一种制备硼化铪粉体的方法
1.本发明涉及硼化铪粉体制备技术领域,尤其涉及一种聚乙二醇辅助制备硼化铪粉体的方法。
背景技术:
2.随着航空、航天科技,空间飞行器技术的不断发展,人类可以探索到更浩瀚的外太空,然而在外太空的极端环境下,很多材料的性能无法达到需求。
3.硼化铪(hfb2)作为一种超高温硼化物材料,具有高熔点、高硬度、高抗热震性能、高电导率和高热导率等特性,因此成为火箭发动机、超声速飞机、耐火材料等极端超高温服役条件下零部件的候选材料。但是,hfb2陶瓷材料的烧结性能受到纯度,粒度等原始粉末特性的严重制约。
4.目前,硼化铪粉体的制备方法主要有:固相合成法、电化学合成法、机械化学合成法、自蔓延高温燃烧合成法、熔盐法等。但是这些制备方法生产周期较长,步骤较多,且制备条件要求较高,产出的硼化铪粉体的颗粒尺寸、纯度、产量无法满足科研和生产的需要。
5.saranya venugopal等人提出一种采用酚醛树脂、氯化铪和硼酸合成硼化铪粉体的凝胶溶胶法(j.am.ceram.soc.,97[1]92
–
99(2014)),但是该方法制备出的硼化铪粉体颗粒粒径较大且不均匀,进一步影响到粉体的质量以及后续陶瓷块材的性能。
[0006]
本技术人提出了一种采用硼酸、山梨醇、氯化铪为原料合成硼化铪粉体的方法(cn113816379a),然而该方法制备的硼化铪粉体仍然存在粒径分布较宽的问题,导致后续烧结成的块状陶瓷体致密度不高。
技术实现要素:
[0007]
针对现有技术中的上述不足之处,本发明提供了一种聚乙二醇辅助制备硼化铪粉体的方法,不仅制备过程简单、反应过程易控制、生产周期短、成本低廉,而且所制备的硼化铪粉体具有较高的纯度、较小的粒径、较窄的粒径分布和良好的微观形貌,能够在后续成型过程中增强烧结体的力学性能和烧结驱动力。
[0008]
本发明的目的是通过以下技术方案实现的。
[0009]
在一个方面,本发明提供一种制备硼化铪粉体的方法,包括以下步骤:
[0010]
步骤a、将铪源化合物溶解于第一溶剂中形成第一溶液;
[0011]
步骤b、将硼源化合物和碳源化合物溶解于第二溶剂中,并加入聚乙二醇,形成第二溶液;
[0012]
步骤c、将第一溶液加入第二溶液中,收集所产生的沉淀;
[0013]
步骤d、将步骤c所收集的沉淀干燥,并研磨成粉末,然后进行高温煅烧,从而制得硼化铪粉体。
[0014]
在上述方法中,步骤a和步骤b的顺序并不受到限制,并且可以互换。
[0015]
在优选的方面,铪源化合物选自氯化铪、正丁醇铪等,优选氯化铪。
[0016]
在其他优选的方面,硼源化合物选自硼酸、无定形硼等,优选硼酸。
[0017]
在又一优选的方面,碳源化合物选自山梨醇、酚醛树脂、葡萄糖等,优选山梨醇。
[0018]
在其他优选的方面,聚乙二醇的用量为第一和第二溶液总质量的0.5wt%~5wt%,更优选为1wt%~2wt%。
[0019]
在其他优选的方面,聚乙二醇的分子量为1,000~20,000,更优选为2,000~10,000。
[0020]
在又一优选的方面,第一溶剂和第二溶剂可以相同或不同,各自独立地选自羧酸和醇类溶剂,更优选乙酸、乙醇等,再更优选第一溶剂和第二溶剂相同且均为乙酸。
[0021]
在另外优选的方面,铪源化合物与硼源化合物的hf与b的原子比范围为1:2~1:6,更优选为1:3~1:4;铪源化合物与碳源化合物的hf与c的原子比范围为1:8~1:13,更优选为1:9.5~1:11。
[0022]
优选地,在步骤b中,通过恒温搅拌使得硼源化合物和碳源化合物溶解于第二溶剂中,温度为20~90℃,更优选为50~70℃。
[0023]
优选地,在步骤c中,通过滴加将第一溶液加入第二溶液中,第一溶液的滴入速率为0.5~15ml/min,更优选为5~10ml/min。
[0024]
优选地,在步骤d中,将步骤c所收集的沉淀在100~150℃下干燥4~24小时。
[0025]
优选地,在步骤d中,所述研磨成粉末通过球磨处理进行,球磨转速为50~400rpm,球磨时间为0.5~4小时。
[0026]
优选地,在步骤d中,所述高温煅烧是以惰气(优选高纯氩气)作为保护气,在高温管式炉中以1~5℃/min的速率由室温升温至1000~1200℃,然后以0.5~2℃/min的速率升温至1450~1700℃,更优选1500~1600℃,并保温10~120分钟,再以0.5~2℃/min的速率降温至1000~1200℃,之后以1~5℃/min的速率降温至300~400℃,最后自然降温至室温。
[0027]
在又一方面,本发明提供一种制备硼化铪粉体的方法,包括以下步骤:
[0028]
步骤a、将乙酸、四氯化铪混合在一起,并恒温搅拌,使四氯化铪完全溶于乙酸,得到第一溶液;
[0029]
步骤b、将乙酸、硼酸、山梨醇混合在一起,并恒温搅拌,使硼酸和山梨醇完全溶于乙酸,然后加入分子量为1,000~20,000的聚乙二醇,并冷却至室温,形成第二溶液,其中聚乙二醇的加入量为第一和第二溶液总质量的1wt%~2wt%;
[0030]
步骤c、将第一溶液滴入第二溶液中,使得hf与b的原子比范围为1:3~1:4,hf与c的原子比范围为1:9.5~1:11,收集所产生的沉淀,即为硼化铪前驱体;
[0031]
步骤d、将步骤c所收集的硼化铪前驱体干燥,并研磨成粉末,然后进行高温煅烧,从而制得硼化铪粉体。
[0032]
本发明所提供的聚乙二醇辅助制备硼化铪粉体的方法采用液相沉淀法,以硼源化合物(优选硼酸)、碳源化合物(优选山梨醇)、铪源化合物(优选四氯化铪)为原料在优选乙酸的体系中形成溶液并且产生沉淀,并加入聚乙二醇作为分散剂调控粒度与形貌,产生沉淀后充分干燥并研磨成粉末,然后放入高温管式炉中高温煅烧,从而制得高纯超细的硼化铪粉体。该硼化铪粉体纯度高、粒径小(平均粒径在400nm以下)、粒径分布窄(粒径分布跨度在300nm以下)、微观形貌呈球形颗粒,能够在后续成型过程中增强烧结体的力学性能和烧结驱动力,而且制备过程简单、不需要特殊仪器和药品、反应过程易控制、生产周期短、成本
低廉、适合大批量生产,十分适合用于制备在超高温条件下服役的陶瓷材料。
附图说明
[0033]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域的普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他附图。
[0034]
图1a-1e为采用x射线衍射分析仪分别对本发明实施例1~4及对比例1所制备的硼化铪粉体进行物质检测,从而得到的xrd图谱。
[0035]
图2a-2e为采用扫描电镜分别对本发明实施例1~4及对比例1所制备的硼化铪粉体在20k放大倍率下进行形貌检测,从而得到的扫描电镜照片。
[0036]
图3a-3e为采用扫描电镜分别对本发明实施例1~4及对比例1所制备的硼化铪粉体在60k放大倍率下进行形貌检测,从而得到的扫描电镜照片。
具体实施方式
[0037]
下面结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明的保护范围。
[0038]
下面对本发明所提供的聚乙二醇辅助制备硼化铪粉体的方法进行详细描述。本发明实施例中未作详细描述的内容属于本领域专业技术人员公知的现有技术。
[0039]
本发明提供一种制备硼化铪粉体的方法,包括以下步骤:
[0040]
步骤a、将铪源化合物溶解于第一溶剂中形成第一溶液;
[0041]
步骤b、将硼源化合物和碳源化合物溶解于第二溶剂中,并加入聚乙二醇,形成第二溶液;
[0042]
步骤c、将第一溶液加入第二溶液中,收集所产生的沉淀;
[0043]
步骤d、将步骤c所收集的沉淀干燥,并研磨成粉末,然后进行高温煅烧,从而制得硼化铪粉体。
[0044]
在上述方法中,步骤a和步骤b的顺序并不受到限制,并且可以互换。
[0045]
在优选的方面,铪源化合物选自氯化铪、正丁醇铪等,优选氯化铪。
[0046]
在其他优选的方面,硼源化合物选自硼酸、无定形硼等,优选硼酸。
[0047]
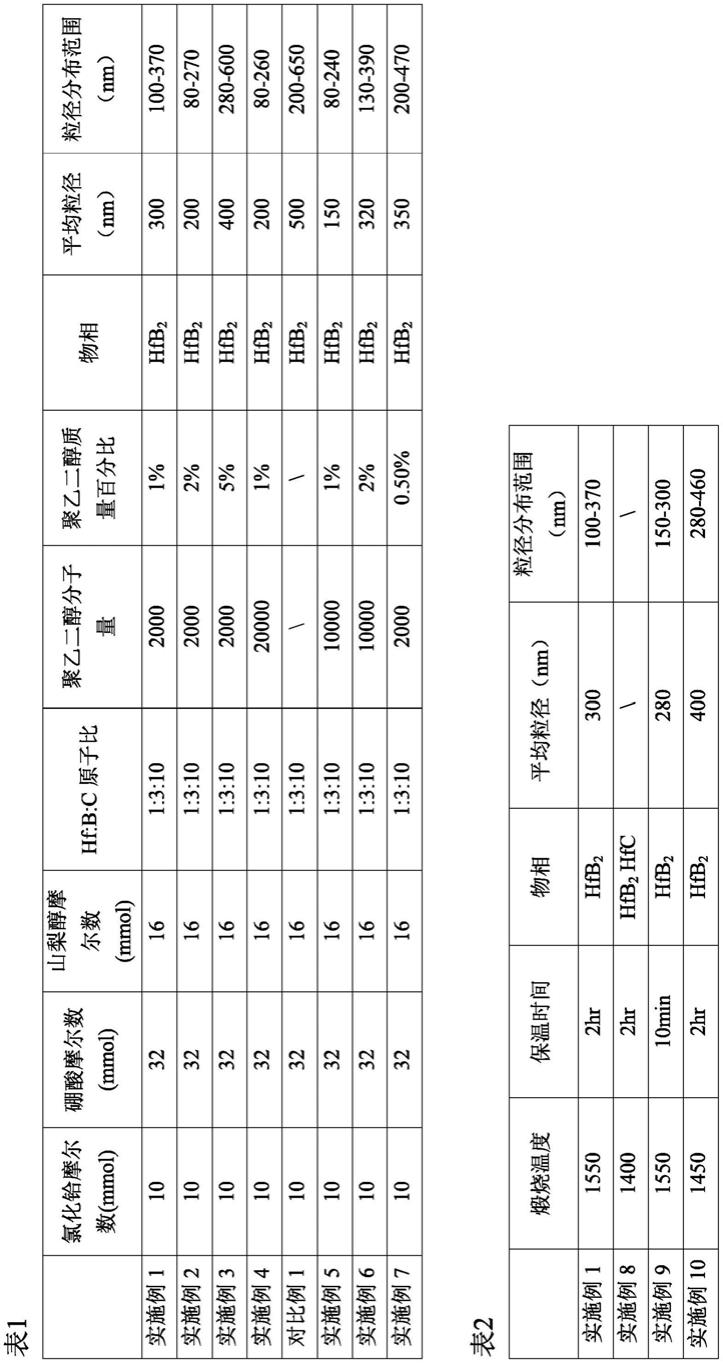
在又一优选的方面,碳源化合物选自山梨醇、酚醛树脂、葡萄糖等,优选山梨醇。
[0048]
在其他优选的方面,聚乙二醇的用量为第一和第二溶液总质量的0.5wt%~5wt%,更优选为1wt%~2wt%。
[0049]
在其他优选的方面,聚乙二醇的分子量为1,000~20,000,更优选为2,000~10,000。
[0050]
本发明通过在反应体系中加入分散剂聚乙二醇,能够有效地调整硼化铪粉体的微观形貌及粒径大小和分布,得到高纯超细的硼化铪粉体。特别地,通过聚乙二醇辅助制备的硼化铪粉体纯度高、粒径小(平均粒径在400nm以下,更优选在300nm以下)、粒径分布窄(粒
径分布跨度在300nm以下,更优选在200nm以下)、微观形貌呈球形颗粒,能够在后续成型过程中增强烧结体的力学性能和烧结驱动力。
[0051]
在又一优选的方面,第一溶剂和第二溶剂可以相同或不同,各自独立地选自羧酸和醇类溶剂,更优选乙酸、乙醇等,再更优选第一溶剂和第二溶剂相同且均为乙酸。
[0052]
在另外优选的方面,铪源化合物与硼源化合物的hf与b的原子比范围为1:2~1:6,更优选为1:3~1:4;铪源化合物与碳源化合物的hf与c的原子比范围为1:8~1:13,更优选为1:9.5~1:11。
[0053]
优选地,在步骤a中,通过恒温搅拌(例如:磁力搅拌)使得铪源化合物溶解于第一溶剂中,温度为20~90℃,更优选为室温,得到淡黄色的溶液。
[0054]
优选地,在步骤b中,通过恒温搅拌(例如:磁力搅拌)使得硼源化合物和碳源化合物溶解于第二溶剂中,温度为20~90℃,更优选为50~70℃。得到澄清的溶液后,加入聚乙二醇,然后冷却到室温。
[0055]
优选地,在步骤c中,通过滴加将第一溶液加入第二溶液中,第一溶液的滴入速率为0.5~15ml/min,更优选为5~10ml/min,产生白色沉淀。
[0056]
优选地,在步骤d中,将步骤c所收集的沉淀在100~150℃下干燥4~24小时。
[0057]
优选地,在步骤d中,所述研磨成粉末通过球磨处理进行,球磨转速为50~400rpm,球磨时间为0.5~4小时。
[0058]
优选地,在步骤d中,所述高温煅烧是以惰气(优选高纯氩气,例如ar≥99.999%)作为保护气,在高温管式炉中以1~5℃/min的速率由室温升温至1000~1200℃,然后以0.5~2℃/min的速率升温至1450~1700℃,更优选1500~1600℃,并保温10~120分钟,再以0.5~2℃/min的速率降温至1000~1200℃,之后以1~5℃/min的速率降温至300~400℃,最后自然降温至室温。这一高温煅烧可以使碳热还原反应充分进行,从而得到高纯度硼化铪粉体。
[0059]
在又一方面,本发明提供一种聚乙二醇辅助制备硼化铪粉体的方法,包括以下步骤:
[0060]
步骤a、将乙酸、四氯化铪混合在一起,并恒温搅拌,使四氯化铪完全溶于乙酸,得到第一溶液;
[0061]
步骤b、将乙酸、硼酸、山梨醇混合在一起,并恒温搅拌,使硼酸和山梨醇完全溶于乙酸,然后加入分子量为1,000~20,000的聚乙二醇,并冷却至室温,形成第二溶液,其中聚乙二醇的加入量为第一和第二溶液总质量的1wt%~2wt%;
[0062]
步骤c、将第一溶液滴入第二溶液中,使得hf与b的原子比范围为1:2~1:6,更优选为1:3~1:4,hf与c的原子比范围为1:8~1:13,更优选为1:9.5~1:11,收集所产生的沉淀,即为硼化铪前驱体;
[0063]
步骤d、将步骤c所收集的硼化铪前驱体干燥,并研磨成粉末,然后进行高温煅烧,从而制得硼化铪粉体。
[0064]
本发明的方法通过使用聚乙二醇作为反应体系中的分散剂,起到良好的分散作用,使得制得的硼化铪前驱体充分分散,避免或减少颗粒之间的相互团聚粘结现象而导致的硼化铪颗粒变大。另外,通过将铪源、硼源和碳源的比例以及煅烧过程的控制,也有利于制备较小粒径、较窄的粒径分布和优异形貌的硼化铪粉体。
[0065]
进一步地,本发明提供的聚乙二醇辅助制备硼化铪粉体的方法至少具有以下优点:
[0066]
(1)本发明提供的聚乙二醇辅助制备硼化铪粉体的方法所制备的硼化铪粉体具有较高的纯度,可以直接使用,在xrd图谱中不显示任何杂质峰,无需再进行除杂处理。
[0067]
(2)本发明提供的聚乙二醇辅助制备硼化铪粉体的方法所制备的硼化铪粉体具有较细的粒径、较窄的粒径分布和较好的微观形貌,其平均粒径多在200~400nm,粒径分布范围的跨度通常为300nm以下,微观形貌为球形颗粒,具有较高的烧结活性。
[0068]
(3)本发明提供的聚乙二醇辅助制备硼化铪粉体的方法仅需实验室常用的普通设备,不需专用设备,也不需要特殊药品,工艺过程简单易操作。
[0069]
(4)本发明提供的聚乙二醇辅助制备硼化铪粉体的方法可以大批量生产,十分适合相关陶瓷材料的宏量、低成本、规模化生产制备。
[0070]
综上可见,本发明实施例不仅制备过程简单、反应过程易控制、生产周期短、成本低廉,而且所制备的硼化铪粉体具有较高的纯度、较细的粒径、较窄的粒径分布和良好的微观形貌,能够在后续成型过程中增强烧结体的力学性能和烧结驱动力。
[0071]
为了更加清晰地展现出本发明所提供的技术方案及所产生的技术效果,下面以具体实施例对本发明实施例所提供的聚乙二醇辅助制备硼化铪粉体的方法进行详细描述。
[0072]
实施例1
[0073]
称取2g(32mmol)硼酸和3g(16mmol)山梨醇,放入同一烧杯中混合,然后向其中倒入20ml乙酸(分析纯,21g),并利用油浴磁力搅拌器使其逐渐升温至60℃,恒温搅拌,直到硼酸和山梨醇完全溶于乙酸,溶液完全澄清,从而得到澄清溶液1。向所述澄清溶液1中加入0.4g聚乙二醇(分子量为2000),并停止加热,持续搅拌直到冷却至室温,然后继续保持搅拌状态。
[0074]
称取3.2g(10mmol)四氯化铪放入烧杯,向其中倒入10ml乙酸(分析纯,10.5g),在常温下使用磁力搅拌器恒温搅拌,直至四氯化铪完全溶于乙酸得到溶液2,溶液呈淡黄色。以10ml/min的速率将淡黄色溶液匀速缓慢滴入到溶液1中(用时1分钟),产生白色沉淀,从而制得硼化铪前驱体溶液。
[0075]
将所述硼化铪前驱体放入恒温干燥箱中,在120℃下烘干12小时,使其完全干燥,从而制得硼化铪前驱体。利用行星球磨机将所述硼化铪前驱体干凝胶研磨至粉末状,球磨转速为400rpm,球磨时间为0.5h。再装进石墨舟中,放入高温管式炉内,以高纯氩气(ar≥99.999%)作为保护气,以5℃/min的速率从室温升温至1000℃,然后以2℃/min的速率升温至1550℃,并保温120分钟,然后以2℃/min的速率降温至1000℃,再以5℃/min的速率降至300℃,最后自然冷却至室温,从而制得实施例1的硼化铪粉体。
[0076]
实施例2
[0077]
称取2g(32mmol)硼酸和3g(16mmol)山梨醇,放入同一烧杯中混合,然后向其中倒入20ml乙酸(分析纯,21g),并利用油浴磁力搅拌器使其逐渐升温至60℃,恒温搅拌,直到硼酸和山梨醇完全溶于乙酸,溶液完全澄清,从而得到澄清溶液1。向所述澄清溶液中加入0.8g聚乙二醇(分子量为2000),并停止加热,持续搅拌直到冷却至室温,然后继续保持搅拌状态。
[0078]
称取3.2g(10mmol)四氯化铪放入烧杯,向其中倒入10ml乙酸(分析纯,10.5g),在
常温下使用磁力搅拌器恒温搅拌,直至四氯化铪完全溶于乙酸得到溶液2,溶液呈淡黄色。以10ml/min的速率将淡黄色溶液匀速缓慢滴入到溶液1中(用时1分钟),产生白色沉淀,从而制得硼化铪前驱体溶液。
[0079]
将所述硼化铪前驱体放入恒温干燥箱中,在120℃下烘干12小时,使其完全干燥,从而制得硼化铪前驱体。利用行星球磨机将所述硼化铪前驱体干凝胶研磨至粉末状,球磨转速为400rpm,球磨时间为0.5h。再装进石墨舟中,放入高温管式炉内,以高纯氩气(ar≥99.999%)作为保护气,以5℃/min的速率从室温升温至1000℃,然后以2℃/min的速率升温至1550℃,并保温120分钟,然后以2℃/min的速率降温至1000℃,再以5℃/min的速率降至300℃,最后自然冷却至室温,从而制得实施例2的硼化铪粉体。
[0080]
实施例3
[0081]
称取2g硼酸(32mmol)和3g山梨醇(16mmol),放入同一烧杯中混合,然后向其中倒入20ml乙酸(分析纯,21g),并利用油浴磁力搅拌器使其逐渐升温至60℃,恒温搅拌,直到硼酸和山梨醇完全溶于乙酸,溶液完全澄清,从而得到澄清溶液1。向所述澄清溶液中加入2g聚乙二醇(分子量为2000),并停止加热,持续搅拌直到冷却至室温,然后继续保持搅拌状态。
[0082]
称取3.2g(10mmol)四氯化铪放入烧杯,向其中倒入10ml乙酸(分析纯,10.5g),在常温下使用磁力搅拌器恒温搅拌,直至四氯化铪完全溶于乙酸得到溶液2,溶液呈淡黄色。以10ml/min的速率将淡黄色溶液匀速缓慢滴入到溶液1中(用时1分钟),产生白色沉淀,从而制得硼化铪前驱体溶液。
[0083]
将所述硼化铪前驱体放入恒温干燥箱中,在120℃下烘干12小时,使其完全干燥,从而制得硼化铪前驱体。利用行星球磨机将所述硼化铪前驱体干凝胶研磨至粉末状,球磨转速为400rpm,球磨时间为0.5h。再装进石墨舟中,放入高温管式炉内,以高纯氩气(ar≥99.999%)作为保护气,以5℃/min的速率从室温升温至1000℃,然后以2℃/min的速率升温至1550℃,并保温120分钟,然后以2℃/min的速率降温至1000℃,再以5℃/min的速率降至300℃,最后自然冷却至室温,从而制得实施例3的硼化铪粉体。
[0084]
实施例4
[0085]
称取2g硼酸(32mmol)和3g山梨醇(16mmol),放入同一烧杯中混合,然后向其中倒入20ml乙酸(分析纯,21g),并利用油浴磁力搅拌器使其逐渐升温至60℃,恒温搅拌,直到硼酸和山梨醇完全溶于乙酸,溶液完全澄清,从而得到澄清溶液1。向所述澄清溶液中加入0.4g聚乙二醇(分子量为20000),并停止加热,持续搅拌直到冷却至室温,然后继续保持搅拌状态下。
[0086]
称取3.2g(10mmol)四氯化铪放入烧杯,向其中倒入10ml乙酸(分析纯,10.5g),在常温下使用磁力搅拌器恒温搅拌,直至四氯化铪完全溶于乙酸得到溶液2,溶液呈淡黄色。以10ml/min的速率将淡黄色溶液匀速缓慢滴入到溶液1中(用时1分钟),产生白色沉淀,从而制得硼化铪前驱体溶液。
[0087]
将所述硼化铪前驱体放入恒温干燥箱中,在120℃下烘干12小时,使其完全干燥,从而制得硼化铪前驱体。利用行星球磨机将所述硼化铪前驱体干凝胶研磨至粉末状,球磨转速为400rpm,球磨时间为0.5h。再装进石墨舟中,放入高温管式炉内,以高纯氩气(ar≥99.999%)作为保护气,以5℃/min的速率从室温升温至1000℃,然后以2℃/min的速率升温
至1550℃,并保温120min,然后以2℃/min的速率降温至1000℃,再以5℃/min的速率降至300℃,最后自然冷却至室温,从而制得实施例4的硼化铪粉体。
[0088]
对比例1
[0089]
称取2g硼酸(32mmol)和3g山梨醇(16mmol),放入同一烧杯中混合,然后向其中倒入20ml乙酸(分析纯,21g),并利用油浴磁力搅拌器使其逐渐升温至60℃,恒温搅拌,直到硼酸和山梨醇完全溶于乙酸,溶液完全澄清,从而得到澄清溶液1。停止加热,持续搅拌直到冷却至室温,然后继续保持搅拌状态下。
[0090]
称取3.2g(10mmol)四氯化铪放入烧杯,向其中倒入10ml乙酸(分析纯,21g),在常温下使用磁力搅拌器恒温搅拌,直至四氯化铪完全溶于乙酸得到溶液2,溶液呈淡黄色。以10ml/min的速率将淡黄色溶液匀速缓慢滴入溶液1中(用时1分钟),产生白色沉淀,从而制得硼化铪前驱体溶液。
[0091]
将所述硼化铪前驱体放入恒温干燥箱中,在120℃下烘干12小时,使其完全干燥,从而制得硼化铪前驱体。利用行星球磨机将所述硼化铪前驱体干凝胶研磨至粉末状,球磨转速为400rpm,球磨时间为0.5h。再装进石墨舟中,放入高温管式炉内,以高纯氩气(ar≥99.999%)作为保护气,以5℃/min的速率从室温升温至1000℃,然后以2℃/min的速率升温至1550℃,并保温120min,然后以2℃/min的速率降温至1000℃,再以5℃/min的速率降至300℃,最后自然冷却至室温,从而制得对比例1的硼化铪粉体。
[0092]
纯度检测及形貌观察
[0093]
对本发明实施例1~4及对比例1所制得的硼化铪粉体进行纯度检测和形貌观察,从而得到以下结果:
[0094]
(1)采用x射线衍射分析仪分别对本发明实施例1~4及对比例1中所制备的硼化铪粉体进行物质检测,从而得到如图1a-1e所示的x射线衍射图谱;其中,图1a为本发明实施例1所制得的硼化铪粉体的xrd图谱,图1b为本发明实施例2所制得的硼化铪粉体的xrd图谱,图1c为本发明实施例3所制得的硼化铪粉体的xrd图谱,图1d为本发明实施例4所制得的硼化铪粉体的xrd图谱,图1e为本发明对比例1所制得的硼化铪粉体的xrd图谱。由图1可以看出:本发明实施例1~4及对比例1所制得的硼化铪粉体在xrd图谱上都表现为高纯,无任何杂质峰。
[0095]
(2)采用扫描电镜sem分别对本发明实施例1~4及对比例1中所制备的硼化铪粉体进行形貌检测,从而得到如图2a-2e和图3a-3e所示的扫描电镜照片。其中,图2a为本发明实施例1所制得的硼化铪粉体在放大倍率为20k下的fesem照片;图2b为本发明实施例2所制得的硼化铪粉体在放大倍率为20k下的fesem照片;图2c为本发明实施例3所制得的硼化铪粉体在放大倍率为20k下的fesem照片;图2d为本发明实施例4所制得的硼化铪粉体在放大倍率为20k下的fesem照片;图2e为本发明对比例1所制得的硼化铪粉体在放大倍率为20k下的fesem照片;图3a为本发明实施例1所制得的硼化铪粉体在放大倍率为60k下的fesem照片;图3b为本发明实施例2所制得的硼化铪粉体在放大倍率为60k下的fesem照片;图3c为本发明实施例3所制得的硼化铪粉体在放大倍率为60k下的fesem照片;图3d为本发明实施例4所制得的硼化铪粉体在放大倍率为60k下的fesem照片;图3e为本发明对比例1所制得的硼化铪粉体在放大倍率为60k下的fesem照片。由图2a-2e和图3a-3e可以看出:未加聚乙二醇的对比例1所制得的硼化铪粉体的微观形貌多呈不均匀、不规则的相互粘结的球体,而本发明
实施例3所制得的硼化铪粉体的微观形貌呈稍有粘结的球体,本发明实施例1所制得的硼化铪粉体的微观形貌多呈分散开的球体,本发明实施例2和例4所制得的硼化铪粉体的微观形貌基本呈均匀且微小的球形颗粒。
[0096]
(3)采用扫描电镜分别对本发明实施例1~4及对比例1所制备的硼化铪粉体进行形貌检测,从而得到硼化铪粉体的平均粒径和粒径分布。各实施例和比较例的硼化铪粉体的平均粒径及粒径分布范围结果见下表1。由表1的结果可以看出:未添加聚乙二醇的对比例1所制备的硼化铪粉体的粒径约为500nm,而本发明实施例1所制备的硼化铪粉体的平均粒径约为300nm,本发明实施例3所制备的硼化铪粉体的平均粒径约为400nm,本发明实施例2和例4所制备的硼化铪粉体的平均粒径最佳,约为200nm。更为明显地,由本发明的方法制备的硼化铪粉体的粒径分布更为均匀,粒径分布窄:实施例1的粉体的粒径在100~370nm的范围内;实施例2的粉体的粒径在80~270nm的范围内;实施例3的粉体的粒径在280~600nm的范围内;实施例4的粉体的粒径在80~260nm的范围内;而对比例1的粉体的粒径分布较宽,在200~650nm的范围内。
[0097]
实施例5-7
[0098]
为了探究不同聚乙二醇分子量及添加量对于制备的硼化铪粉体的粒径及粒径分布的影响,申请人还按照实施例1的方法步骤制备硼化铪粉体,不同之处仅在于采用了不同的聚乙二醇分子量及添加量,具体参数及结果请见表1。
[0099]
实施例8-10
[0100]
为了探究步骤d中高温煅烧的温度及保温时间对于制备的硼化铪粉体的粒径及粒径分布的影响,申请人还按照实施例1的方法步骤制备硼化铪粉体,不同之处仅在于在步骤d中采用了不同的煅烧温度和/或保温时间,具体参数及结果请见表2。
[0101]
综上可见,本发明不仅制备过程简单、反应过程易控制、生产周期短、成本低廉,而且所制备的硼化铪粉体具有较高的纯度、较细的粒径、较窄的粒径分布和良好的微观形貌,能够在后续成型过程中增强烧结体的力学性能和烧结驱动力。
[0102]
以上仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求书限定的保护范围为准。
[0103]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1