一种伏立康唑及其中间体的制备方法与流程
本发明涉及药物化学领域,具体涉及用于治疗急性或慢性深部真菌感染的第二代三唑类抗真菌药伏立康唑及其中间体一种新的制备方法。
背景技术:
伏立康唑为一手性药物,分子中含有两个手性中心,有四个异构体。分别为(2R,3S/2S,3R)与(2S,3S/2R,3R)。构效关系研究(SAR)证实,其中(2R,3S/2S,3R)对映体的抗菌活性为(2S,3S/2R,3R)对映体的200倍以上。进一步对具有良好活性的(2R,3S/2S,3R)一对对映体进行拆分的试验研究,结果表明,单个异构体(2R,3S),即伏立康唑。对曲霉菌(真菌)的抑制活性为(2S,3R)异构体的200倍以上,且相比较于(2S,3R),异构体(2R,3S)具有更广泛的抗菌谱。目前临床上使用的活性成分(API)为具有绝对构型的(2R,3S)单一异构体。
文献报道的制备伏立康唑的方法归纳起来主要由两条途径,其中一条为先制备(2R,3S/2S,3R)消旋体,然后拆分制备。第二条途径为采用手性试剂(催化剂或配体)进行不对称缩合直接制备。现对以上两条制备伏立康唑的途径进行详细的阐述。
(1)拆分法:文献[1]_WO2012114273A1,[2]_CN102516233A,[3]_ CN102190628A,[4]_WO2009084029A2,[5]_CN1814597A,[6]_CN1488630A,[7]_CN1473825A,[8]_Organic Process Research&Development(2001),5(1),28-36,[9]_WO9706160A1,[10]_WO2008075205A2报道的方法都是先制备获得伏立康唑消旋体(2R,3S/2S,3R混合物,各占50%),然后通过与手性樟脑磺酸形成两对非对映异构体,利用两对非对映异构体物理性质(溶解度)的不同,进而达到拆分的目的。采用此种办法制备伏立康唑,反应条件简单,适合于工业化生产,但是也存在明显的缺陷,主要表现在制备伏立康唑消旋体(2R,3S/2S,3R)过程中,不可避免的会生成(2S,3S/2R,3R)一对异构体,(2S,3S/2R,3R)作为伏立康唑的非对映异构体虽可以通过重结晶去除,但终究是降低了目标产物的转化率,进而最终降低了收率。另一方面,在对伏立康唑消旋体(2R,3S/2S,3R)进行光学拆分制备过程中,不可避免的又要舍弃(2S,3R)异构体。导致制备伏立康唑产品的过程中总收率又至少降低50%。(2)不对称合成法:文献[11]_CN1919846A,[12]_CN104987325A,[13]_WO2015041148A1,[14]_CN103788073A,[15]_Journal of Organic Chemistry(2013),78(22),11396-11403,[16]_WO2014060900A1报道的方法主要是在伏立康唑的嘧啶环结构片段与2',4'-二氟-2-[1-(1H-1,2,4-三唑基)]苯乙酮在进行羰基加成反应过程中,要么在嘧啶环结构片段引入手性辅助试剂,或者通过在缩合过程中,加入过渡金属催化剂,手性配体(试剂)进行不对称诱导反应制备伏立康唑(2R,3S)异构体。采用此种策略制备伏立康唑,优势是(2R,3S)异构体占比较高(一般大于50%),但是目前也存在以下几个问题,一是在不对称合成制备伏立康唑或其中间体过程中,需要使用到价格昂贵及难于大量获得的过渡金属催化剂(如双三苯基磷二氯化钯(Ⅱ), CuF(PPh3)2)及手性配体((S)-1-((SP)-2-[2-(二苯基膦)苯基]二茂铁基)乙基二[3,5-二(三氟甲基)苯基]膦),目前,此种方法仍然处于试验室小规模制备伏立康唑样品阶段,无法满足工业化生产对成本控制的要求。二是虽然采用不对称合成制备伏立康唑,使(2R,3S)异构体的含量占比提高,但由于不对称合成的选择性问题,提高幅度有限,制备的伏立康唑当中,异构体杂质(2S,3R)的含量仍然可以占到(5%~30%),因此最终如需要获得异构体杂质含量合格的伏立康唑产品,后续纯化仍然需要进行拆分处理。需要提及的是文献[11],[14]报道的方法只是提高了(2R,3S/2S,3R)异构体对与(2S,3S/2R,3R)异构体对的比值,因而不是严格意义上的不对称合成,只是对拆分方法的优化。
综上,使用上述方法制备式I化合物,由于理论及大生产对成本控制的限制,虽可以对部分参数进行优化,但工业化生产过程中,仍然无法大幅提高原辅料利用率,进而获得收率较高,生产成本更低的伏立康唑原料药,为此亟待一种新的技术方案,以提供更优的工艺条件,提高原辅料利用率,减少资源浪费和保护环境,最大限度的降低生产成本,使其更有利于大规模工业生产。
技术实现要素:
本发明的目的是提供一种由伏立康唑异构体制备伏立康唑及其中间体的制备方法,该方法具有原辅料利用率高,反应条件温和,原料与试剂毒性低,分离纯化过程简单,非常适合于大规模工业化循环生产。
本发明提供一种伏立康唑中间体的制备方法,其特征在于,包括以下步骤:以伏立康唑异构体为原料,于碱性条件下,经反应并后处理后得化合物 Ⅱ与化合物Ⅲ;
所述的伏立康唑异构体是指伏立康唑分子中的两个手性中心具有2S,3S或2R,3R或2S,3R构型的分子或其药学上可接受的盐或上述物质的混合物;
所述碱性条件是指碱性水溶液或碱性水溶液与有机溶剂的混合液;
其中,所述有机溶剂选自烃、卤代烃、酯或醚;烃或卤代烃选自甲苯、1,2-二氯乙烷、二氯甲烷、三氯甲烷;优选1,2-二氯乙烷或二氯甲烷;酯选自甲酸乙酯、乙酸甲酯、乙酸乙酯、乙酸丁酯或乙酸丙酯;醚选自乙醚、异丙醚或甲基叔丁基醚;
其中,所述用于配制碱性水溶液的碱选自碱金属或碱土金属阳离子与OH-,CO32-,HCO3-,H-,-OCH3,-OC2H5,-OC4H9;优选NaOH,KOH,Na2CO3,K2CO3,NaH,NaOCH3,KOCH3,Ca(OH)2等;
其中,所述碱性水溶液的浓度为0.01~12mol/L;优选的,浓度为0.5~10mol/L;较优选的,浓度为1~8mol/L;较优选的,浓度为2~6mol/L;较优选的,浓度为2.5~5mol/L;最优选的,浓度为3mol/L;
其中,所述伏立康唑异构体与碱的投料摩尔比为1:0.5~20.0;较优选的,比例为1:1.0~15.0;较优选的,比例为1:1.2~10.0;较优选的,比例为1:1.2~7.0;最优选的,比例为1:4.5;
其中,所述伏立康唑异构体与碱性水溶液进行反应的温度为25~100℃;优选的,反应温度为30~80℃;较优选的,反应温度为40~70℃;更优选的,反应温度为50~60℃;
其中,所述伏立康唑异构体与碱性水溶液进行反应的时间为1~24小时;优选的,反应时间为2~12小时;优选的,反应时间为4~6小时;
所述的反应后处理步骤包括萃取、浓缩、蒸馏;优选的,所述的后处理包括先萃取分离出包含化合物Ⅱ与化合物Ⅲ的萃取液,然后对萃取液进行浓缩,获得残渣固体,对所述残渣固体进行重结晶或不进行重结晶即可获得化合物Ⅲ;同时收集浓缩获得的浓缩液,再对浓缩液进行蒸馏,获得化合物Ⅱ;
另外,本发明还提供了一种伏立康唑的制备方法,其特征在于,包括以下步骤:以伏立康唑异构体为原料,在碱性条件下,经反应并后处理后得化合物Ⅱ与化合物Ⅲ,将化合物Ⅱ与化合物Ⅲ进行缩合反应,制备获得伏立康唑外消旋体,对伏立康唑外消旋体进行光学拆分,精制后获得伏立康唑,即化合物Ⅰ;
所述的缩合反应是指将化合物Ⅱ溴代后与化合物Ⅲ进行雷福尔马茨基反 应,或者在二异丙氨基锂或六甲基二硅氨基锂等存在下进行缩合反应;
所述的对伏立康唑外消旋体进行手性拆分的方法为本领域的常见方法;优选的,所述的拆分试剂为酸性拆分试剂;进一步优选的,所述的拆分试剂为:所述化学拆分方法的拆分试剂为酸性拆分试剂;优选的,所述的拆分试剂为:(+)-酒石酸、(+)-樟脑酸、L-(+)-甘氨酸或L-樟脑-10-磺酸;更优选的,所述的拆分试剂为L-樟脑-10-磺酸。
本发明的有益效果
与现有技术相比,本发明体现的技术优点体现在以下几个方面:
(1)本发明提供的技术方案是以伏立康唑异构体为起始原料,采用简单的反应条件即可制备获得化合物Ⅱ与化合物Ⅲ,无需使用制备色谱或层析柱分离,相较于现有技术制备化合物Ⅱ与化合物Ⅲ的方案具有合成路线步骤短(只需一步即可同时获得化合物Ⅱ与化合物Ⅲ),收率高,成本低,更易于规模化工业生产的优势。
(2)本发明提供的技术方案采用的起始原料伏立康唑异构体,是现有技术制备或拆分伏立康唑消旋体过程中产生的固废或杂质,是一种“变废为宝”的清洁生产技术方案,可以大幅提高现有技术制备伏立康唑原辅料利用率,节约资源及保护环境,不但可以降低成本,亦可以使伏立康唑的生产可循环化。
(3)本发明提供的技术方案制备获得的化合物Ⅱ与化合物Ⅲ,后续用于进行伏立康唑的制备无需使用钯碳进行脱氯氢解,较现有技术方案制备伏立康唑不可避免的需要使用钯碳进行脱氯氢解又节约了一步化学反应,不但简 化了后续操作,同时进一步降低了制备伏立康唑的成本。
附图说明
图1为化合物Ⅰ伏立康唑的化学结构式;
图2为以伏立康唑异构体为原料,制备出伏立康唑中间体化合物Ⅱ、Ⅲ的反应路线图;
图3为以伏立康唑异构体为原料,制备出化合物Ⅰ伏立康唑的反应路线图;
图4实施例2的反应路线图;
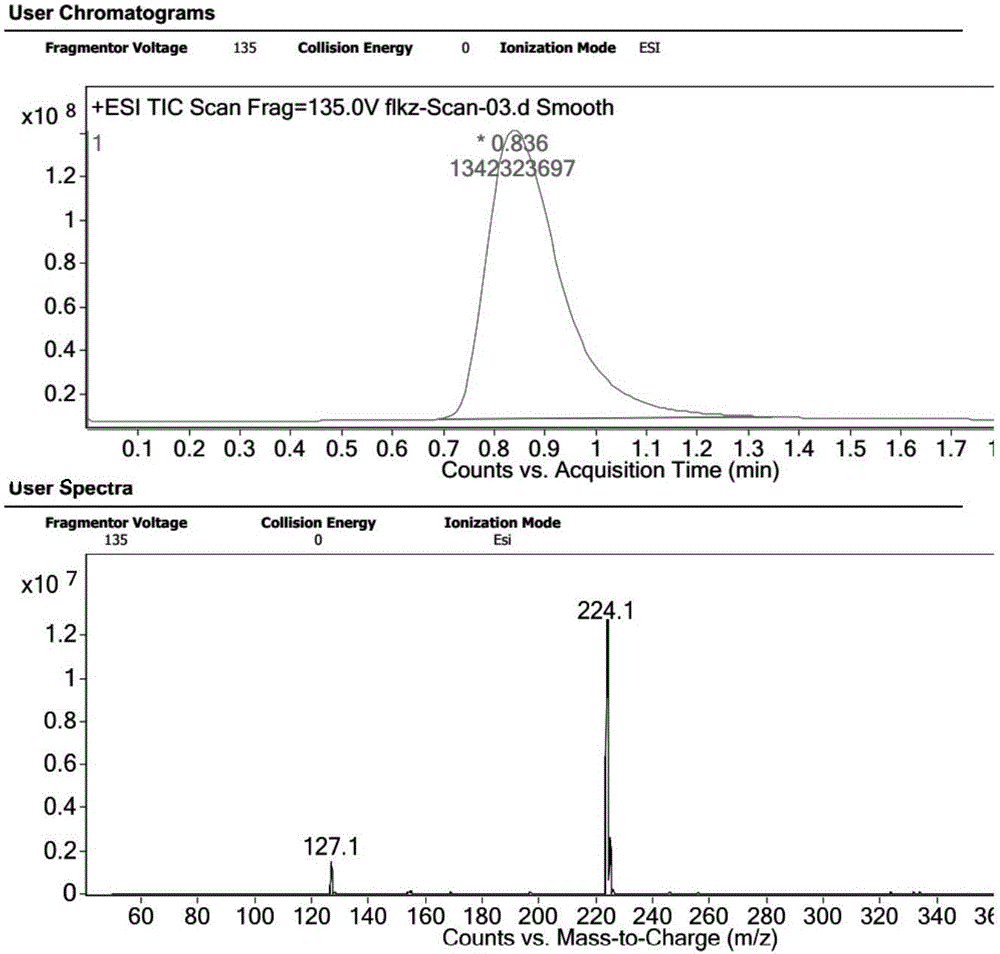
图5为化合物Ⅱ的液质联用(LC-MS)图谱;
图6为化合物Ⅲ的液质联用(LC-MS)图谱;
图7为化合物Ⅰ伏立康唑的1HNMR图谱。
具体实施方式
实施例1:
一种伏立康唑中间体的制备方法,其特征在于,包括以下步骤:以伏立康唑异构体(2S,3R)为原料,取原料157.5g,在NaOH水溶液中进行反应,NaOH的浓度为2mol/L,反应温度为50℃,反应时间为5小时,反应结束后,将反应混合液冷却,先使用乙酸乙酯萃取分离出化合物Ⅱ与化合物Ⅲ的混合物,而后浓缩获得化合物Ⅲ,然后再对收集的浓缩液进行常压蒸馏,以获得化合物Ⅱ。化合物Ⅱ与化合物Ⅲ的液质联用图谱见附图5和6。
实施例2:
一种伏立康唑的制备方法,其特征在于,包括以下步骤:以伏立康唑异构体(2S,3R)为原料,取原料157.5g,在NaOH水溶液中进行反应,NaOH的浓度为1mol/L,反应温度为50℃,反应时间为7小时,反应结束后,将反应混合液冷却,经过乙酸乙酯萃取,得化合物Ⅱ与化合物Ⅲ的混合物,而后浓缩获得化合物Ⅲ,然后再对收集的浓缩液进行常压蒸馏,而获得化合物Ⅱ。将化合物Ⅱ使用NBS溴代(可参考相应文献),获得4-(1-溴乙基)-5-氟嘧啶。再加入反应试剂,与化合物Ⅲ进行雷福尔马茨基反应缩合反应,制备获得伏立康唑外消旋体,然后加入手性试剂(L)-樟脑-10-磺酸对上述外消旋体进行光学拆分,将拆分产物结晶,游离获得伏立康唑,即化合物Ⅰ,反应路线图见附图4。伏立康唑的1HNMR图谱见附图7。
实施例3:
一种伏立康唑中间体的制备方法,其特征在于,包括以下步骤:以伏立康唑异构体(2R,3R/2S,3S/2S,3R)混合物为原料,取原料174.5g,在NaOH水溶液中进行反应,NaOH的浓度为5mol/L,反应温度为45℃,反应时间为3小时,反应结束后,将反应混合液冷却,经过二氯甲烷萃取,得化合物Ⅱ与化合物Ⅲ的混合物,而后浓缩获得化合物Ⅲ,然后再对收集的浓缩液馏分进行常压蒸馏,而获得化合物Ⅱ。化合物Ⅱ与化合物Ⅲ的质谱图见附图5和6。
实施例4:
一种伏立康唑的制备方法,其特征在于,包括以下步骤:以伏立康唑异 构体(2S,3R)樟脑磺酸盐为原料,取原料77.6g,在NaOH/乙酸乙酯水溶液中进行反应,NaOH的浓度为1mol/L,反应温度为60℃,反应时间为4小时,反应结束后,将反应混合液冷却,经过乙酸乙酯萃取,得化合物Ⅱ与化合物Ⅲ的混合物,而后浓缩获得化合物Ⅲ,然后再对收集的浓缩液馏分进行常压蒸馏,而获得化合物Ⅱ。将化合物Ⅱ使用NBS溴代(可参考相应文献),获得4-(1-溴乙基)-5-氟嘧啶。再加入反应试剂,与化合物Ⅲ进行雷福尔马茨基反应缩合反应,制备获得伏立康唑外消旋体,然后加入手性试剂(L)-樟脑-10-磺酸对上述外消旋体进行光学拆分,将拆分产物结晶,游离获得伏立康唑,即化合物Ⅰ。伏立康唑的1HNMR图谱见附图7。
- 还没有人留言评论。精彩留言会获得点赞!