一种具有增强植物基因表达活性的5′UTR序列及其应用的制作方法
本发明涉及生物技术领域,具体地说,涉及一种具有增强基因表达能力的马铃薯花粉特异表达基因SBgLR的5′UTR序列。
背景技术:
基因表达调控一直是分子生物学研究的热点和前沿,真核基因表达调控可以在染色体、转录、转录后、翻译、翻译后等多个层次上进行。在植物基因工程研究中,目的基因表达水平偏低成为限制转基因植物应用和基因功能研究的主要因素。目前提高基因表达的主要方式是使用强启动子或利用增强子提高基因的转录水平。除了启动子和增强子序列对基因转录的贡献,5′非翻译区(5′untranslated region,5′UTR)也可以影响真核生物基因的表达水平,5′UTR具有稳定mRNA、形成翻译起始复合体和促进蛋白质合成等作用(Barrett et al.,2013)。很多研究都确定了在5′UTR的内含子可提高基因表达(Akua et al.,2010;Kamo et al.,2012;Xiao et al.,2014),这种上调作用定义为内含子介导的增强(intron-mediated enhancement,IME),IME增加了成熟mRNA的表达量,却不影响mRNA的稳定性(Gallegos and Rose,2015)。除了IME外,在一些基因中也发现内含子序列能抑制基因在转录水平的表达(Pan and Simpson,1999;Zheng et al.,2009)。
5′UTR中存在的上游起始密码子(Upstream AUG,uAUG)或上游开放阅读框(uORFpstream open reading frame u,uORF)一般对翻译有抑制作用。uAUG和uORF可以提供不同于主要ORF的翻译起始位点,核糖体结合uORF中的uAUG可以使主要ORF的翻译效率下降,由于核糖体具有重新起始翻译的作用,从而位于uORF终止密码子之后的主要ORF可以重新翻译;而uAUG之后不存在终止密码子则会导致核糖体不能重新起始翻译,因而uAUG对于翻译的抑制作用要强于uORF(Barrett et al.,2013;Wilkie et al.,2003)。
研究发现花粉特异基因中也存在着5′UTR对基因表达的调控。LAT59基因的5′UTR在瞬时表达体系中可以抑制基因表达20倍,而在稳定表达的转基因植株中可以抑制报告基因表达300倍,非翻译区的抑制作用是在+62~+108区段存在一个茎环结构,降低了mRNA积累,而对翻译效率和mRNA的稳定性没有影响(Curie et al.,1997)。在LAT52的5′UTR存在着增强基因表达的顺式作用元件,可以提高报告基因表达13~60倍(Bate et al.,1996)。
已有的研究发现,5′UTR的长度、起始位点的序列、内含子、二级结构、uAUG、uORF以及内部核糖体进入位点(internal ribosome entry sites,IRES)等都可能影响基因的表达(Wilkie et al.,2003)。不同基因的5′UTR对下游基因的作用是不同的,有的可以增强基因表达,而有的则起抑制作用。
技术实现要素:
为了解决现有技术中存在的问题,本发明的目的是提供一种具有增强基因表达能力的马铃薯花粉特异表达基因SBgLR的5′UTR序列及其应用。
为了实现本发明目的,本发明的技术方案如下:
第一方面,本发明提供了一种具有增强植物基因表达活性的5′UTR序列,其核苷酸序列为:1)SEQ ID NO.1所示的DNA序列;或2)在高严谨条件下可与SEQ ID NO.1所示的DNA序列杂交的核苷酸序列。所述高严谨条件为在0.1×SSPE(或0.1×SSC)、0.1%SDS的溶液中,65℃条件下杂交并洗膜。
本发明从马铃薯中克隆了花粉特异表达基因SBgLR 5′UTR DNA序列,研究发现SBgLR基因启动子+5′UTR可以驱动GUS基因在转基因烟草花粉中特异表达。而当该5′UTR缺失后,SBgLR基因启动子则不能驱动GUS基因表达。
进一步地,本发明提供了含有所述5′UTR序列的载体。
作为优选,所述载体为植物表达载体。
进一步地,所述载体还含有启动子序列和目的基因序列,经试验研究发现,当所述5′UTR序列位于启动子序列与目的基因序列之间,能显著提高目的基因的表达量。
在本发明的一个具体实施方式中,所构建的表达载体中启动子与5′UTR之间的距离是一个酶切位点(6个碱基),5′UTR与目的基因(GUS基因)起始密码子的距离是24个碱基。应当理解,上述距离发生改变却不影响本发明技术方案的方案均在本发明的保护范围之内。
更为优选地,所述启动子可选自马铃薯中SBgLR基因启动子、CaMV35S启动子、玉米种子特异启动子19Z或谷子种子特异启动子F128。
当该5′UTR序列位于CaMV35S启动子和GUS基因之间时能增加GUS基因在转基因烟草根、茎、叶、花药和花粉中的表达。检测到烟草幼苗中的GUS基因的转录水平提高了3-5倍,GUS酶活性提高了12-15倍。花药和花粉中GUS酶活性提高了2.5倍以上。
当该5′UTR序列位于玉米种子特异启动子19Z和GUS基因之间时能增加GUS基因在转基因烟草种子中的表达,检测到未成熟种子中GUS酶活性提高了约10倍。
当该5′UTR序列位于谷子种子特异启动子F128和GUS基因之间时能增加GUS基因在转基因烟草种子中的表达,检测到未成熟种子中GUS酶活性分别提高了10倍。
本发明所述的5′UTR序列来源于马铃薯SBgLR基因,能增强自身启动子和异源启动子的表达活性。5′UTR可以直接影响真核生物中基因的表达,很多研究都确定了在5′UTR的内含子可提高基因表达。而本发明的5′UTR序列不包含内含子。
更进一步地,本发明还提供了含有前述载体的转基因细胞系和宿主菌。
此外,用于扩增所述5′UTR序列的引物以及用于检测所述5′UTR序列的引物,由于其设计需要基于所述5′UTR序列的特异性,也同样在本发明的保护范围之内。
第二方面,本发明提供了所述5′UTR序列在增强植物基因表达方面的应用。
所述应用具体为:将启动子、5′UTR序列和目的基因构建得到重组质粒,所述5′UTR序列位于启动子序列与目的基因序列之间;将所得重组质粒利用农杆菌介导转化植物植株,增强目的基因的表达。
经试验对比发现,当所述启动子为种子特异启动子时,5′UTR序列能更加显著的发挥增强植物基因表达的作用。
本发明的有益效果在于:
SBgLR是马铃薯花粉发育晚期特异表达基因,编码一个细胞骨架相关蛋白。对SBgLR基因的调控序列的研究发现,SBgLR基因5′UTR中不含有上游AUG(uAUG)和上游开放阅读框(uORF),也不包含内含子。本发明首次揭示了SBgLR基因的5′UTR序列可以提高不同类型启动子的表达活性,这种增强作用具有通用性,可在基因工程和基因功能研究中用于提高目标基因的表达。
附图说明
图1为5′RACE方法确定转录起始位点。
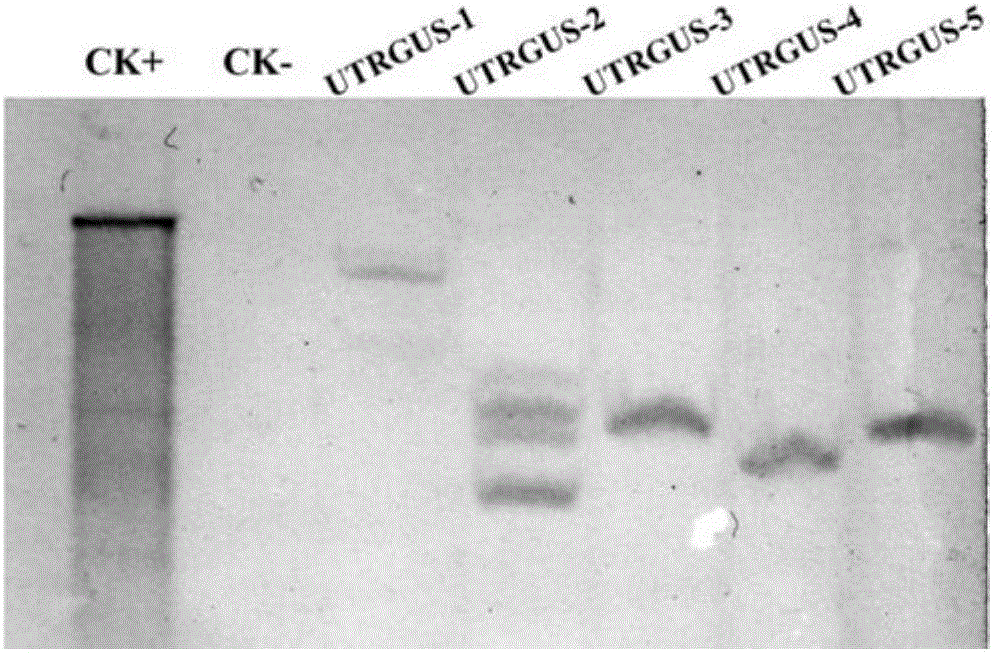
图2为转p35SUTR-GUS烟草植株的Southern杂交;CK+:HindIII单酶切后的质粒pBI121;CK-:野生型烟草DNA。
图3为SBgLR 5′UTR增强GUS基因在转基因烟草中的表达;A.p175-GUS、p35SUTR-GUS和p35S-GUS载体示意图;B.T1转基因烟草不同组织GUS组织化学染色分析。
图4为转p35SUTR-GUS烟草幼苗中GUS表达水平分析。
图5为转p35SUTR-GUS烟草幼苗GUS酶活性分析。
图6为转p35SUTR-GUS烟草不同组织GUS酶活性分析。
图7为表达载体p19ZUTR-GUS和pF128UTR-GUS构建策略。
图8为转基因烟草种子GUS活性分析;A.烟草种子GUS组织化学染色;B.烟草种子GUS酶活性定量分析。
具体实施方式
下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、SBgLR基因转录起始位点的确定
利用5′RACE方法确定SBgLR基因的转录起始位点,操作步骤参照Invitrogen公司的Invitrogen GeneRacer Kit说明书。取野生型马铃薯花粉,Trizol(Invitrogen)法提取花粉总RNA。取5μg总RNA,用CIP酶进行去磷酸化,再用TAP酶对去磷酸化的RNA进行去5′帽子结构处理。已去帽子结构的RNA 5′端连接已知的接头(RNA oligo)作为模板,以oligodT为引物进行反转录合成第一链cDNA。以cDNA为模板进行PCR,第一轮PCR引物:GeneRacerTM5′Primer:5′-CGACTGGAGCACGAGGACACTGA-3′,UTR-GSP:5′-ACAGCCACCACTTTCGGTGCAATC-3′,反应程序如下:94℃,5min;94℃,30s;60℃,30s;68℃,30s;扩增循环数:30。最后68℃,延伸10min。以第一轮PCR产物为模板,进行第二轮PCR,引物:GeneRacerTM5′Nested Primer:5′-GGACACTGACATGGACTGAAGGAGTA-3′,UTR-NGSP:5′-ACTGCGTGCTTTGATTCCCCACAAC-3′,反应程序同上。第二轮PCR扩增出一条约300bp条带。测序结果显示扩增产物包含RNA Oligo接头和5′UTR序列(图1),确定转录起始位点是位于起始密码子上游-184bp位置的A,SBgLR基因5′UTR全长184bp(序列表),不含有上游AUG(uAUG)和上游开放阅读框(uORF)。与基因组序列比对显示,基因5′UTR中也不包含内含子。
实施例2、SBgLR基因5′UTR增强自身启动子和组成型启动子CaMV35S的表达活性
1、表达载体构建所用引物
1.1 缺失5′UTR的SBgLR启动子(175bp)引物序列:
Futr-d:AAGCTTAACTTTAAATCTTGAATTCCGTCACG(含HindⅢ位点)
Rutr-d:GGATCCTTTTCACCCTTTGGTTTTTCTTGGC(含BamHI位点)
1.2 获得5′UTR(184bp)的引物序列:
Futr-1:CTGCAGAAAGGGTGAAAAGAAAAAAAG(含XbaI位点)
Rutr-1:GGATCCTTTTTATTATGAAGAATTGGAG(含BamHI位点)
2、表达载体的构建
2.1 表达载体p175-GUS(缺失5′UTR的SBgLR启动子驱动GUS基因)的构建:
以含有完整SBgLR基因序列的质粒[Lang,Z.,Zhou,P.,Yu,J.,Ao,G.,and Zhao,Q.Functional characterization of the pollen-specific SBgLR promoter from potato(Solanumtuberosum L.).Planta 2008,227,387-396]为模板,用引物Futr-d和Rutr-d进行PCR扩增,扩增出缺失了5′UTR的SBgLR核心启动子片段,该片段长175bp。将PCR产物用HindⅢ和BamHI双酶切,酶切片段连接到载体pBI121的HindⅢ和BamHI位点,替换载体上的CaMV35S启动子序列(载体示意图见图3A)。
2.2 表达载体p35SUTR-GUS的构建:
以含有SBgLR基因序列的质粒为模板,用引物Futr-1和Rutr-1进行PCR扩增,扩增出长184bp的SBgLR基因的5′UTR序列。PCR产物用XbaI和BamHI双酶切,酶切片段插入到载体pBI121的CaMV35S启动子和GUS基因之间(载体示意图见图3A)。
3、表达载体转化烟草
利用冻融法将构建的植物表达载体p175-GUS、p35SUTR-GUS和对照质粒pBI121(p35S-GUS)转入农杆菌LBA4404菌株,转化方法参照Hofen等[Hofen R,Willmitzer L,Storage of competent cells for Agrobacteriun transformation.Nucleic Acids Research,1988.16,9877]。烟草的转化参照Horsch等的方法[Horsch RB,Fry JE,Horffman N.A simple and general method for transferring genes into plants.Science,1985.227:1229-1231]。具体操作如下,将含有植物表达载体的农杆菌接种于YEB液体培养液(含100μg/ml Km,125μg/ml Sm)中,28℃振荡培养至OD600为0.6-0.8;以10,000rpm室温离心1min,用MS盐溶液(pH 7.0)重新悬浮菌体,使用时采用MS盐溶液稀释至原体积的20~50倍;无菌的烟草叶片切去边缘和主要叶脉,切成0.4-0.6cm2大小,在农杆菌菌液中浸泡10min;用无菌滤纸吸干植物材料表面的菌液,转入上铺一层滤纸的MS基本培养基,28℃暗培养;三天后,将材料转到含有100μg/ml Km和500μg/ml Cb的MS分化培养基中(MS培养基+3mg/L 6-BA,0.2mg/L NAA,进行培养;待抗性芽生长至2-3cm高时,切下小芽转入MS生根培养基(MS培养基含有100μg/ml Km和500μg/ml Cb)中诱导生根。
4、转基因烟草的分子检测
PCR检测:提取再生烟草叶片基因组DNA,利用GUS基因的特异引物(GUSP1:CTGCGACGCTCACACCGATACC;GUSP2:TCACCGAAGTTCATGCCAGTCCAG)进行PCR检测,转入外源基因的烟草植株可以扩增出500bp的目的条带,而非转基因植株则无相应条带。
Southern杂交检测:选取PCR阳性的转基因烟草,进行基因组DNA Southern杂交检测,取20μg DNA,用限制性内切酶HindⅢ完全酶切,电泳,转膜。以地高辛标记的GUS片段为探针,探针标记和杂交按照Roche DIG High Prime DNA Labeling and Detection Starter Kit I说明书进行。Southern杂交结果显示,在检测的5个转p35SUTR-GUS烟草株系中,除了一个株系为多拷贝外,其余4个株系均为单拷贝的GUS基因插入,而且来自不同的转化事件(图2)。
5、转基因烟草GUS活性的组织化学分析
GUS活性的组织化学分析方法参考Bradford方法[Bradford HM,A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding.Anal Biochem.,1976,72:248-254.]。将待检测的植物材料加入GUS染色液中(50mM磷酸钠缓冲液(PH7.0),0.5mM K3[Fe(CN)6],0.5mM K4[Fe(CN)6],10mM EDTA,0.5mg/ml X-Gluc),37℃保温数小时或过夜;叶片等绿色材料用70%乙醇脱色2-3次,至阴性对照材料呈白色;肉眼或显微镜观察,白色背景下的蓝色即为GUS表达位点。
对转化不同表达载体的T1代烟草幼苗、花粉和花药进行GUS组织化学染色分析,发现在p175-GUS转基因烟草中,当具有花粉特异启动活性的-163~+180区段缺失了5′UTR的167bp后,检测不到花粉表达活性,其它各组织中也未检测到GUS的表达(图3B)。在对照p35S-GUS转基因烟草中,CaMV35S启动子能够驱动GUS在各组织中表达,但在子叶叶柄、根尖和花粉中却未检测到GUS活性。然而,在p35SUTR-GUS转基因烟草中,将SBgLR基因5′UTR的184bp与CaMV35S启动子融合后,可以驱动报告基因GUS在烟草各组织中表达,子叶叶柄、根尖中均有GUS表达,花粉中也有表达,而且GUS染色的程度比p35S-GUS转基因烟草深(图3B)。结果表明5′UTR可能具有增强表达的功能。
6、Real-time PCR分析转基因烟草中GUS表达水平
Trizol法提取转基因烟草植株幼苗总RNA,以OligodT为引物反转录合成cDNA第一条链。设计Real-time PCR扩增引物,GUS-RT-F:CGTCGTCGGTGAACAGGTAT和GUS-RT-R:TCATTGTTTGCCTCCCTGCT。以cDNA为模板,在荧光定量PCR仪(qTOWER2.2,Analytik Jena,德国)中反应。反应条件:95℃,10min;95℃,10sec,60℃,1min,40个循环。
对4个单拷贝插入的纯合转p35SUTR-GUS载体的烟草株系进行了GUS表达的定量分析。qRT-PCR分析T2代转p35SUTR-GUS烟草幼苗中GUS的相对表达量,数值为三次生物学重复的平均值,对照与转基因株系间的显著性分析依照t检验(*p<0.05,**p<0.01)。
各p35SUTR-GUS转基因株系中GUS基因的表达量比转p35S-GUS烟草中均明显提高,其中1个株系提高3倍左右,其余3个株系均提高了5倍以上(图4),说明SBgLR 5′UTR对转录有影响,能够增加基因的表达量。
7、转基因烟草的GUS酶活性定量检测
7.1 植物总蛋白的提取及浓度测定
(1)取0.1~0.2g植物样品在液氮中研磨成粉末,加入3倍体积的蛋白提取缓冲液(0.05M磷酸缓冲液(pH 7.0),0.1%SDS,0.01M EDTA(pH 8.0),20%甲醇,0.1%Triton X-100,0.1%β-巯基乙醇),冰浴振荡5min;4℃,12000rpm离心10min,取上清4℃保存备用。
(2)制作蛋白标准曲线:配制BSA浓度梯度溶液,分别与考马斯亮蓝染液G-250反应后,用紫外分光光度计(UV2300,日立,日本)测定595nm的光吸收值,制作标准曲线。
(3)样品蛋白含量测定:取适量提取的蛋白溶液,加入蛋白提取缓冲液至总体积400μl,混匀,加入1.1ml考马斯亮蓝染液G-250,混匀,室温放置2min,测定595nm的光吸收值,根据蛋白标准曲线计算样品蛋白含量。
7.2 GUS荧光测定
(1)制作4-MU标准曲线:用反应终止液(0.2M Na2CO3)配制4-MU(4-Methylumbelliferone,4-甲基伞形酮,Sigma)梯度浓度溶液:0nM,10nM,100nM 500nM,1000nM,2500nM;用荧光分光光度计(F4500,日立,日本)在狭缝5nm,激发光365nm,发射光455nm下分别测定样品的荧光值,绘制标准曲线。
(2)GUS酶活检测:取100μl蛋白提取液,加入400μl 37℃预热的反应缓冲液(蛋白提取缓冲液中含2mM MUG),立即取50μl加入到950μl反应终止液作为零时的空白对照,其余在37℃温浴,反应30min,60min,90min和120min后各取50μl加入到950μl反应终止液。测定荧光值,根据标准曲线,计算各样品酶活(单位:nmol 4-MU/min.mg protein)。
选取了3个株系的T2代p35SUTR-GUS转基因烟草幼苗(萌发后1周)进行了GUS酶活性的检测,以p35S-GUS转基因烟草为对照。
萌发后1周的T2代转基因烟草幼苗的GUS酶活性检测,数值为三次生物学重复的平均值,对照与转基因株系间的显著性分析依照t检验(***p<0.001)。结果显示p35SUTR-GUS转基因烟草幼苗中GUS酶活性比对照中增强了12倍以上(图5)。
对萌发后生长4周的T2代转基因烟草的根、茎和叶进行取样,检测了不同组织器官中GUS酶活性。T2代转基因烟草各组织(根、茎、叶和花药)的GUS酶活性检测,数值为三次生物学重复的平均值,对照与转基因株系间的显著性分析依照t检验(***p<0.001)。结果也显示5′UTR使不同组织中的GUS酶活性增强了15倍以上(图6A、B和C)。此外,摘取T2代转基因烟草的花药,分析花粉和花药中的GUS表达情况,结果显示转p35SUTR-GUS烟草花粉和花药中的GUS表达也明显增强了2.5倍以上(图6D)。以上结果表明,CaMV35S与SBgLR 5′UTR的融合明显地增强了GUS基因在花粉中的表达,烟草幼苗和不同器官(根、茎、叶和花药)中的GUS表达也显著增强,说明SBgLR 5′UTR具有增强基因表达的作用。
实施例3、SBgLR基因5′UTR增强种子特异启动子19Z和F128的表达活性
1、表达载体构建所用引物
1.1 扩增19Z启动子引物序列:
F19Z:AAGCTTTTTGTTGTGATTGAGTCG(含HindⅢ位点)
R19Z-1:GGATCCTGTTGGTACACTATTGTGC(含BamHI位点)
R19Z-2:CTGCAGTGTTGGTACACTATTGTGC(含PstⅠ位点)
1.2 扩增F128启动子引物序列:
FF128:AAGCTTTGTGGAGAAGCAGAGAGAAG(含HindⅢ位点)
RF128-1:GGATCCGCTATTAGATCTACAAGTAG(含BamHI位点)
RF128-2:CTGCAGGCTATTAGATCTACAAGTAG(含PstⅠ位点)
1.3 扩增5′UTR引物序列:
Futr-2:CTGCAGAAAGGGTGAAAAGAAAAAAAG(含PstⅠ位点)
Rutr-1:GGATCCTTTTTATTATGAAGAATTGGAG(含BamHI位点)
2、表达载体p19Z-GUS和pF128-GUS的构建
玉米19KD Zein是玉米胚乳特异表达蛋白。根据玉米19KD Zein基因(GenBank:X05911.1)启动子序列,设计引物。以玉米自交系B73基因组DNA为模板,用引物F19Z和R19Z-1扩增出长677bp的19Z启动子片段。F128是谷子种子特异启动子,其驱动的目的基因主要在授粉后15-25天表达。根据F128启动子序列设计引物,以质粒pBIpF128(CN101063139B中国专利2007 1 0099169.7)为模板,用引物FF128和RF128-1扩增出长1056bp的F128启动子片段。将PCR产物用HindⅢ和BamHI双酶切,酶切片段连接到载体pBI121的HindⅢ和BamHI位点,替换载体上的CaMV35S启动子序列,构建成对照载体p19Z-GUS和pF128-GUS。
3、表达载体p19ZUTR-GUS、pF128UTR-GUS的构建
分别以质粒p19Z-GUS和pF128-GUS为模板,用引物F19Z和R19Z-2,FF128和RF128-2,扩增得到带有HindⅢ和PstⅠ酶切位点的19Z与F128启动子序列。以p35SUTR-GUS为模板,以引物Futr-2和Rutr-1扩增带有PstⅠ和BamHⅠ酶切位点的UTR序列。将各序列分别与pMD19T连接,构建载体pMD19T-F128、pMD19T-19Z和pMD19T-UTR。
利用HindⅢ和PstⅠ分别双酶切pMD19T-F128、pMD19T-19Z和pMD19T-UTR。将双酶切得到的F128和19Z启动子片段与pMD19T-UTR连接,得到启动子与UTR融合片段。再利用HindⅢ和BamHⅠ对pMD19T-F128UTR、pMD19T-19ZUTR和pBI121进行双酶切,回收启动子与UTR融合片段后与表达载体pBI121进行连接,替换了pBI121上原有的CaMV35S启动子,最终构建得到pF128UTR-GUS和p19ZUTR-GUS。载体构建策略如图7所示。
4、转基因烟草的GUS酶活性分析
将构建的植物表达载体p19ZUTR-GUS和pF128UTR-GUS及对照质粒p19Z-GUS和pF128-GUS转入农杆菌LBA4404菌株,利用农杆菌介导的叶盘法转化烟草,对获得的再生植株进行PCR和Southern杂交检测,分析外源基因单拷贝插入的烟草植株中的GUS酶活性。转基因烟草未成熟种子的GUS组织化学染色结果显示,4种载体的转化植株的种子中都检测到GUS活性(图8A),而其它组织包括根、茎和叶中都未检测到GUS表达,表明SBgLR基因5′UTR没有改变组织特异启动子的表达模式。从GUS染色结果还可以看出,转p19ZUTR-GUS和pF128UTR-GUS载体的烟草种子比转其对照载体p19Z-GUS和pF128-GUS的烟草种子染色更深。进一步对4种转基因烟草种子进行GUS酶活性的定量分析,每种载体分别取6个不同转基因株系的未成熟种子,测定GUS酶活性,结果显示,转p19ZUTR-GUS载体的烟草种子中的GUS酶活性比转p19Z-GUS载体烟草种子的GUS酶性提高10倍左右;转了pF128UTR-GUS载体的烟草种子中的GUS酶活性比转了pF128-GUS载体的烟草种子中的GUS酶活性也提高10倍左右(图8B)。
研究结果表明,SBgLR基因5′UTR不仅提高了组成型启动子CaMV35S的表达活性,也可以提高种子特异启动子的表达活性。每个载体分析6棵植株,数值为6棵植株的平均值,对照与转基因株系间的显著性分析依照t检验(***p<0.001)
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!