一种Na+/Pi协同转运蛋白启动子和终止子及其应用的制作方法
本发明属于基因工程技术领域,具体涉及圆红冬孢酵母(Rhodosporidium toruloides)的启动子终止子及其用途,包括基因工程菌株构建所必需的转化方法等。
背景技术:
微生物是自然界中分布最广泛的物种之一,具有卓越的生物合成能力,几乎能合成地球上所有的有机化学品。与多细胞生物相比,微生物的代谢途径虽然相对简单,但其化合物的生产高效、快捷,具有反应条件温和、可控性强、易于大规模生产等特点,可作为一个优良的细胞工厂。
自然界中一部分微生物在特定条件下(如氮源缺乏)能在胞内贮存超过其细胞干重20%的油脂,其中以甘油三酯为主,具有这种表型的微生物称为产油微生物,包括细菌、酵母、霉菌、藻类等(Ratledge,C.and Wynn,J.P.Adv Appl Microbiol,2002,51,1-51.)。利用微生物转化生物质资源生产油脂,可发展成基本不依赖耕地、可连续生产、降低农业污染、资源综合利用的新技术,是形成化学品的石化资源替代品新的生产途径(赵宗保.中国生物工程杂志,2005,25,8-11.)。做为某一化学品的天然生产菌株或环境治理应用菌株,其特定的生产性能往往并非最优化。如何优化或改变工业菌株的代谢网络和表达调控网络,以提高生物基产品的积累速度或定向控制靶产品的质量,是当今生物技术领域基因工程改造的热点和难点。虽然对于一些常规酵母的基因工程改造已经较为成熟(Alper H,Stephanopoulos G.Nat Rev Microbiol,2009,7,715-723.),但是对于这些非常规产油酵母的遗传操作尚处于起步阶段,许多非常规产油酵母没有合适的基因操作平台。开发合适的基因操作方法,对于这些非常规微生物的应用意义重大。
圆红冬孢酵母属于担子菌门异宗配合型真菌,是发酵工业中一种极为重要的微生物,可利用源于生物质的己糖和戊糖为原料生产重要的生物基产品:微生物油脂,胞内油脂可达细胞干重的60%以上(Ratledge C,Wynn J P.Adv.Appl.Microbiol.2002,51:1-51;Li Y,Zhao Z,Bai F.Enzyme Microb.Technol.2007,41(3):312-317);工业用酶或药物合成用酶如磷酸二酯酶、苯丙氨酸解氨酶(Hodgins D S.J Biol.Chem.1971,246(9):2977-2985;Gilbert H J,Clarke I N,Gibson R K,et al.J Bacteriol.1985,161(1):314-320)、D氨基酸氧化酶(Gadda G,Negri A,Pilone M S.J Biol.Chem.1994,269(27):17809-17814;Liao G J,Lee Y J,Lee Y H,et al.Biotechnol.Appl.Biochem.1998,27(Pt 1):55-61)等以及β-胡萝卜素和胞外多糖;并在污水处理和生物制药中有较广泛的应用。实验结果表明,该菌可同时利用五碳糖和六碳糖为底物,抗逆性好,能直接以玉米秸秆酸水解液为碳源积 累油脂,可实现生物质到生物基产品的高效转化(李永红,刘波,孙艳,等.中国生物工程杂志,2005,25(12):39-44)。目前R.toruloides功能基因的研究主要通过基因克隆、异源表达和酿酒酵母功能互补分析来进行(Gilbert H J,Clarke I N,Gibson R K,et al.J Bacteriol.1985,161(1):314-320;Liao G J,Lee Y J,Lee Y H,et al.Biotechnol.Appl.Biochem.1998,27(Pt 1):55-61)。但要从分子水平上进行R.toruloides油脂积累机制、遗传发育、生长代谢和菌株遗传改造研究,必须具备相应的遗传操作系统。然而对于广泛分布、工业应用前景良好的R.toruloides而言,由于其特殊分类地位和生化特性以及遗传操作系统的缺乏,使得其遗传改良和分子机制研究进展缓慢,目前有效的遗传操作系统是基于其自身3-磷酸甘油酸激酶启动子pPGK和甘油醛-3-磷酸脱氢酶GPD启动子的农杆菌介导转化体系(Lin XP,Wang YN,Zhang SF,et al.Fems Yeast Res.2014,14:547–555)。但使用的组成型启动子无法选择性调控某一基因转录,不适合细胞毒性基因的过表达;同时,代谢工程也需要更多的启动子和终止子元件可供选择,以实现某一代谢途径各基因表达水平的精确调控。
诱导型启动子在特定的物理或化学信号的刺激下,调控下游基因转录的启动和关闭,对一些细胞毒性基因的遗传操作尤为重要。Na+/Pi协同转运蛋白PHO89,是一种高亲和力磷酸盐转运子,受无机磷阻遏和去阻遏的严格控制,在磷酸盐浓度较低(0.25g/L左右)时表达上调,而磷酸盐浓度正常时则表达受阻。PHO89启动子Ppho89的诱导无需加入诱导剂,前期研究中所用的限磷油脂发酵培养基中磷酸盐浓度恰恰就是Ppho89诱导激活浓度。非常适用于一些关键限速酶在油脂积累期的过表达调控,比如过表达ME以增加NADPH还原力供应。虽然曾有人分离酿酒(Saccharomyces cerevisiae)的Na+/Pi协同转运蛋白启动子、以及其它磷酸盐饥饿诱导启动子用于自身的基因工程操作(Hiraoka E,Murai M,Yano J,et al.Journal of Fermentation and Bioengineering.77(4):376–381.;Lee KM,DaSilva NA.Yeast.2005,22(6):431–440.),但未见该类启动子用于红冬孢酵母属(Rhodosoporidium)、锁掷酵母属(Sporidiobolus)、掷孢酵母属(Sporobolomyces)和红酵母属(Rhodotorula)中基因表达、遗传工程操作和菌株改良的基因工程操作的报道。同时,本领域技术人员周知“不同的微生物在遗传背景、基因表达模式、生理生化特征方面存在大的差异,即使同为酵母,不同的种属间也存在很大的差异,例如,同属子囊菌门的酿酒酵母、毕赤酵母、耶氏解脂酵母,必须分别构建其遗传体系,选择自身来源启动子”。然而,启动子对于遗传操作系统来说必不可少。因此,分离能够在这些红酵母中启动报告基因表达的Na+/Pi协同转运蛋白启动子成为目前研究的焦点。
终止子序列,是给予RNA聚合酶转录终止信号的DNA序列,同时也决定mRNA的稳定性、转录效率和mRNA从转录复合体的释放。商业化酵母表达载体和报道的合成生物学文献中,多选择高表达基因的终止子做为转录终止元件。
技术实现要素:
鉴于上述现有技术瓶颈,本发明的主要目的是提供可通用于红冬孢酵母属(Rhodosoporidium)、锁掷酵母属(Sporidiobolus)、掷孢酵母属(Sporobolomyces)和红酵母属(Rhodotorula)中基因表达、遗传工程操作和菌株改良的中外源基因表达的启动子和终止子,及采用合适的转化技术对这些红酵母菌株进行改良的方法。
为实现本发明的目的,本发明通过对圆红冬孢酵母的基因组序列进行分析,获得响应磷酸盐饥饿诱导的Na+/Pi协同转运蛋白(Pho89)的启动子序列,并进一步通过PCR技术从圆红冬孢酵母染色体DNA中成功分离了包含有效启动子的DNA片段,采用合适的转化方法将含有圆红冬孢酵母Na+/Pi协同转运蛋白(Pho89)启动子的外源DNA片段分别导入红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属中,成功实现了外源基因的表达,完成了本发明。
本发明成功分离了能够在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母中启动报告基因表达的圆红冬孢酵母Na+/Pi协同转运蛋白(Pho89)启动子,并构建了其表达载体。
具体讲,本发明包含下述技术方案(A)到(H):
(A)本发明涉及一种具有红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属转录启动子活性的DNA片段,所述DNA片段:
(1)具有如SEQ ID NO:1所示DNA序列的全部序列或包含该DNA序列自3’-末端起500bp以内的部分序列,
(2)具有可与如SEQ ID NO:1所示序列的全部或其DNA序列3’-末端起500bp以内的部分序列杂交的、且保持转录启动子活性的序列,或
(3)对SEQ ID NO:1所示的脱氧核苷酸序列进行一个或50个碱基以内的取代、缺失、插入或添加所获得的,与SEQ ID NO:1所示序列具有70%以上同源性、且具有启动子活性的序列。
(B)一种来自圆红冬孢酵母的DNA片段,所述DNA片段可作为终止子,并且:(1)具有如SEQ ID NO:2所示的DNA序列的全部或包含该DNA序列5’-末端的部分序列;或(2)具有可与如(1)所示序列杂交的、且保持如(1)所述序列活性的序列。
(C)一种可完成靶基因在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母中转录起始和转录终止的DNA分子,它具有上述(A)所述的具有红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母转录启动子活性的DNA序列,或同时具有上述(A)所述的具有红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母转录启动子活性的DNA序列,和(B)所述的DNA片段,且(B)所述的DNA片段位于(A)所述的DNA序列的下游,与其相邻1-10000个核苷酸的DNA片段。
(D)一种可将靶基因与(A)-(C)任一种所述的DNA分子连接的DNA表达盒,以便于所述靶基因能够在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母中表达重组的DNA。所述靶基因为蛋白编码核酸或反义核酸编码 核酸。优选地,所述目的基因的cDNA序列具有如SEQ ID NO:4所示的核苷酸序列(LsNa+/Pi协同转运蛋白cDNA)。
(E)一种携带(A)-(D)任一所述的的DNA分子中的任意一个的重组载体。所述载体可以是游离型载体或整合型载体,所述游离型载体例如,但不限于,pMD18-T、pUC18、pYES2c/t或pYX212等,所述整合型载体为农杆菌介导的双元表达载体,例如,但不限于,PZPK或pZP2000等。
(F)一种将(D)所述的DNA分子或如(E)所述的载体转入红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属菌株的转化方法。
(G)一种转入了如(D)所述的DNA表达盒或如(E)所述的重组载体的红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属的基因工程菌株。
(H)Na+/Pi协同转运蛋白(Na+/Pi协同转运蛋白)启动子,其特征在于:其来源为圆红冬孢酵母,可启动目的基因在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母中的转录和表达,所述启动子:(1)具有如SEQ ID NO:1所示DNA序列的全部序列或包含该DNA序列自3’-末端起1500bp以内的部分序列,(2)具有可与如SEQ ID NO:1所示序列的全部或其DNA序列3’-末端起1500bp以内的部分序列杂交的、且保持转录启动子活性的序列,或(3)对SEQ ID NO:1所示的脱氧核苷酸序列进行一个或50个碱基以内的取代、缺失、插入或添加所获得的,与SEQ ID NO:1所示序列具有70%以上同源性、且具有启动子活性的序列;
Na+/Pi协同转运蛋白终止子,其特征在于:其来源为圆红冬孢酵母,可终止目的基因在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母中的转录和表达,所述终止子:(1)具有如SEQ ID NO:2所示的DNA序列的全部或包含该DNA序列5’-末端的部分序列,(2)具有可与如SEQ ID NO:2所示序列的全部或其DNA序列5’-末端的部分序列杂交的、且保持转录终止子活性的序列,或(3)对SEQ ID NO:2所示的脱氧核苷酸序列进行一个或50个碱基以内的取代、缺失、插入或添加所获得的,与SEQ ID NO:2所示序列具有50%以上同源性、且具有终止子活性的序列。
使用本发明的具有启动子活性的DNA分子和具有转录终止子活性的DNA,可实现外源基因或内源基因在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母中的表达。
本发明提供了用于红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母菌株遗传工程改造的启动子、终止子和载体。为红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母菌株开启了一条育种新途径,并因此可以提供具有工业用途的新型酵母菌株。
综上所述,本发明提供下述技术方案:
1.Na+/Pi协同转运蛋白启动子,其核苷酸序列如SEQ ID NO:1所示。
2.第1项所述的Na+/Pi协同转运蛋白(Pho89)启动子用于构建能够在c(Rhodosoporidium)、锁掷酵母属(Sporidiobolus)、掷孢酵母属(Sporobolomyces) 和红酵母属(Rhodotorula)的酵母菌株中表达的重组表达载体的应用,其中克隆了包含所述Na+/Pi协同转运蛋白(Pho89)启动子的重组表达载体的红冬孢酵母属、锁掷酵母属和红酵母属酵母菌株构成红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属遗传操作系统。
3.一种DNA表达盒,其含有SEQ ID NO:1所示的Na+/Pi协同转运蛋白启动子的核苷酸序列。
4.第3项所述的表达盒,其还含有SEQ ID NO:2所示的核苷酸序列作为终止子,其中SEQ ID NO:1所示的序列位于SEQ ID NO:2所示的序列的上游,SEQ ID NO:1和SEQ ID NO:2之间为编码目的基因的开放阅读框架。
5.第4项所述的DNA表达盒,其特征在于:所述编码目的基因的开放阅读框架的cDNA序列具有如SEQ ID NO:4所示的核苷酸序列。
6.一种包含第3-5项中任一项所述的DNA表达盒的重组载体。
7.第6项所述的重组载体,其特征在于:所述载体为游离型或者整合型载体。
8.第7项所述的重组载体,所述整合型载体为农杆菌介导的双元表达载体,例如PZPK或pZP2000,或者为携带有靶基因翼侧1500-4000碱基同源重组臂的同源重组载体,所述同源重组载体骨架或所述游离型载体选自大肠杆菌克隆载体或酵母穿梭载体,优选pMD18-T、pUC18、pYES2c/t或pYX212。
9.包含第6-8项中任一项所述的重组载体的宿主细胞为大肠杆菌细胞或酵母细胞。
10.一种用于产油酵母遗传表达的表达系统,所述表达系统包含:
(1)能够在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属属菌株中表达的改良载体,所述改良载体通过在能够在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属中整合表达或游离表达的质粒中插入SEQ ID NO:1所示的启动子序列和SEQ ID NO:2所示的终止子序列构建获得,其中SEQ ID NO:1所示的启动子序列位于SEQ ID NO:2所示的终止子序列的上游,SEQ ID NO:1所示的序列与SEQ ID NO:2所示的序列之间为多克隆位点,并且所述载体还包含选择性标记基因;
(2)目的基因开放阅读框序列,其能够可操作性地插入到(1)所述的改良载体中,并使所述目的基因与(1)所述的改良载体中的启动子和终止子符合阅读框的连接,和
(3)红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属菌株。
11.第10项的用于产油酵母遗传表达的表达系统,其中(1)所述的改良载体通过在能够在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属中表达的质粒中插入SEQ ID NO:1所示的启动子序列和SEQ ID NO:2所示的终止子序列构建获得。
12.第11项的用于产油酵母遗传表达的表达系统,其中(1)所述质粒为游离型或者整合型质粒,并且,其中所述整合型质粒为农杆菌介导的双元表达 质粒,例如PZPK或pZP2000,并且,所述游离型质粒选自pMD18-T、pUC18、pYES2c/t或pYX212。
13.第10项的用于产油酵母遗传表达的表达系统,其中(3)所述的圆红冬孢酵母菌株选自圆红冬孢酵母(Rhodosporidium toruloides)、贝吉维红冬孢酵母(Rhodosporidium babjevae),所述锁掷酵母属(Sporidiobolus)菌株选自拟粉红锁掷孢酵母(Sporidiobolus pararoseus),所述掷孢酵母属(Sporobolomyces)菌株选自粉红掷孢酵母(Sporobolomyces roseus),所述红酵母属(Rhodotorula)菌株选自深红酵母(Rhodotorula rubra)、胶红酵母(Rhodotorula mucilaqinosa)、海滨红酵母(Rhodotorula marina)、禾本红酵母(Rhodotorula graminis)和粘红酵母(Rhodotorula glutinis)。
14.第10项的用于产油酵母遗传表达的表达系统,其中(2)所述的目的基因开放阅读框序列具有如SEQ ID NO:4所示的核苷酸序列。
本领域技术人员应该理解,术语“表达系统”是指包括重组载体、待表达的目标蛋白的编码核苷酸序列、适合的宿主细胞或宿主菌株等的组合物体系,用来在所述宿主细胞或宿主菌株中表达所述目标蛋白。
本发明的有益效果是:
为红冬孢酵母属、锁掷酵母属和红酵母属的酵母菌提供了启动子、终止子遗传转化方法,将有力促进今后的红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属酵母菌的菌株改良和代谢工程研究。
附图说明
图1、PHO89简并PCR产物(泳道1)的琼脂糖凝胶电泳结果,泳道M为分子量标准。
图2、PZPK-PHO89p-hyg-PHO89t载体的结构示意图,LB,T-DNA左边界;RB,T-DNA右边界。
图3、使用HYG基因转化R.babjevae NCYC 2630获得重组子的PCR鉴定结果,泳道M为分子量标准,泳道1-6分别表示不同的Hyg抗性转化子,WT表示野生型。
图4、表示HYG的表达检测——Western blot分析结果图,泳道1-6为R.babjevae NCYC 2630潮霉素转化子1-6号总蛋白;泳道7为作为阴性对照的出发菌株R.babjevae NCYC 2630总蛋白样品。
图5、BLE抗性Ura3营养缺陷圆红冬孢酵母重组菌株的转录水平表达分析——RT-PCR结果图,泳道1-2为BLE抗性Ura3营养缺陷圆红冬孢酵母ATCC 10788重组菌株1-2,泳道3为对照菌株圆红冬孢酵母ATCC 10788。
图6、PZPK-PHO89p-hyg-Thsp载体的结构示意图,LB,T-DNA左边界;RB,T-DNA右边界。
图7、pZPK-Ppho89-MCS-Thsp载体的结构示意图,LB,T-DNA左边界;RB,T-DNA右边界。
图8、PZPK-HYG-Ppho89-MCS-Thsp载体的结构示意图,LB,T-DNA左边界; RB,T-DNA右边界。
图9、pZPK-HYG-Ppho89-CpFAH-Thsp载体的结构示意图,LB,T-DNA左边界;RB,T-DNA右边界。
图10、表示CpFAH的表达检测——Western blot分析结果图,泳道1-6为拟粉红锁掷孢酵母JCM 3765潮霉素转化子1-6号总蛋白;泳道7为作为阴性对照的出发菌株拟粉红锁掷孢酵母JCM 3765总蛋白样品。
图11、pZPK-HYG-Ppho89-ME-Thsp载体的结构示意图,LB,T-DNA左边界;RB,T-DNA右边界。
图12、表示RtME的表达检测——Western blot分析结果图,泳道1-3为粉红掷孢酵母S.roseus JCM 8242潮霉素转化子1-3号总蛋白;泳道4为作为阴性对照的出发菌株粉红掷孢酵母S.roseus JCM 8242总蛋白样品。
图13、pZPK-HYG-Ppho89-GFP-Thsp载体的结构示意图,LB,T-DNA左边界;RB,T-DNA右边界。
图14、表示GFP的表达检测——Western blot分析结果图,泳道1-3为重组深红酵母CGMCC 2.279潮霉素转化子1-3号总蛋白;泳道4为作为阴性对照的出发菌株重组深红酵母CGMCC 2.279总蛋白样品。
图15、表示GFP的表达检测——Western blot分析结果图,泳道1-3为重组胶红酵母CGMCC 2.22潮霉素转化子1-3号总蛋白;泳道4为作为阴性对照的出发菌株重组胶红酵母CGMCC 2.22总蛋白样品。
图16、pZPK-HYG-Ppho89-INU-Thsp载体的结构示意图,LB,T-DNA左边界;RB,T-DNA右边界。
序列表说明
具体实施方式
在本文中,“启动子”是指能够被RNA聚合酶识别、结合并能启动基因转录的DNA序列。术语“启动子”还可理解为:包括5’非编码区、顺式作用元件(如增强子)以及其它能与转录因子结合的核苷酸序列。
启动子的存在或强度通常是通过启动子活性表示,其测定方法:将报告基因(如抗性基因)连接于所述启动子的下游,并将该DNA构建体转化相应宿主细胞,检测报告基因是否表达。如果人们观察到连接于所述启动子下游报告基因的表达,就可以认为所述启动子在它所转化的宿主细胞内有活性。
在本文中,“终止子”是指染色体上提供终止信号使RNA聚合酶与DNA模板分离而使转录终止的一段DNA序列。可以通过“启动子-报告基因-终止子”构建体使报告基因有效表达而确定终止子的活性。
本发明中的“圆红冬孢酵母”,包括属于该“物种”的任何二倍体和单倍体,野生型菌株和营养缺陷型菌株。本发明中的“红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属”,没有具体限制,其实例包括圆红冬孢酵母(Rhodosporidium toruloides)、贝吉维红冬孢酵母(Rhodosporidium babjevae),粉红锁掷孢酵母(Sporidiobolus pararoseus),粉红掷孢酵母(Sporobolomyces roseus),深红酵母(Rhodotorula rubra),胶红酵母(Rhodotorula mucilaqinosa),海滨红酵母(Rhodotorula marina),禾本红酵母(Rhodotorula graminis)和粘红酵母(Rhodotorula glutinis)。
本发明的“目的基因”,包括能够在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属属菌株中表达的蛋白编码序列、反义RNA编码序列和核酸酶编码序列。能够在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属菌株中表达的蛋白编码序列的例子包括源于这两类菌属中的蛋白或者核酸序列,且并不局限于此,还包括来源于其它微生物、植物和动物的蛋白或者核酸序列。本领域技术任意应该理解,当使用来源于其它微生物、植物和动物的蛋白编码序列作为目的基因(即,外源目的基因)时,为优化在红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属菌株中的表达,通常需要针对红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属菌株的密码子优先性对所述目的基因进行密码子优化。而密码子优化属于本领域的常规技术手段。
本发明中的启动子:(1)具有如SEQ ID NO:1所示DNA序列的全部序列或包含该DNA序列自3’-末端起1500bp以内的部分序列,(2)具有可与如SEQ ID NO:1所示序列的全部或其DNA序列3’-末端起1500bp以内的部分序列杂交的、且保持转录启动子活性的序列,或(3)对SEQ ID NO:1所示的脱氧核苷酸序列进行一个或多个碱基的取代、缺失、插入或添加所获得的,与SEQ ID NO:1所示序列具有50%以上同源性、且具有启动子活性的序列。
本发明中的终止子:(1)具有如SEQ ID NO:2所示的DNA序列的全部或包含该DNA序列5’-末端的部分序列,(2)具有可与如SEQ ID NO:2所示序列的全部或其DNA序列5’-末端的部分序列杂交的、且保持转录终止子活性的 序列,或(3)对SEQ ID NO:2所示的脱氧核苷酸序列进行一个或多个碱基的取代、缺失、插入或添加所获得的,与SEQ ID NO:2所示序列具有50%以上同源性、且具有终止子活性的序列。
本发明中的启动子-目的基因的构建体、目的基因-终止子的构建体或启动子-目的基因-终止子的构建体,可以直接或经载体介导转化红冬孢酵母属、锁掷酵母属、掷孢酵母属和红酵母属菌株,以便于目的基因表达,可以优选质粒载体作为介导载体。
下面结合附图及实施例对本发明作进一步说明,将有助于本领域的普通技术人员理解本发明,但不以任何形式限制本发明。下述实施例中所有引物合成及测序工作,如无特别说明则均由大连TakaRa公司完成。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的实验材料,如无特殊说明,均为自常规生化试剂公司购买得到的。
R.toruloides CGMCC 2.1389:中国普通微生物菌种保藏中心(CGMCC),源自东京大学应用微生物研究所(IFO 8766),由IFO 0559和IFO 0880经配合而成的二倍体,等同于CBS 6016或NBRC 8766或NRRL Y-6987。
R.toruloides ATCC 10788:美国标准生物品收藏中心(ATCC),源自CBS-KNAW fungal biodiversity centre,等同于IFO 0559或JCM 3792或CCRC 20306或DBVPG 6740或IAM 13469或IGC 4416或MUCL 30249或NCYC 921或NRRL Y-1091或VKM Y-334或PYCC 4416。
Rhodosporidium babjevae NCYC 2630购自英国国家酵母菌种保藏中心(National Collection of Yeast Cultures,NCYC)。
拟粉红锁掷孢酵母(Sporidiobolus pararoseus)JCM 3765购自于购自中国普通微生物菌种保藏管理中心(CGMCC)。
粉红掷孢酵母(Sporobolomyces roseus)JCM 8242购自于日本微生物菌种保藏中心(JCM)。
深红酵母(Rhodotorula rubra)CGMCC 2.279购自于酵母菌种保藏中心(购自中国普通微生物菌种保藏管理中心(CGMCC)。
胶红酵母CGMCC 2.22购自于酵母菌种保藏中心(购自中国普通微生物菌种保藏管理中心(CGMCC)。
海滨红酵母(Rhodotorula marina)CGMCC 2.4203购自于酵母菌种保藏中心(购自中国普通微生物菌种保藏管理中心(CGMCC)。
粘红酵母(Rhodotorula glutinis)NCYC 2666购自于英国国家酵母菌种保藏中心(National Collection of Yeast Cultures,NCYC)。
实施例1:圆红冬孢酵母(Rhodosporidium toruloides)CGMCC 2.1389总RNA的提取
将圆红冬孢酵母(R.toruloides)CGMCC 2.1389(购自中国普通微生物菌种保藏管理中心(China General Microbiological Culture Collection Center,CGMCC))由斜面接种到10mL YEPD液体培养基(葡萄糖20.0g/l,酵母提取 物10.0g/L,蛋白胨20.0g/L,pH 6.0)中,于30℃摇床培养24h,再以1:50的体积比例将菌液分别转接到100mL YEPD液体培养基中,于30℃摇床培养14h达对数生长期。在4℃下,5000rpm离心4min,收集菌体,用液氮迅速冷冻菌体,研磨破壁(Yang F,Tan HD,Zhou YJ,et al.Mol.Biotechnol.2010,47(2):144–151.)。使用TakaRa公司RNAiso试剂盒,并按照其标准步骤提取总RNA。
RNA进行1.5%(质量/体积浓度)琼脂糖凝胶电泳,使用荧光-紫外分析仪观察鉴定,可见清晰的两条带。用紫外/可见光光谱仪分析总RNA样品,测得OD260/OD280=1.99,表明总RNA质量很好。总RNA样品冻存于-80℃,备用。
实施例2:圆红冬孢酵母CGMCC 2.1389cDNA第一链合成和pho89简并PCR
以圆红冬孢酵母NRRL Y-11557总RNA为模板,反转录合成cDNA第一链。首先,将1.0μL总RNA(约2μg),1.0μL引物SMART IV:5′-AAGCAGTGGTATCAACGCAGAGTGGCCATTACGGCCGGG-3′和1.0μLoligo dT-接头引物CDSⅢ/3':5′-ATTCTAGAGGCCGAGGCGGCCGACATG-d(T)30N-1N-3′,2.0μL DEPC处理水(焦碳酸二乙酯处理水,购自大连TakaRa公司),加入到PCR管中混匀,于72℃保温2min,立即置于冰上冷却2min,将2.0μL 5×第一链缓冲液(Clontech公司),1.0μL DTT(20mM),1.0μL dNTP(10mM),1.0μL powerscript反转录酶(Clontech公司)加入到体系中,混匀。于42℃延伸反应60min,最后4℃结束反应,存于-20℃,备用。
设计合成两条简并引物PHO89-sense:5′-ATGCC(AGCT)ATGCA(CT)CA(AG)TA(CT)GA(CT)TAC-3′(括号内碱基在此位置随机出现,为简并碱基)和PHO89-anti:5′-CTA(AGCT)TG(CT)GA(AG)GG(AG)AA(AG)TG(AG)GGCGC-3′(括号内碱基在此位置随机出现,为简并碱基),以反转录合成的cDNA第一链为模板,进行PHO89基因的简并PCR扩增,5×PCR缓冲液10.0μL,dNTPs(10mM)1.0μL,上游引物(50mmol/l)1.0μL,下游引物(50mmol/L)1.0uL,PrimeSTAR DNA聚合酶(大连TakaRa)0.5μL,合成的cDNA第一链模板1.0μL,ddH2O补加至50μL,于94℃保温3min,然后于98℃保温10s,62℃10s,72℃1min,35个循环,72℃10min,4℃结束反应。扩增产物进行1%(质量/体积浓度)琼脂糖凝胶电泳,观察到1.8kb左右的条带(图1),利用DNA回收试剂盒(购自上海生工),按照供应商建议步骤纯化PCR产物。PCR产物参照大连TakaRa公司提供的方法克隆到pMD18-T载体(购自大连TakaRa),转化入E.coli DH5α感受态细胞,其中感受态细胞按氯化钙法(分子克隆实验指南第三版,萨姆布鲁克著,黄培堂等译,科学出版社出版)制备。挑选Amp抗性转化子进行增菌培养、质粒提取。重组质粒样品送至大连TakaRa公司测序,序列结果推测出的氨基酸序列经Blastp分析,证实为Na+/Pi协同转运蛋白序列(RtPHO89氨基酸序列),如SEQ ID NO:5所示。Na+/Pi协同转运蛋白cDNA序列(RtPHO89cDNA)如SEQ ID NO:4序列所示。
实施例3:圆红冬孢酵母CGMCC 2.1389基因组DNA的扩增
圆红冬孢酵母CGMCC 2.1389的基因组DNA提取采用玻璃珠破壁法(精编分子生物学实验指南第三版第13章,奥斯伯等著,颜子颖等译,科学出版社出版)。制备好的基因组DNA,利用Nanodrop 1000测定,测得OD260/OD280=1.85,表明基因组DNA质量很好。浓度为280ng/μL,共500μL,基因组DNA样品冻存于-20℃,备用。
根据实施例2中获得的Na+/Pi协同转运蛋白(RtPHO89)cDNA序列,设计1对基因特异性引物,PHO89-p1:5’-atgcctatgcaccaatacgactacctc-3’和PHO89-p2:5’-tcactgcgaggggaagtgaggggcatt-3’,以圆红冬孢酵母CGMCC 2.1389的基因组DNA为模板,按照常规方法进行PCR扩增,得到约2.4kb的PCR产物(未显示)。PCR扩增产物按照实施例2的操作步骤回收、克隆到pMD18-T载体,并进行测序,得到如序列表SEQ ID NO:3所示的DNA序列(RtPHO89编码区)。经与实施例2中获得的Na+/Pi协同转运蛋白cDNA序列比对,证实该基因片段为其Na+/Pi协同转运蛋白编码区序列,其中含有10个内含子和11个外显子。
实施例4:染色体步移获得RtPHO89基因5’翼侧序列(启动子)
本实施例利用Genome Walking Kit(购自大连TakaRa)完成。
根据实施例3中得到的RtPHO89DNA序列,设计3条Specific Primer(基因特异性引物),分别为PHO89-SP1:5’-ggcggaggcgcgggaggggaaactcac-3’,PHO89-SP2:5’-ggtaggcagggggcgaatcagcagctg-3’和PHO89-SP3:5’-cagcggcgcgagcaggcggaaaacgggcg-3’,做为下游引物,按照试剂盒说明书进行以下操作。
1.1stPCR反应
以实施例3中精制的基因组DNA为模板,进行第一轮扩增。反应体系50μL:10×LA PCR buffer II(Mg2+plus,大连TakaRa)5.0μL,dNTPs(2.5mmol/l)8.0μL,LA Taq DNA聚合酶(5U/μL,大连TakaRa)1.0μL,AP1Primer(100μmol/L,大连TakaRa)1.0μL,PHO89-SP1(10μmol/L)1.0μL,圆红冬孢酵母NRRL Y-11557基因组DNA模板(120ng/μL)1.0μL,ddH2O加至50μL。反应条件:先进行5个高温退火温度的高特异性反应,然后进行1个极低退火温度的低特异性反应;然后进行热不对称PCR:2个高退火温度(65℃)的高特异性反应和1个低退火温度(44℃)的低特异性反应交替循环,共15次。具体参数如下:94℃1min,98℃1min;94℃30s,65℃1min,72℃2min,共5个循环;94℃30s,25℃3min,72℃2min;94℃30s,65℃1min,72℃2min,94℃30s,65℃1min,72℃2min,94℃30s,44℃1min,72℃2min,共15个循环;72℃10min,结束反应。
2.2nd巢式PCR反应
反应体系50μL:10×LA PCR buffer II(Mg2+plus,大连TakaRa)5.0μL,dNTPs(2.5mmol/L)8.0μL,LA Taq DNA聚合酶(5U/μL,大连TakaRa)1.0μL, AP1 Primer(100μmol/L,大连TakaRa)1.0μL,1stPCR反应产物1.0μL,PHO89-SP2(10μmol/L)1.0μL,ddH2O加至50μL。反应条件:94℃30s,65℃1min,72℃2min,94℃30s,65℃1min,72℃2min,94℃30s,44℃1min,72℃2min,共15个循环;72℃10min,结束反应。
3.3rd巢式PCR反应
反应体系50μL:10×LA PCR buffer II(Mg2+plus,大连TakaRa)5.0μL,dNTPs(2.5mmol/L)8.0μL,LA Taq DNA聚合酶(5U/μL,大连TakaRa)1.0μL,AP1Primer(100μmol/L,大连TakaRa)1.0μL,2nd巢式PCR反应产物1.0μL,PHO89-SP3(10μmol/L)1.0μL,ddH2O加至50μL。反应条件:94℃30s,65℃1min,72℃2min,94℃30s,65℃1min,72℃2min,94℃30s,44℃1min,72℃2min,共15个循环;72℃10min,结束反应。
3rd巢式PCR反应产物经1%(质量/体积浓度)琼脂糖凝胶电泳后切割目的条带,利用DNA片段凝胶纯化试剂盒(购自碧云天)进行纯化。纯化后的DNA片段经TA克隆插入pMD18-T载体(购自大连TakaRa公司),转化DH5α感受态细胞;其中感受态细胞按氯化钙法(分子克隆实验指南第三版,萨姆布鲁克著,黄培堂等译,科学出版社出版)制备。挑选Amp抗性转化子进行增菌培养、质粒提取。重组质粒样品送至大连TakaRa公司测序,得到如SEQ ID NO:1所示的DNA序列,证实为预期的RtPHO89启动子序列。
实施例5:染色体步移获得RtPHO89基因3’翼侧序列(终止子)
本实施例也是利用Genome Walking Kit(购自大连TakaRa)完成。
根据实施例3中得到的PHO89DNA序列,设计3条Specific Primer(基因特异性引物)分别为PHO89-SP11:5’-gtttcgtacatgcaggcatcactggttc-3’,PHO89-SP22:5’-gggtcatcaccgtccccatcgtcggcac-3’和PHO89-SP33:5’-ccctcgctctccaccaggcatcgttct-3’,做为上游引物,按照试剂盒说明书进行3’翼侧染色体步移操作,除Specific Primer分别由PHO89-SP1,PHO89-SP2,PHO89-SP3依次更换为PHO89-SP11,PHO89-SP22,PHO89-SP33外,其它同实施例4。
3rd巢式PCR反应产物(未显示)利用DNA片段凝胶纯化试剂盒(购自碧云天)进行纯化,经TA克隆插入pMD18-T载体(购自大连TakaRa公司),转化DH5α感受态细胞;其中感受态细胞按氯化钙法(分子克隆实验指南第三版,萨姆布鲁克著,黄培堂等译,科学出版社出版)制备。挑选Amp抗性转化子进行增菌培养、质粒提取。重组质粒样品送至大连TakaRa公司测序,得到如SEQ ID NO:2所示的DNA序列,证实为预期的Na+/Pi协同转运蛋白编码基因的终止子序列。
实施例6:Rt PHO89启动子-开放阅读框架-终止子全长基因获得
根据实施例4和实施例5中获得的启动子和终止子序列,重新设计一对引物进行RtPHO89“启动子-开放阅读框架-终止子”全长基因的扩增。Ppho89-p1:5’-gcgggatcttccctcgcttcgaccacc-3’,PHO89t-p2:5’-ccgcgacaacccctccctccctcaatc-3’。以实施例3中制备的圆红冬孢酵母CGMCC 2.1389基因组DNA为模板进行PCR扩增。PCR体系(50μL):10×Speed buffer(大连TakaRa)5.0μL,dNTPs(10mmol/L)1.0μL,上游引物(10μmol/L)2.0μL,下游引物(10μmol/L)2.0μL,SpeedSTARTM HS DNA聚合酶(扩增速度快,1kb/10s,购自大连TakaRa公司)0.5μL,基因组DNA模板(120ng/μL)2μL,ddH2O加至50μL。反应条件:98℃1min,98℃10s,65℃1.0min,35个循环,72℃10min,4℃结束反应。PCR产物经1%(质量/体积浓度)琼脂糖凝胶电泳分析后利用PCR片段纯化试剂盒(购自碧云天)进行纯化。片段经TA克隆插入pMD18-T载体(购自大连TakaRa公司),转化DH5α感受态细胞;其中感受态细胞按氯化钙法(分子克隆实验指南第三版,萨姆布鲁克著,黄培堂等译,科学出版社出版)制备。挑选Amp抗性转化子进行增菌培养、质粒提取。重组质粒样品送至大连TakaRa公司测序,证实为预期的Na+/Pi协同转运蛋白基因全长序列Ppho89t,该重组载体命名为T-PHO89。
实施例7:RF克隆法构建潮霉素蛋白表达盒PHO89p-hyg-PHO89t
根据NCBI报道的HYG蛋白质序列委托上海生工公司全基因合成HYG基因(如SEQ ID NO:6所示)。以合成的基因为模板,参照文献方法(van den Ent,F.,and Lowe,J.J Biochem Biophys Methods,2006,67,67-74.),设计RF克隆引物:HYG-RF-p1:5’-CTTCCGCCAGCAGCCAACGTCGTCAAGatgccggagctcacggcgacgtcggtc-3′和HYG-RF-p2:5’-CGACAAGAGAGAAAGACGAAGGGAGACGActattctttcgcccgcgggcgcgtcga-3’为引物,进行PCR扩增。进行RF第一轮扩增。体系(50μL):5×Prime buffer(大连TakaRa)10.0μL,dNTPs(2.5mmol/l)4.0μL,上游引物(10μmol/l)2.0μL,下游引物(10μmol/l)2.0μL,PrimeSTAR HS DNA聚合酶(大连TakaRa)1.0μL,T-GFPuv质粒(100ng/μL)1μL,ddH2O加至50μL。反应条件:95℃3min,98℃8s,49℃15s,72℃1min,35个循环,72℃10min,4℃结束反应。RF I反应产物利用DNA片段胶回收纯化试剂盒纯化,-20℃保存备用。
RF II反应:5×Prime buffer(大连TakaRa)10.0,dNTPs(2.5mmol/l)4.0μL,实施例6中构建的T-PHO89质粒(100ng/μL)1.0μL,本实施例前述步骤中RF I反应产物(200ng/μL)5.0μL,PrimeSTAR HS DNA聚合酶(大连TakaRa)1.0μL,ddH2O加至50μL。反应条件:95℃3min,68℃12min,之后95℃30s,65℃45s(-1℃/cyc),68℃12min,15个循环,接下来再进行一轮:95℃30s,55℃45s,68℃12min,20个循环,72℃10min,4℃结束反应。
DpnI消化和电击转化:取8μL RF II反应产物加入1μL DpnI(购自TaKaRa)和1μL DpnI buffer,混匀后在37℃作用120min去除原T-PHO89质粒后,取2μL电击转化DH5α感受态细胞,感受态细胞按标准方法制备(分子克隆实验指南第三版,萨姆布鲁克著,黄培堂等译,科学出版社出版),电 击转化参数:2200-2500V,400Ω,25μF,0℃,4-8ms。挑选Amp抗性转化子进行增菌培养、质粒提取,并利用RF I反应所用引物HYG-RF-p1和HYG-RF-p2进行菌落PCR鉴定,鉴定阳性的重组载体送大连TakaRa进行测序,得到5’端和3’端分别为PHO89启动子和PHO89终止子的“PHO89p-hyg-PHO89t”表达盒,同时,该重组载体命名为T-PHO89p-hyg-PHO89t。完整的“PHO89p-hyg-PHO89t”表达盒如SEQ ID NO:7所示。
实施例8:hyg在贝吉维红冬孢酵母(Rhodosporidium babjevae)NCYC 2630中的磷酸盐饥饿诱导表达
根癌农杆菌(Agrobacterium tumefaciens)在自然条件下可以通过病斑或伤口进入寄主组织,将其体内的一段DNA转入植物基因组中,最终刺激寄主在侵染部位形成冠瘿瘤。农杆菌这种特性最早被应用于植物基因组的改造,称为ATMT技术。随后ATMT技术广泛应用于多种真菌和酵母遗传改造和T-DNA插入突变文库构建。
ATMT转化过程大致可以分为载体构建、农杆菌活化、宿主材料制备、共转化和转化子筛选这五个步骤。首先在双元载体的T-DNA区域间插入合适的选择标记,并转入农杆菌;含有双元载体的农杆菌工程菌株活化后用乙酰丁香酮(AS)诱导;宿主菌株经过活化后稀释到一定的浓度;将活化并诱导后的农杆菌与宿主材料混合,涂布于铺有载体介质的共培养平板上,在适宜的温度下进行共转化;将共培养的混菌转移到筛选平板上,置于宿主菌最适宜的温度下培养,至转化子出现。
1.双元载体PZPK-PHO89p-hyg-PHO89t的构建
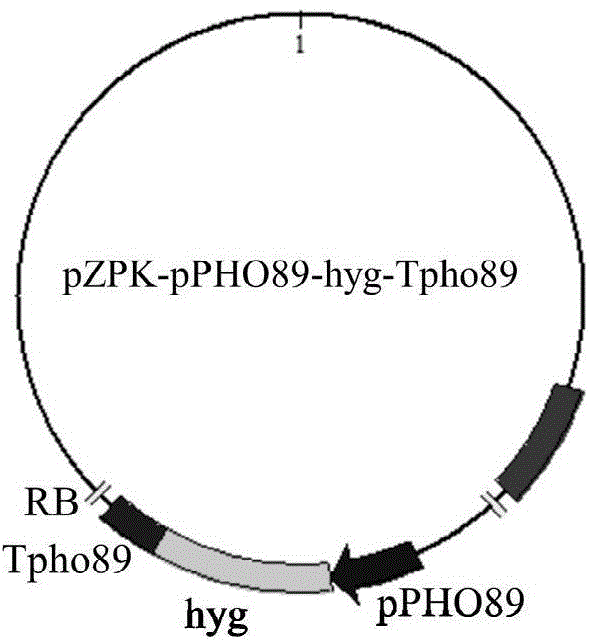
PZPK骨架载体来源于中科院大连化学物理研究所赵宗保研究员实验室(Lin XP,Wang YN,Zhang SF,et al.Fems Yeast Res.2014,14:547–555),PZPK-PHO89p-hyg-PHO89t载体的构建则采用PHO89-HYG-RF-p1:5’-CCGAATTGAATTCGAGCTCGCTCGGTACCCGGgcgggatcttccctcgcttcgaccacc-3′和PHO89-HYG-RF-p2:5’-GCTTGCATGCTGCAGGTCGACTCTAGA ccgcgacaacccctccctccctcaatc-3′为引物,以实施例7构建的T-PHO89p-hyg-PHO89t为模板扩增获得“PHO89p-hyg-PHO89t”片段,PCR胶回收后采用RF克隆方法(如实施例7中所示)将“PHO89p-hyg-PHO89t”片段插入PZPK双元载体的LB和RB之间。所获得的载体命名为PZPK-PHO89p-hyg-PHO89t,结构如图2所示。
2.含有PZPK-PHO89p-hyg-PHO89t质粒的农杆菌工程菌株的构建
所获得的PZPK-PHO89p-hyg-PHO89t双元载体采用电击转化法转化至根癌农杆菌(Agrobacterium tumefaciems)AGL1(购自美国标准生物品收藏中心(ATCC))中,于含有50ng/μL卡那霉素的LB平板上挑取转化子。农杆菌转化子首先采用菌落PCR的方法进行验证。验证正确的转化子,提取其中的质粒,转化至大肠杆菌中。双元载体通过大肠杆菌大量富集后送测序验证。含有 测序正确质粒的农杆菌菌株为工程菌株,保存备用。
3.HYG基因在Rhodosporidium babjevae NCYC 2630中的诱导表达
R.babjevae NCYC 2630购自英国国家酵母菌种保藏中心(National Collection of Yeast Cultures,NCYC)。取一环活化后的R.babjevae NCYC 2630,接于5mL YEPD(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)培养液中,30℃,200r/min过夜培养10h。用无菌水洗涤一遍后,调节至OD600=0.1-0.8,备用。含有PZPK-PHO89p-hyg-PHO89t质粒的农杆菌活化后,接于5mL含有卡那霉素(50ng/μL)和利福平(50ng/μL)的LB液体中,30℃,200r/min培养8h。用无菌水洗涤一遍,调节至OD600=0.1-1.6,备用。
取上述酵母及农杆菌稀释液各400μL,混匀,直接滴于诱导平板(5mmol/L葡萄糖,0.5%甘油,1.45g/L磷酸二氢钾,2.05g/L磷酸氢二钾,0.15g/L氯化钠,0.5g/L七水硫酸镁,66mg/L二水氯化钙,2.48g/L七水硫酸铁,0.5g/L硫酸铵,40mmol/L MES(2-(N-吗啡啉)乙磺酸),2%琼脂粉,200μmol/L乙酰丁香酮)上(Bundock,P.,den Dulk-Ras,A.,Beijersbergen,A.,et al.EMBO J.,1995,14,3206-3214.),24-25℃,培养4天。用10mL左右的无菌水将混合菌苔洗下,3000r/min离心5min,弃去上层主要含有农杆菌的液体,剩余的细胞用800μL无菌水重悬,取50-200μL涂布于磷酸盐限制平板(50ng/μL潮霉素、300μg/mL头孢菌素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,琼脂粉1.5g/L,PH 6.0)上,于30℃进行培养,直至转化子出现。
4.R.babjevae NCYC 2630潮霉素转化子的PCR鉴定
随机挑取6个潮霉素抗性R.babjevae转化子,接入含有50ng/μL潮霉素的磷酸盐限制培养基(30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁pH 6.0)中,于30℃摇床培养36h。参考实施例3中所述玻璃珠破壁法提取重组R.babjevae菌株基因组DNA,采用HYG-RF-p1和HYG-RF-p2为引物,进行PCR鉴定。PCR体系(50μL):10×PCR buffer(大连TakaRa)5.0μL,dNTPs(2.5mmol/l)4.0μL,上游引物(10μmol/l)2.0μL,下游引物(10μmol/l)2.0μL,rTaq DNA聚合酶(大连TakaRa)1.0μL,基因组DNA(30ng/μL)1μL,ddH2O加至50μL。反应条件:95℃3min,98℃10s,55℃15s,72℃1min,35个循环,72℃10min,4℃结束反应。PCR产物经1%(质量/体积浓度)琼脂糖凝胶电泳分析。结果如图3所示。可见,外源HYG片段成功整合进入R.babjevae NCYC 2630基因组中。
5.R.babjevae NCYC 2630潮霉素转化子的Western blot分析
除了直接利用潮霉素抗性表型来确定圆红冬孢酵母PHO89启动子可以启动潮霉素抗性基因在R.babjevae NCYC 2630的表达外,由于潮霉素抗性基因表达产物的C-端携带6×His Tag,还可利用抗His Tag抗体通过Western blot分析 确定相应蛋白的表达。上述PCR鉴定正确的6个转化子,接入10mL含有50ng/μL潮霉素的磷酸盐限制培养基中,于30℃摇床培养48h,4000r/min离心10min收集菌株。获得的菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,再加入同等体积的酸洗玻璃珠。采用玻璃珠破壁法破碎细胞提取细胞总蛋白(精编分子生物学实验指南第三版第13章,奥斯伯等著,颜子颖等译,科学出版社出版)。细胞总蛋白在12%SDS-PAGE胶上进行分离后,转移到硝酸纤维素膜上(Solarbio,Beijing,China),采用小鼠抗His-tag抗体(购自上海碧云天生物技术有限公司)和辣根过氧化物酶标记山羊抗小鼠IgG(购自上海碧云天生物技术有限公司)作为一抗和二抗,DAB辣根过氧化物酶显色试剂盒(购自上海碧云天生物技术有限公司)进行显色,均可观察到潮霉素基因的表达(图4)。
实施例9:利用PHO89p-BLE-PHO89t表达盒进行圆红冬孢酵母ATCC 10788的URA3基因敲除兼BLE的诱导表达
在此利用圆红冬孢酵母URA3基因作为整合位点,通过RF克隆方法,使得“PHO89p-BLE-PHO89t”表达盒的5’和3’末端携带有约1500bp的圆红冬孢酵母URA3基因同源重组臂,利用BLE抗性筛选标记进行转化子的筛选。
1.圆红冬孢酵母URA3基因的获得
根据圆红冬孢酵母URA3(orotidine-5'-phosphate decarboxylase)基因序列(NCBI登录号:KB722658.1,EU693529.1),设计特异引物:URA3-P1:5’-TGGCTCCAATGAGTCGTTGCTTCCAGCGC-3′和URA3-P2:5’-CAAGGAGAGAGGCGTTAAGCCTAAAG-3’,以圆红冬孢酵母ATCC 10788基因组为模板,扩增获得含有URA3启动子、阅读框架和终止子的全长基因组序列。利用DNA回收试剂盒纯化PCR产物后,克隆到pMD18-T载体,转化入大肠杆菌(E.coli)DH5α感受态细胞。挑选Amp抗性转化子进行增菌培养、质粒提取。重组质粒样品送至大连TakaRa公司测序,序列结果与基因组测序结果比对,证实包含URA3启动子,终止子和阅读框架的DNA序列(如SEQ ID NO:8所示,URA3p-URA3-URA3t)。该质粒命名为T-URA3。
2.RF克隆方法构建PHO89p-BLE-PHO89t表达盒
以pPICZαA(购自Invitrogen)为模板,利用一对引物BLE-RF-p1:5’-CTCCTGCCAGCAGCCAACCGTCGTCAAGatggccaagttgaccagtgccg-3′和BLE-RF-p2:5’-CCAAGGACCGGAAGACGAATGGAACCAAGtcagtcctgctcctcggccacg-3’,进行PCR扩增。依照实施例7中所述的RF克隆方法,以T-PHO89p-hyg-PHO89t为骨架,将BLE(SEQ ID NO:9)插入pRtPHO89和RtPHO89t之间,构建成的质粒命名为T-PHO89p-BLE-PHO89t。
3.URA3-BLE敲除盒的构建
采用引物URA3-PHO89-BLE-RF-P1:5’-CTTGTCTTCTACAGTATATCCCTAAATTgcgggatcttccctcgcttcgaccacc-3′和 URA3-PHO89-BLE-RF-P2:5’-CTGCCCTTCACTCATCAATTACCAccgcgacaacccctccctccctcaatc-3’为引物,进行PCR扩增。依照实施例3中所述的RF克隆方法,以T-URA3为骨架,将“PHO89p-BLE-PHO89t”表达盒插入RtURA3中,所得的序列经过Takara测序后如SEQ ID NO:10所示(pURA3-PHO89-BLE-URA3t),构建成的质粒命名为T-URA3-PHO89-BLE,结构如图7所示。重组质粒样品送至大连TakaRa公司测序,序列结果与基因组测序结果比对,证实该基因片段为两端带有1500bp URA3重组臂的“URA3-PHO89-BLE-URA3”敲除盒。
4.URA3-PHO89-BLE-URA3敲除盒的大量制备
以T-URA3-PHO89-BLE质粒为模板,以URA3-P1和URA3-P2为引物,进行“URA3-PHO89-BLE-URA3”敲除盒的大量制备。PCR体系(500μL):10×Speed buffer(大连TakaRa)50.0μL,dNTPs(10mmol/L)10.0μL,上游引物(10μmol/L)20.0μL,下游引物(10μmol/L)20.0μL,SpeedSTAR HS DNA聚合酶(扩增速度快,1kb/10s,购自大连TakaRa公司)5.0μL,基因组DNA模板(120ng/μL)15.0μL,ddH2O加至500μL,混匀后分装。反应条件:98℃1min,98℃10s,65℃60s,35个循环,72℃10min,4℃结束反应。
PCR产物经1%(质量/体积浓度)琼脂糖凝胶电泳分析后利用PCR片段纯化试剂盒(购自生工)进行纯化。纯化后的DNA片段浓度为500ng/μL,共60μL,-20℃保存备用。
PCR产物经1%(质量/体积浓度)琼脂糖凝胶电泳分析后利用PCR片段纯化试剂盒(购自生工)进行纯化。纯化后的DNA片段浓度为500ng/μL,共60μL,-20℃保存备用。
5.圆红冬孢酵母ATCC 10788感受态细胞制备
圆红冬孢酵母ATCC 10788感受态细胞的制备:圆红冬孢酵母ATCC 10788挑菌落接种10ml YEPD培养基(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0),30℃,200rpm,培养20h;培养物1:50比例转接新鲜YEPD培养基,100mL(500mL锥形瓶,装液量100mL),30℃,200rpm,培养6-9h,OD值达到0.6-1.2;培养物冰浴10-30min,4℃,4000r/min离心5min,弃上清;0℃无菌Milli-Q水洗1次;0℃1mol/L山梨醇洗涤2次;冰浴,备用。取100μL圆红冬孢酵母ATCC 10788感受态细胞,加入“URA3-PHO89-BLE-URA3”敲除盒10μL(总共5μg),混匀后移入预冷至0℃的电击杯中,参数:电压0.8-2.0千伏,电阻200Ω,电容25μF,时间4-8ms;电击后立即加入1mL含1M山梨醇的YEPD,30℃温育1-2h;涂布含1M山梨醇YEPD-Zecine平板(含博来霉素50ng/μL),30℃培养5天以上,待转化子出现。
6.转化子的菌落PCR鉴定
博来霉素抗性转化子接种10ml YEPD液体培养基。参照实施例3中玻璃珠破壁法提取基因组DNA,采用BLE-RF-p1和BLE-RF-p2进行PCR鉴定。 PCR体系(25μL):10×PCR buffer(大连TakaRa)2.5μL,dNTPs(2.5mmol/l)2.0μL,上游引物(10μmol/l)1.0μL,下游引物(10μmol/l)21.0μL,rTaq DNA聚合酶(大连TakaRa)0.5μL,基因组DNA(20ng/μL)1μL,ddH2O加至25μL。反应条件:95℃3min,95℃30s,55℃30s,72℃1min,35个循环,72℃10min,4℃结束反应。PCR产物经1%(质量/体积浓度)琼脂糖凝胶电泳分析。结果显示,所有圆红冬孢酵母ATCC 10788重组菌株均可扩增出外源BLE基因片段,而对照菌株圆红冬孢酵母ATCC 10788则无相应PCR产物(未显示)。
进一步对上述鉴定正确的转化子进行表型验证。5’-FOA抗性表型分析结果显示,此尿嘧啶营养缺陷型圆红冬孢酵母工程菌株在含有5’-FOA的限磷培养基(0.1%5’-FOA,葡萄糖70g/L,(NH4)2SO40.1g/L,酵母粉0.75g/L,KH2PO40.06g/L,MgSO4·7H2O 1.5g/L,pH 6.0)上能够正常生长,而野生型的R.toruloides ATCC 10788则在含有5’-FOA的培养基中无法生长。
因此,从基因型到表型,均验证圆红冬孢酵母ATCC 10788重组菌株URA3基因的缺失。
7.Ble基因的表达分析(转录水平表达分析,RT-PCR)
除了直接利用博莱霉素抗性表型来确定圆红冬孢酵母PHO89启动子可以启动博莱霉素抗性基因在圆红冬孢酵母的表达(博莱霉素抗性表型决定于莱霉素抗性基因的表达)外,还利用RT-PCR方法检测了博莱霉素抗性基因ble的转录水平的表达。
随机挑取2株菌落PCR鉴定和表型分析均正确的博来霉素抗性转化子接种10mL磷酸盐限制培养基(30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁pH 6.0),诱导培养48h。参照实施例1方法提取细胞总RNA,参照实施例2方法进行反转录(RT),以所合成的cDNA第一链为模板,利用BLE-p1:ATGGCCAAGTTGACCAGTGCCG和BLE-p2:TCAGTCCTGCTCCTCGGCCACG为引物进行PCR扩增。PCR体系(25μL):10×Ex Taq buffer(大连TakaRa)2.5μL,dNTPs(10mmol/l)0.5μL,上游引物BLE-p1(10μmol/l)1.0μL,下游引物BLE-p2(10μmol/l)1.0μL,Ex Taq DNA聚合酶0.25μL,cDNA第一链模板1.0μL,ddH2O加至25μL。反应条件:98℃1min,98℃10s,65℃60s,35个循环,72℃10min,4℃结束反应。结果显示,所有圆红冬孢酵母ATCC 10788重组菌株均可扩增出0.3kb BLE基因片段,而对照菌株圆红冬孢酵母ATCC 10788则无相应PCR产物(图5)。
实施例10:双表达盒PHO89启动子载体的构建
1.pZPK-Ppho89-hyg-Thsp单表达盒载体的构建
以实施例3中制备的R.toruloides CGMCC2.1389基因组DNA为模板,利用引物HSPt-H1:CGAAAGAACACCACCATCACCATCACTAGacgattccgccccgtctcacctcgcat和HSPt-H2:GCATGCTGCAGGTCGACTCTAGAGGATCC cgcgcacttctctgcactgcatctttg,扩增获得Thsp终止子序列(SEQ ID NO:11),PCR产物经DNA胶回收试剂盒(购自生工)纯化后采用RF克隆方法(如实施例7中所示)将Thsp终止子片段置换PZPK-PHO89p-hyg-PHO89t载体的Tpho89终止子,所获得的载体命名为PZPK-PHO89p-hyg-Thsp,结构如图6所示。
2.多克隆位点的引入和pZPK-Ppho89-MCS-Thsp单表达盒载体的构建
选择pZPK-pPGK-hyg-Tnos载体(Lin XP,Wang YN,Zhang SF,et al.Fems Yeast Res.2014,14:547–555)和上一步骤构建的PZPK-PHO89p-HYG-Thsp载体中均不含有的3个酶切位点EcoR V、Nco I、Spe I设计多克隆位点MCS(Multiple Cloning Site)。参考RF克隆原理,直接设计两条完全反向互补且含EcoR V、Nco I、Spe I酶切位点的长链引物PHO89-HSPt-H1:5’-agcagccaacgtcgtcaag GATATCCCATGGACTAGTacgattccgccccgtctca-3’和PHO89-HSPt-H1:5’-cttgacgacgttggctgctACTAGTCCATGGGATATCcttgacgacggttggctgc-3’作为大引物(mega-primer),等摩尔比加入RFII反应体系,以pZPK-pPGK-hyg-Tnos载体为出发载体,直接进行RF II克隆(如实施例7中所示)将MCS片段置换上一步骤构建的PZPK-PHO89p-HYG-Thsp载体的hyg ORF,所构建的载体命名为pZPK-Ppho89-MCS-Thsp。结构如图7所示。
3.基于Ppho89启动子的双表达盒诱导表达载体的构建
以pZPK-Ppho89-MCS-Thsp载体为模板,利用引物PHO89-HSPt-p1:5’-AGCTTGAGCTTGGATCAGATTGTCGTTTCGAGGGATCTTCCCTCGCTTTGAC-3’和PHO89-HSPt-p2:5’-CAAACACTGATAGTTTAAACTGAAGGCGGCGCGCACTTCTCTGCACTGCATC-3’进行“Ppho89-MCS-Thsp”表达盒的扩增,PCR产物经DNA胶回收纯化试剂盒(购自生工)纯化后采用RF克隆方法(如实施例7中所示)将Thsp终止子片段同方向插入在pZPK-pPGK-hyg-Tnos载体(Lin XP,Wang YN,Zhang SF,et al.Fems Yeast Res.2014,14:547–555)的“pPGK-hyg-Tnos”表达盒与RB之间,“pPGK-hyg-Tnos”表达盒(简称HYG)和“Ppho89-MCS-Thsp”之间有100bp左右的间隔序列,所获得的载体命名为PZPK-HYG-Ppho89-MCS-Thsp,结构如图8所示。该载体的“pPGK-hyg-Tnos”表达盒用于筛选重组子,“Ppho89-MCS-Thsp”则用于目的基因的插入克隆和诱导表达。
实施例11:脂肪酸羟化酶在锁掷孢酵母属S.pararoseus JCM 3765中的表达
蓖麻油酸(12-hydroxy-octadeca-cis-9-enoic acid:C18:1-OH)是一类十分有价值的重要工业原料。现有来源主要为植物榨取,但是资源受到限制。来源于病原性真菌——麦角菌(Claviceps purpurea)油酸羟化酶基因(CpFAH,Genbank登记号:EU661785)在微生物中的表达可为蓖麻油酸的供应提供一条新的途径。利用Ppho89启动子进行油酸羟化酶基因CpFAH的诱导表达,以实现在圆红冬孢酵母中合成蓖麻油酸的目的。
1.CpFAH磷酸盐饥饿诱导表达载体的构建
依据NCBI上报道的CpFAH(EU661785)基因委托上海生工全基因合成此基因,序列如SEQ ID NO:12所示(CpFAH)。引物CpFAH-NcoI-E1:5’-cggCCATGGACatggcttccgctactcctgcaatg-3′和CpFAH-NcoI-E2:5’-CCGACTAGTctaGTGGTGGTGGTGGTGGTGctgagtcttcattgaaat-3’为引物,进行PCR扩增。PCR扩增产物利用DNA胶回收纯化试剂盒(购自生工)进行纯化,并利用Nco I、Spe I进行双酶切,并连接入同样Nco I、Spe I处理的pZPK-HYG-Ppho89-MCS-Thsp,插入在启动子Ppho89和终止子Thsp之间,成功构建脂肪酸羟化酶的磷酸盐饥饿诱导表达载体pZPK-HYG-Ppho89-CpFAH-Thsp,结构如图9所示。重组表达脂肪酸羟化酶的C端引入6×His Tag,可用于Western blot操作。
2.含有pZPK-HYG-Ppho89-CpFAH-Thsp载体的农杆菌工程菌株的构建
所构建的pZPK-HYG-Ppho89-CpFAH-Thsp载体采用电击转化法转化至农杆菌AGL1中,于含有50ng/μL卡那霉素的LB平板上挑取转化子。卡那霉素抗性农杆菌转化子首先采用菌落PCR的方法进行验证。验证正确的转化子,命名为AGL1/ZPK-HYG-Ppho89-CpFAH-Thsp,保存备用。
3.CpFAH经ATMT整合入锁掷孢酵母属S.pararoseus JCM 3765染色体
拟粉红锁掷孢酵母(S.pararoseus)JCM 3765购自中国普通微生物菌种保藏管理中心(CGMCC)。取一环活化后的S.pararoseus JCM 3765,接种于5ml YEPD(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中,25℃,200r/min过夜培养。用无菌水洗涤一遍后,调节至OD600=1-2,备用。
步骤2获得的重组农杆菌AGL1/ZPK-HYG-Ppho89-CpFAH-Thsp接种于5mL含有卡那霉素(100ng/μL)和利福平(80ng/μL)的LB液体中,250℃,200r/min培养过夜。用无菌水洗涤一遍,调节至OD600=1-3,备用。
取上述拟粉红锁掷孢酵母JCM 3765及农杆菌稀释液各200μL,混匀,直接滴于诱导平板(5mmol/L葡萄糖,0.5%甘油,1.45g/L磷酸二氢钾,2.05g/L磷酸氢二钾,0.15g/L氯化钠,0.5g/L七水硫酸镁,66mg/L二水氯化钙,2.48g/L七水硫酸铁,0.5g/L硫酸铵,40mmol/L MES(2-(N-吗啡啉)乙磺酸),2%琼脂粉,200μmol/L乙酰丁香酮)表面的滤膜上,25℃,培养2天。无菌镊子将滤膜从IM诱导平板上转移到诱导筛选平板(50ng/μL潮霉素、300μg/mL头孢菌素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,琼脂粉1.5g/L,PH 6.0)上,30℃倒置培养48h,直至转化子出现。
4.拟粉红锁掷孢酵母JCM 3765潮霉素抗性转化子的菌落PCR鉴定
随机挑取6个遗传霉素抗性S.pararoseus JCM 3765转化子接入50mL含有50ng/μL遗传霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)上,参考实施例3中所述玻璃珠破壁法提取基因组DNA,采用CpFAH-NcoI-E1和CpFAH-NcoI-E2为引物,进行PCR鉴定。结果证明, CpFAH成功整合进入S.pararoseus JCM 3765基因组(未显示)。
5.重组拟粉红锁掷孢酵母JCM 3765生产蓖麻油酸的发酵实验
挑取3个菌落PCR鉴定正确的拟粉红锁掷孢酵母JCM 3765转化子单菌落接于5ml YEPD培养基(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中。培养24h,以1:50的接种量接于50mL含有50ng/μL潮霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,作为二级种子。二级种子培养24h后,取5mL加入45mL的限磷诱导培养基中。培养96h后结束发酵。取40mL发酵菌液置于50mL圆底离心管,8000r/min室温离心5min收集菌体,用去离子水洗涤2次,105℃烘24h至恒重。取出后于干燥器中冷却,然后称量记录,称量后按1g干菌体加入6ml 4mol/L盐酸的比例,每管加入4mL 4mol/L盐酸,78℃水浴消化1h,冷却后加入2倍体积的氯仿/甲醇(1:1,v/v),充分振荡混匀,萃取1h后离心,取氯仿层放入一个新的离心管中。水相以等体积氯仿再次萃取一次,充分振荡后离心,取氯仿层放入上述新的离心管中合并,并加入等体积0.1%氯化钠溶液,振荡混匀后离心。用注射器将下层有机相小心抽取并注入事先准备好的玻璃漏斗中(漏斗预先塞入脱脂棉花,并加入适量无水硫酸钠),待漏斗中有机溶液基本流入下方的玻璃油瓶中,加入适量的氯仿冲洗漏斗4遍,一并流入下方油瓶中,萃取有油脂的氯仿采用旋转蒸发仪进行油脂收集,将收集了油脂的油瓶放入烘箱烘干24h。
对菌油进行甲酯化:称取70.0mg的待测油脂,加入含5%KOH的CH3OH溶液0.5mL,加热回流50min,然后加入BF3的甲醇溶液(VBF3乙醚溶液:VCH3OH=4:10)0.7mL,继续回流10min。冷却后加入1mL去离子水和0.7mL正己烷,混匀后吸出有机相,经去离子水洗涤两遍后用于气相色谱分析。
6.重组拟粉红锁掷孢酵母JCM 3765生产蓖麻油酸的检测
将步骤7转酯化得到的样品溶解于正己烷中,采用文献(Meesapyodsuk,D.,and Qiu,X.Plant Physiol.,2008,147,1325-1333.)中的方法进行检测,标准品为蓖麻油酸甲酯(CAS:141-24-2),负对照为野生型拟粉红锁掷孢酵母JCM 3765的油脂转酯化产物。结果显示,转入CpFAH基因的拟粉红锁掷孢酵母JCM 3765中确有蓖麻油酸产生,含量为280μg/mL,总量占总体游离脂肪酸含量的62%以上。
7.重组拟粉红锁掷孢酵母JCM 3765中CpFAH表达分析——Western blot
除了直接通过蓖麻油酸生产表型来确定圆红冬孢酵母Ppho89启动子可以启动油酸羟化酶基因在锁掷孢酵母属的表达外,由于油酸羟化酶基因表达产物的C-端携带6×His Tag,还可利用抗His Tag抗体通过免疫印迹分析油酸羟化酶基因的表达。上述步骤4中PCR鉴定正确的6个转化子,接入10mL的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,于30℃摇床培养48h,4000r/min离心10min收集菌株。菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,按实施例8的步骤5进行SDS-PAGE和Western blot分析,均可观察到油酸羟化酶的表达(图10)。
实施例12:苹果酸酶在掷孢酵母属Sporobolomyces roseus中的表达
苹果酸酶(Malic enzyme,ME)是NADPH的主要产生酶,为油脂合成提供还原力。我们利用磷限制策略进行粉红掷孢酵母(S.roseus)JCM 8242的油脂发酵和代谢组分析时发现,在限磷恒化培养时,粉红掷孢酵母JCM 8242可积累42%的胞内油脂,代谢组分析结果提示NADPH较之磷充足的丰富培养基恒化培养物下调了上万倍。如果在磷限制培养时过表达苹果酸酶增加NADPH的供应,理论上可提高菌株的胞内油脂含量。
1.圆红冬孢酵母苹果酸酶RtME磷酸盐饥饿诱导表达载体的构建
根据圆红冬孢酵母ME基因所在Scaffold序列(NCBI登录号:KB722664.1),设计特异引物:ME-P1:5’-atgcccgcacactttgccccctcccagc-3′和ME-P2:5’-atgcccgcacactttgccccctcccagccc-3’,以实施例1获得的圆红冬孢酵母CGMCC 2.1389总RNA为模板,按实施例2所示方法进行RT-PCR,扩增获得含有ME的全长cDNA序列。利用DNA回收试剂盒纯化PCR产物后,克隆到pMD18-T载体,转化入大肠杆菌(E.coli)DH5α感受态细胞。挑选Amp抗性转化子进行增菌培养、质粒提取。重组质粒样品送至大连TakaRa公司测序,序列结果与基因组测序结果比对,证实包含ME编码基因全长,其序列如SEQ ID NO:13所示,(RtME)。该质粒命名为T-ME。
设计引物ME-NcoI-E1:5’-cggCCATGGACatgcccgcacactttgccccctcccagcccctcca-3′和ME-NcoI-E2:5’-CCGACTAGTctaGTGATGGTGATGGTGGTGctgcgcctgctgct-3’,以T-ME为模板,进行PCR扩增。PCR扩增产物利用DNA胶回收纯化试剂盒(购自生工)进行纯化,并利用Nco I、Spe I进行双酶切,并连接入同样Nco I、Spe I处理的pZPK-HYG-Ppho89-MCS-Thsp,插入在启动子Ppho89和终止子Thsp之间,成功构建苹果酸酶的磷酸盐饥饿诱导表达载体pZPK-HYG-Ppho89-ME-Thsp,所示(pZPK-HYG-Ppho89-ME-Thsp),结构如图11所示。重组表达脂肪酸羟化酶的C端引入6×His Tag,可用于Western blot操作。
2.含有pZPK-HYG-Ppho89-ME-Thsp载体的农杆菌工程菌株的构建
所构建的pZPK-HYG-Ppho89-ME-Thsp载体采用电击转化法转化至农杆菌AGL1中,于含有50ng/μL卡那霉素的LB平板上挑取转化子。卡那霉素抗性农杆菌转化子首先采用菌落PCR的方法进行验证。验证正确的转化子,命名为AGL1/ZPK-HYG-Ppho89-ME-Thsp,保存备用。
3.ME经ATMT整合入掷孢酵母属粉红掷孢酵母JCM 8242染色体
粉红掷孢酵母(Sporobolomyces roseus)JCM 8242购自于日本微生物菌种 保藏中心(JCM)。取一环活化后的S.roseus JCM 8242,接种于5mLYEPD(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中,25℃,200r/min过夜培养。用无菌水洗涤一遍后,调节至OD600=1-2,备用。
步骤2获得的重组农杆菌AGL1/ZPK-HYG-Ppho89-ME-Thsp接种于5mL含有卡那霉素(100ng/μL)和利福平(80ng/μL)的LB液体中,250℃,200r/min培养过夜。用无菌水洗涤一遍,调节至OD600=1-3,备用。
取上述粉红掷孢酵母JCM 8242及农杆菌稀释液各200μL,混匀,按实施例13步骤3进行ATMT操作,直至转化子出现。
4.粉红掷孢酵母JCM 8242潮霉素抗性转化子的菌落PCR鉴定
随机挑取6个遗传霉素抗性S.roseus JCM 8242转化子接入50mL含有50ng/μL遗传霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)上,参考实施例3中所述玻璃珠破壁法提取基因组DNA,采用ME-NcoI-E1和ME-NcoI-E2为引物,进行PCR鉴定。结果证明,ME编码基因成功整合进入S.roseus JCM 8242基因组(未显示)。
5.重组粉红掷孢酵母S.roseus JCM 8242油脂发酵
挑取3个菌落PCR鉴定正确的粉红掷孢酵母JCM 8242转化子单菌落,按实施例11步骤5所示方法进行发酵、胞内油脂提取和分析。结果显示,较之野生菌株S.roseus JCM 8242,过表达了圆红冬孢酵母苹果酸酶的重组S.roseus JCM 8242胞内油脂含量由42%提高到了68%,增加了62%。
6.重组S.roseus JCM 8242中ME表达分析——Western blot
除了直接通过油脂积累增加性能来确定圆红冬孢酵母Ppho89启动子可以启动圆红冬孢酵母苹果酸酶(RtME)基因在锁掷孢酵母属的表达外,由于RtME表达产物的C-端携带6×His Tag,还可利用抗His Tag抗体通过免疫印迹分析ME的表达。上述步骤4中PCR鉴定正确的6个转化子,随机挑取3个,接入10mL的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,于30℃摇床培养48h,4000r/min离心10min收集菌株。菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,按实施例8的步骤5进行SDS-PAGE和Western blot分析,均可观察到RtME的表达(图12)。
实施例13:GFP在红酵母属深红酵母CGMCC 2.279中的磷去阻遏诱导表达
鉴于深红酵母遗传背景不清晰,无法分离自身启动子和终止子元件进行目的基因的表达,本技术发明利用圆红冬孢酵母Ppho89启动子和Thsp终止子,建立了深红酵母遗传操作体系,实现了GFP在深红酵母CGMCC 2.279中的诱导表达。
1.GFP磷酸盐饥饿诱导表达载体的构建
依据NCBI上报道的GFP的氨基酸序列(AFA52654.1),按圆红冬孢酵母密码子偏好性进行优化,委托上海生工全基因合成其基因,序列如SEQ ID NO:16所示(GFP)。引物GFP-NcoI-E1:5’-cggCCATGGACatgtcgaagggtgaggagcttttc-3′和GFP-NcoI-E2:5’-CCGACTAGTctaGTGGTGGTGGTGGTGGTGcttgtagagttcgtccatgccgtg-3’为引物,进行PCR扩增。PCR扩增产物利用DNA胶回收纯化试剂盒(购自生工)进行纯化,并利用Nco I、Spe I进行双酶切,并连接入同样Nco I、Spe I处理的pZPK-HYG-Ppho89-MCS-Thsp,插入在启动子Ppho89和终止子Thsp之间,成功构建绿色荧光蛋白的磷酸盐饥饿诱导表达载体pZPK-HYG-Ppho89-GFP-Thsp,结构如图13所示。重组表达绿色荧光蛋白的C端引入6×His Tag,可用于Western blot操作。
2.含有pZPK-HYG-Ppho89-GFP-Thsp载体的农杆菌工程菌株的构建
所构建的pZPK-HYG-Ppho89-GFP-Thsp载体采用电击转化法转化至农杆菌AGL1中,于含有50ng/μL卡那霉素的LB平板上挑取转化子。卡那霉素抗性农杆菌转化子首先采用菌落PCR的方法进行验证。验证正确的转化子,命名为AGL1/ZPK-HYG-Ppho89-GFP-Thsp,保存备用。
3.GFP经ATMT整合入深红酵母CGMCC 2.279染色体
深红酵母(Rhodotorula rubra)CGMCC 2.279购自于酵母菌种保藏中心(购自中国普通微生物菌种保藏管理中心(CGMCC)。取一环活化后的深红酵母CGMCC 2.279,接种于5mL YEPD(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中,25℃,200r/min过夜培养。用无菌水洗涤一遍后,调节至OD600=1-2,备用。
步骤2获得的重组农杆菌AGL1/ZPK-HYG-Ppho89-GFP-Thsp接种于5mL含有卡那霉素(100ng/μL)和利福平(80ng/μL)的LB液体中,250℃,200r/min培养过夜。用无菌水洗涤一遍,调节至OD600=1-3,备用。
取上述深红酵母CGMCC 2.279及农杆菌稀释液各200μL,混匀,按实施例13步骤3进行ATMT操作,直至转化子出现。
4.深红酵母CGMCC 2.279潮霉素抗性转化子的菌落PCR鉴定
随机挑取6个遗传霉素抗性深红酵母CGMCC 2.279转化子接入50mL含有50ng/μL遗传霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)上,参考实施例3中所述玻璃珠破壁法提取基因组DNA,采用GFP-NcoI-E1和GFP-NcoI-E2为引物,进行PCR鉴定。结果证明,GFP基因成功整合进入深红酵母CGMCC 2.279基因组(未显示)。
5.深红酵母CGMCC 2.279潮霉素抗性转化子的荧光观察
挑取3个菌落PCR鉴定正确的深红酵母CGMCC 2.279潮霉素抗性转化子单菌落接于5mL YEPD培养基(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中。培养24h,以1:50的接种量接于50ml含有50ng/μL 潮霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,作为二级种子。二级种子培养24h后,取5mL加入45mL的限磷诱导培养基中。培养48h后结束发酵。取菌液10μL点于载玻片,盖上盖玻片后置于荧光显微镜进行绿色荧光观察,激发波长498nm,发射波长516nm,GFP重组深红酵母可观察到绿色荧光,而对照野生型深红酵母CGMCC 2.279则无荧光出现(未显示)。
6.重组深红酵母CGMCC 2.279中GFP表达分析——Western blot
除了直接通过绿色荧光表型来确定圆红冬孢酵母Ppho89启动子可以启动GFP在红酵母属深红酵母的表达外,由于重组GFP的C-端携带6×His Tag,还可利用抗His Tag抗体通过免疫印迹分析GFP的表达。上述步骤4中PCR鉴定正确的6个转化子,随机挑取3个,接入10mL的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,于30℃摇床培养48h,4000r/min离心10min收集菌株。菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,按实施例8的步骤5进行SDS-PAGE和Western blot分析,均可观察到GFP的表达(图14)。
实施例14:GFP在红酵母属胶红酵母CGMCC 2.22中的磷去阻遏诱导表达
鉴于胶红酵母(Rhodotorula mucilaqinosa)遗传背景不清晰,无法分离自身启动子和终止子元件进行目的基因的表达,本技术发明利用圆红冬孢酵母Ppho89启动子和Thsp终止子,建立了深红酵母遗传操作体系,实现了GFP在胶红酵母CGMCC 2.22中的诱导表达。
1.GFP经ATMT整合入胶红酵母CGMCC 2.22染色体
实施例13步骤2获得的重组农杆AGL1/ZPK-HYG-Ppho89-GFP-Thsp接种于5mL含有卡那霉素(100ng/μL)和利福平(80ng/μL)的LB液体中,250℃,200r/min培养过夜。用无菌水洗涤一遍,调节至OD600=1-3,备用。
胶红酵母CGMCC 2.22购自于酵母菌种保藏中心(购自中国普通微生物菌种保藏管理中心(CGMCC)。取一环活化后的胶红酵母CGMCC 2.22,接种于5mLYEPD(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中,25℃,200r/min过夜培养。用无菌水洗涤一遍后,调节至OD600=1-2,备用。
取上述胶红酵母CGMCC 2.22及农杆菌稀释液各200μL,混匀,按实施例13步骤3进行ATMT操作,直至转化子出现。
2.胶红酵母CGMCC 2.22潮霉素抗性转化子的菌落PCR鉴定
随机挑取6个遗传霉素抗性深红酵母CGMCC 2.279转化子接入50mL含 有50ng/μL遗传霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)上,参考实施例3中所述玻璃珠破壁法提取基因组DNA,采用GFP-NcoI-E1和GFP-NcoI-E2为引物,进行PCR鉴定。结果证明,GFP基因成功整合进入胶红酵母CGMCC 2.22基因组(未显示)。
3.胶红酵母CGMCC 2.22潮霉素抗性转化子的荧光观察
挑取3个菌落PCR鉴定正确的胶红酵母CGMCC 2.22潮霉素抗性转化子单菌落按实施例13步骤5进行菌株培养和显微镜观察,GFP重组胶红酵母可观察到绿色荧光,而对照野生型胶红酵母CGMCC 2.22则无荧光出现(未显示)。
4.重组胶红酵母CGMCC 2.22中GFP表达分析——Western blot
除了直接通过绿色荧光表型来确定圆红冬孢酵母Ppho89启动子可以启动GFP在红酵母属胶红酵母CGMCC 2.22的表达外,由于重组GFP的C-端携带6×His Tag,还可利用抗His Tag抗体通过免疫印迹分析GFP的表达。上述步骤3中PCR鉴定正确的6个转化子,随机挑取3个,接入10mL的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,于30℃摇床培养48h,4000r/min离心10min收集菌株。菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,按实施例8的步骤5进行SDS-PAGE和Western blot分析,均可观察到GFP的表达(图15)。
实施例15:脂肪酸羟化酶在红酵母属海滨红酵母中的表达
来源于麦角菌的油酸羟化酶基因(CpFAH,Genbank登记号:EU661785)在Ppho89启动子启动下实现了蓖麻油酸在海滨红酵母中的合成。
1.CpFAH经ATMT整合入海滨红酵母CGMCC 2.4203染色体
实施例11步骤2获得的AGL1/ZPK-HYG-Ppho89-CpFAH-Thsp重组农杆菌,接种于5mL含有卡那霉素(100ng/μL)和利福平(80ng/μL)的LB液体中,250℃,200r/min培养过夜。用无菌水洗涤一遍,调节至OD600=1-3,备用。
海滨红酵母(Rhodotorula marina)CGMCC 2.4203购自于酵母菌种保藏中心(购自中国普通微生物菌种保藏管理中心(CGMCC)。取一环活化后的海滨红酵母CGMCC 2.4203,接种于5mL YEPD(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中,25℃,200r/min过夜培养。用无菌水洗涤一遍后,调节至OD600=1-2,备用。
取上述拟海滨红酵母CGMCC 2.4203及农杆菌稀释液各200μL,按实施例13步骤3进行ATMT操作,直至转化子出现。
2.海滨红酵母CGMCC 2.4203潮霉素抗性转化子的菌落PCR鉴定
随机挑取6个遗传霉素抗性海滨红酵母CGMCC 2.4203转化子接入50mL含有50ng/μL遗传霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5 g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)上,参考实施例3中所述玻璃珠破壁法提取基因组DNA,采用CpFAH-NcoI-E1和CpFAH-NcoI-E2为引物,进行PCR鉴定。结果证明,CpFAH成功整合进入海滨红酵母CGMCC 2.4203基因组(未显示)。
3.重组海滨红酵母CGMCC 2.4203生产蓖麻油酸的发酵实验
挑取3个菌落PCR鉴定正确的海滨红酵母CGMCC 2.4203转化子单菌落接于5mL YEPD培养基(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中。培养24h,以1:50的接种量接于50mL含有50ng/μL潮霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH6.0)中,作为二级种子。二级种子培养24h后,取5mL加入45mL的限磷诱导培养基中。培养96h后结束发酵。取40mL发酵菌液置于50mL圆底离心管,8000r/min室温离心5min收集菌体,用去离子水洗涤2次,105℃烘24h至恒重。取出后按实施例11所示方法进行油脂提取和菌油转酯化。
4.重组海滨红酵母CGMCC 2.4203生产蓖麻油酸的检测
将步骤7转酯化得到的样品溶解于正己烷中,采用文献(Meesapyodsuk,D.,and Qiu,X.Plant Physiol.,2008,147,1325-1333.)中的方法进行检测,标准品为蓖麻油酸甲酯(CAS:141-24-2),负对照为野生型海滨红酵母CGMCC 2.4203的油脂转酯化产物。结果显示,转入CpFAH基因的海滨红酵母中确有蓖麻油酸产生,含量为292μg/mL,总量占总体游离脂肪酸含量的63%以上。
5.重组海滨红酵母CGMCC 2.4203中CpFAH表达分析——Western blot
除了直接通过蓖麻油酸生产表型来确定圆红冬孢酵母Ppho89启动子可以启动油酸羟化酶基因在海滨红酵母CGMCC 2.4203的表达外,由于油酸羟化酶基因表达产物的C-端携带6×His Tag,还可利用抗His Tag抗体通过免疫印迹分析油酸羟化酶基因的表达。上述步骤4中PCR鉴定正确的6个转化子,随机挑取3个,接入10mL的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,于30℃摇床培养48h,4000r/min离心10min收集菌株。菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,按实施例8的步骤5进行SDS-PAGE和Western blot分析,均可观察到油酸羟化酶的表达(未显示)。
实施例16:菊粉酶在红酵母属禾本红酵母CGMCC 2.4202中的表达
菊粉为多聚果糖,自然界中约有30000多种植物均含有菊粉多糖,菊粉的世界年产量可达350000吨,是仅次于淀粉的第二大植物碳水化合物。在工业中以及做为生物质原料应用较多的产菊粉植物为菊苣和菊芋。这些生物质经过外切菊粉酶处理生成可被多种微生物利用的果糖和少量的葡萄糖。本技术发明利用圆红冬孢酵母Ppho89启动子和Thsp终止子,建立了禾本红酵母遗传操作体 系,实现了外切菊粉酶在禾本红酵母(Rhodotorula graminis)CGMCC 2.4202中的诱导表达。
1.菊粉酶磷酸盐饥饿诱导表达载体的构建
根据ZL 2007100159198.8专利中马克斯克鲁维酵母外切菊粉酶氨基酸序列,按照圆红冬孢酵母密码子偏好性进行优化,委托上海生工进行全基因合成,序列如SEQ ID NO:17所示(INU)。设计引物INU-NcoI-E1:5’-cggCCATGGACatgcgcttcgcgtactccctcttgctc-3′和INU-NcoI-E2:5’-CCGACTAGTctaGTGATGGTGATGGTGGTGgaggttgaactgggtgacgttg-3’,以全合成INU基因为模板,进行PCR扩增。PCR扩增产物利用DNA胶回收纯化试剂盒(购自生工)进行纯化,并利用Nco I、Spe I进行双酶切,并连接入同样Nco I、Spe I处理的pZPK-HYG-Ppho89-MCS-Thsp,插入在启动子Ppho89和终止子Thsp之间,成功构建菊粉酶的磷酸盐饥饿诱导表达载体pZPK-HYG-Ppho89-INU-Thsp,结构如图16所示。重组表达脂肪酸羟化酶的C端引入6×His Tag,可用于Western blot操作。
2.含有pZPK-HYG-Ppho89-INU-Thsp载体的农杆菌工程菌株的构建
所构建的pZPK-HYG-Ppho89-INU-Thsp载体采用电击转化法转化至农杆菌AGL1中,于含有50ng/μL卡那霉素的LB平板上挑取转化子。卡那霉素抗性农杆菌转化子首先采用菌落PCR的方法进行验证。验证正确的转化子,命名为AGL1/ZPK-HYG-Ppho89-INU-Thsp,保存备用。
3.INU经ATMT整合入禾本红酵母CGMCC 2.4202染色体
禾本红酵母CGMCC 2.4202购自于日本微生物菌种保藏中心(JCM)。取一环活化后的禾本红酵母CGMCC 2.4202,接种于5ml YEPD(葡萄糖20.0g/l,酵母提取物10.0g/l,蛋白胨20.0g/l,pH 6.0)中,25℃,200r/min过夜培养。用无菌水洗涤一遍后,调节至OD600=1-2,备用。
步骤2获得的重组农杆菌AGL1/ZPK-HYG-Ppho89-INU-Thsp接种于5mL含有卡那霉素(100ng/μL)和利福平(80ng/μL)的LB液体中,250℃,200r/min培养过夜。用无菌水洗涤一遍,调节至OD600=1-3,备用。
取上述禾本红酵母CGMCC 2.4202及农杆菌稀释液各200μL,混匀,按实施例13步骤3进行ATMT操作,直至转化子出现。
4.禾本红酵母CGMCC 2.4202潮霉素抗性转化子的菌落PCR鉴定
随机挑取6个遗传霉素抗性禾本红酵母CGMCC 2.4202转化子接入50ml含有50ng/μL遗传霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)上,参考实施例3中所述玻璃珠破壁法提取基因组DNA,采用INU-NcoI-E1和INU-NcoI-E2为引物,进行PCR鉴定。结果证明,ME编码基因成功整合进入禾本红酵母CGMCC 2.4202基因组(未显示)。
5.以菊粉为碳源进行重组禾本红酵母油脂发酵
挑取3个菌落PCR鉴定正确的禾本红酵母CGMCC 2.4202转化子单菌落接 于5ml YEPD培养基(葡萄糖20.0g/l,酵母提取物10.0g/l,蛋白胨20.0g/l,pH 6.0)中。培养24h,以1:50的接种量接于50ml含有50ng/μL潮霉素的限磷诱导培养基(50ng/μL潮霉素、25g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,作为二级种子。二级种子培养24h后,取5mL加入45ml的菊粉限磷诱导培养基(50ng/μL潮霉素、25g/L菊粉(购自sigma)、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中。培养96h后结束发酵。取40ml发酵菌液置于50ml圆底离心管,8000r/min室温离心5min收集菌体,用去离子水洗涤2次,105℃烘24h至恒重。之后按实施例11的步骤5进行油脂提取。结果显示,野生菌株禾本红酵母CGMCC 2.4202在利用菊粉为唯一碳源进行发酵96h时可积累21%的胞内油脂,而过表达了外切菊粉酶的重组禾本红酵母利用菊粉为唯一碳源进行发酵96h时胞内油脂含量则为52%,可直接转换菊粉为胞内油脂。
6.重组禾本红酵母CGMCC 2.4202中INU表达分析——Western blot
除了直接通过菊粉利用表型来确定圆红冬孢酵母Ppho89启动子可以启动外切菊粉酶在红酵母属的表达外,由于重组INU表达产物的C-端携带6×His Tag,还可利用抗His Tag抗体通过免疫印迹分析ME的表达。上述步骤4中PCR鉴定正确的6个转化子,随机挑取3个,接入10ml的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,于30℃摇床培养48h,4000r/min离心10min收集菌株。菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,按实施例8的步骤5进行SDS-PAGE和Western blot分析,均可观察到INU的表达(未显示)。
实施例17:GFP在红酵母属粘红酵母中的磷去阻遏诱导表达
本技术发明利用圆红冬孢酵母Ppho89启动子和Thsp终止子,建立了粘红酵母(Rhodotorula glutinis)遗传操作体系,实现了GFP在粘红酵母NCYC 2666中的诱导表达。
1.GFP经ATMT整合入粘红酵母NCYC 2666染色体
实施例13步骤2获得的重组农杆AGL1/ZPK-HYG-Ppho89-GFP-Thsp接种于5ml含有卡那霉素(100ng/μL)和利福平(80ng/μL)的LB液体中,250℃,200r/min培养过夜。用无菌水洗涤一遍,调节至OD600=1-3,备用。
粘红酵母NCYC 2666购自于英国国家酵母菌种保藏中心(National Collection of Yeast Cultures,NCYC)。取一环活化后的粘红酵母NCYC 2666,接种于5mLYEPD(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中,25℃,200r/min过夜培养。用无菌水洗涤一遍后,调节至OD600=1-2,备用。
取上述粘红酵母NCYC 2666及农杆菌稀释液各200μL,混匀,按实施例13步骤3进行ATMT操作,直至转化子出现。
2.粘红酵母NCYC 2666潮霉素抗性转化子的菌落PCR鉴定
随机挑取6个遗传霉素抗性粘红酵母NCYC 2666转化子接入50mL含有50ng/μL遗传霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)上,参考实施例3中所述玻璃珠破壁法提取基因组DNA,采用GFP-NcoI-E1和GFP-NcoI-E2为引物,进行PCR鉴定。结果证明,GFP基因成功整合进入粘红酵母NCYC 2666基因组(未显示)。
3.粘红酵母NCYC 2666潮霉素抗性转化子的荧光观察
挑取3个菌落PCR鉴定正确的粘红酵母NCYC 2666潮霉素抗性转化子单菌落接于5mLYEPD培养基(葡萄糖20.0g/L,酵母提取物10.0g/L,蛋白胨20.0g/L,pH 6.0)中。培养24h,以1:50的接种量接于50mL含有50ng/μL潮霉素的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,作为二级种子。二级种子培养24h后,取5mL加入45mL的限磷诱导培养基中。培养48h后结束发酵。取菌液10μL点于载玻片,盖上盖玻片后置于荧光显微镜进行绿色荧光观察,激发波长498nm,发射波长516nm,GFP重组粘红酵母NCYC 2666可观察到绿色荧光,而对照野生型粘红酵母NCYC 2666则无荧光出现(未显示)。
4.重组胶红酵母CGMCC 2.22中GFP表达分析——Western blot
除了直接通过绿色荧光表型来确定圆红冬孢酵母Ppho89启动子可以启动GFP在红酵母属胶红酵母CGMCC 2.22的表达外,由于重组GFP的C-端携带6×His Tag,还可利用抗His Tag抗体通过免疫印迹分析GFP的表达。上述步骤4中PCR鉴定正确的6个转化子,随机挑取3个,接入10mL的限磷诱导培养基(50ng/μL潮霉素、30g/L葡萄糖、5g/L硫酸铵、0.64g/L硫酸钾、0.08g/L十二水合磷酸氢二钠、0.94g/L硫酸钠、1.5g/L七水硫酸镁,PH 6.0)中,于30℃摇床培养48h,4000r/min离心10min收集菌株。菌体用无菌水洗涤一遍后,加入400μL的蛋白提取缓冲液(8M尿素,65mM DTT,0.1%Triton X-100,100mM NaCl,50mM Tris-Cl,1mM PMSF,1mM EGTA,1mM EDTA,pH为7.4)重悬,按实施例8的步骤5进行SDS-PAGE和Western blot分析,均可观察到GFP的表达(未显示)。
- 还没有人留言评论。精彩留言会获得点赞!