氧化氢高级氧化中的性能测试
具体制备过程如下:
将30g(150mmol)的硝酸铁[fe(no3)3·9h2o]、16g(80mmol)的三氯化钼(mocl3)、100g葡萄糖、200g碳酸氢铵直接研磨得到完全混合的粉末;将混合粉末在管式炉中以8℃/min的升温速率由室温升高至550℃,在550℃下热解3h,管式炉中通入n2,流速控制在20-25ml/min,自然降温后即得到femo-g-c3n4材料;
将40gfemo-g-c3n4材料在15l的乙醇中超声25min,得到分散良好的悬浮液a;将溶解有200ghccp和200gbps的乙醇溶液900ml加至悬浮液a中;混合20min后,继续缓慢滴加400ml三乙胺和200ml乙醇的混合液,持续反应10h后进行分离操作。反应后的体系先通过静置使材料自然沉降,除去上清液后再用乙醇离心洗涤6次,在75℃的烘箱中干燥14h,得到femo-g-c3n4@pzs材料;
取80gfemo-g-c3n4@pzs材料于管式炉中进行热解,以6℃/min的升温速率,由室温升至920℃,在920℃下停留2h,在管式炉中通入ar和n2的混合气体,流速控制在20-25ml/min,自然降温冷却后即得到氮磷硫共掺杂多孔碳负载的铁钼双金属磷化物纳米复合材料(femop-nps-c)。
材料的表征:
用sem(如图14所示)和tem(如图15所示)对其形貌表征,可以看出材料为纳米薄片的形貌,纳米片直径约为3-4μm,片上高度均匀分散着金属磷化物,其尺寸约为2-10nm。
材料的催化性能测试:
以双酚a(bpa)为底物,测试材料活化h2o2降解bpa溶液的催化性能。体系中bpa的初始浓度为80mg/l,催化剂初始浓度为100mg/l,h2o2初始浓度为1.5g/l。
具体步骤如下:
取上述制备的氮磷硫共掺杂多孔碳负载的铁钼双金属磷化物纳米复合材料2g,超声分散在20l的80mg/l的bpa溶液中,搅拌30min,取样待测。接着,再往体系中加入30gh2o2,每隔一段时间取样待测。取样的操作为:取1ml样液,用1ml无水乙醇进行猝灭,再经0.22μm的滤膜过滤,后取1ml滤液于液相瓶中待测。将待测液通过高效液相色谱(hplc)检测,分析溶液中的bpa的浓度。
测试结果表明(如图16所示),当反应体系中只有bpa和催化剂时,30min后bpa的浓度降低了1.85%,bpa少量吸附在催化剂表面。上述体系加入h2o2后,5min,20min,40min时bpa的降解率分别为44.84%,71.37%和81.78%,40min时bpa被大部分降解。而当体系中只有h2o2对bpa进行氧化降解时,反应40min后依旧有93.77%的bpa未被去除,说明h2o2氧化降解bpa的能力不足。因此,催化剂起到了主要的作用,其能够活化h2o2,实现对bpa的降解。本发明制备的氮磷硫共掺杂多孔碳负载的铁钼双金属磷化物纳米复合材料在活化h2o2降解bpa中具有良好的催化活性。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,对于本领域的普通技术人员来说,在上述说明及思路的基础上还可以做出其它不同形式的变化或变动,这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!