一种构建重组大肠杆菌生物合成2’‑岩藻乳糖的方法与流程
本发明涉及利用重组大肠杆菌合成2'-岩藻乳糖的方法,属于代谢工程领域。更具体地说是一种构建重组大肠杆菌生物合成2'-岩藻乳糖的方法。
背景技术:
母乳中包含婴儿生长和发育所必须的营养物质,但是它同时也含有传统营养物质中所没有的物质,这些物质对身体是有益的。其中一些物质就是人乳寡糖(HMOs)。唾液乳糖是常见的人乳寡糖,具有抗黏附,维持肠道微生物组成及糖组修饰等功能, 在营养和药用方面,也是很有前途的寡糖。
岩藻寡糖,如2'-岩藻乳糖,乳酰-N-岩藻五糖和乳酰-N-岩藻六糖,都是常见的人乳寡糖。岩藻化寡糖作为选择双歧杆菌的生长刺激因子和致病细菌的可溶性受体类似物,从而保护婴儿免受肠道病原体和绑定的毒素的感染。据报道,尤其是α-1,2-链接的岩藻寡糖具有对空肠弯曲杆菌,鼠伤寒沙门氏菌,产肠毒素的大肠杆菌,幽门螺旋杆菌和诺瓦克病毒]等病原体的防护活性。在所有的这些人乳寡糖中,2'-岩藻乳糖是母乳中含量最丰富的,约占人乳寡糖的30%。如果一个母亲在生气的情况下对婴儿进行哺乳,那么奶中所含的较低的2'-FL可能会提供高婴儿的腹泻率。因此,在营养和药用方面,2'-FL是一种很有前途的寡糖。
目前,2'-岩藻乳糖的生产方法主要为化学法,化学法生产存在诸多弊端,如合成步骤过多,生成的产物较多,副产物复杂,其反应液对环境产生污染等,通过生物法来制备2'-岩藻乳糖越来越受到研究者的关注。
本发明中构建的重组工程菌实现了2'-岩藻乳糖的生物合成,为生物法探索生成目标代谢产物提供了新思路。
技术实现要素:
本发明通过构建一株重组大肠杆菌,实现了从乳糖到2'-岩藻乳糖的生物合成。所采取的技术方案如下:
本发明的第一个目的在于提供了一种利用乳糖合成2'-岩藻乳糖的重组大肠杆菌。其特征在于,具有2'-岩藻乳糖的合成途径。名称为E.coli-XYY-1。同时过表达磷酸甘露糖酶基因(manB),磷酸鸟嘌呤转移酶基因(manC),GDP-甘露糖- 4,6-脱水酶基因(gmd),GDP-fucose合成酶基因(fcl),甘露糖-6-磷酸异构酶基因(manA),GDP-fucose的合成的一个正向调控因子(rcsA),β-半乳糖苷透性酶基因(lacY),α-1,2-岩藻糖转移酶基因 (futC)。
所述磷酸甘露糖酶基因manB,基因登录号GI:946574; 磷酸鸟嘌呤转移酶基因manC,基因登录号GI:946580;GDP-甘露糖- 4,6-脱水酶基因gmd,基因登录号GI:946562;GDP-fucose合成酶基因fcl,基因登录号GI:946563;甘露糖-6-磷酸异构酶基因manA,基因登录号GI:944840;GDP-fucose的合成的一个正向调控因子rcsA,基因登录号GI:946467;β-半乳糖苷透性酶基因lacY,基因登录号GI:949083;来源于幽门螺旋杆菌的α-1,2-岩藻糖转移酶基因 futC, 基因登录号GI:CP010436; 所述具有2'-岩藻乳糖合成途径,是指过表达磷酸甘露糖酶,磷酸鸟嘌呤转移酶,GDP-甘露糖- 4,6-脱水酶,GDP-fucose合成酶,甘露糖-6-磷酸异构酶,GDP-fucose合成的一个正向调控因子,β-半乳糖苷透性酶,α-1,2-岩藻糖转移酶。
本发明第二个目的在于提供了大肠杆菌基因工程菌的构建方法,其特征在于步骤如下:
1)分别制备含有磷酸甘露糖酶基因(manB),磷酸鸟嘌呤转移酶基因(manC),GDP-甘露糖- 4,6-脱水酶基因(gmd),GDP-fucose合成酶基因(fcl),甘露糖-6-磷酸异构酶基因(manA),GDP-fucose的合成的一个正向调控因子(rcsA),β-半乳糖苷透性酶基因(lacY),α-1,2-岩藻糖转移酶基因 (futC)的重组质粒,获得构建代谢途径的质粒;
2)将质粒pSim导入转化到宿主菌E.coli BL21(DE3)中,获得携带质粒的宿主菌;
3)分别扩增带有β-半乳糖苷酶基因lacZ,UDP-葡萄糖脂质载体转移酶基因wcaJ,ATP依赖的蛋白酶基因lon同源臂的抗性敲除片段;
4) 先向步骤2所得的携带质粒pSim的宿主菌中转化同一个基因的抗性敲除片段,获得缺失一个基因的重组菌;
5) 将步骤4所得的重组菌进行溶源化处理,利用pCP20质粒进行抗性消除;
6) 以步骤5所得的缺失一个基因的重组菌为宿主菌,重复步骤5)的操作,获得缺失两个基因的重组菌,再重复步骤5)的操作,每次操作均以上一次操作获得的重组菌为宿主菌,直至将步骤3)所述的基因全部敲除,获得缺失个基因的重组大肠杆菌;
7) 将步骤6)中基因敲除的大肠杆菌进行溶源化处理,再将步骤1)所得的代谢途径构建质粒转化到溶源菌中,获得能够利用乳糖合成2'-岩藻乳糖的重组大肠杆菌;
其中所述构建β-半乳糖苷酶基因lacZ的缺失引物和鉴定引物的核苷酸序列如SEQ ID NO.1-SEQ ID NO.4 所示;构建UDP-葡萄糖脂质载体转移酶基因wcaJ的缺失引物和鉴定引物的核苷酸序列如SEQ ID NO.5-SEQ ID NO.8所示;构建ATP依赖的蛋白酶基因lon的缺失引物和鉴定引物的核苷酸序列如SEQ ID NO.9-SEQ ID NO.12所示;
本发明第三个目的在于提供了采用所获得的大肠杆菌基因工程菌进行发酵合成2'-岩藻乳糖,培养基和发酵方法如下:
LB培养基(1L):Tryptone(胰蛋白胨):10g,Yeast Extract(酵母提取物):5g,NaCl(氯化钠):5g。若配置固体培养基,则再加入15g Agar(琼脂)。
M9培养基(1L):Na2HPO4·7H2O(七水合磷酸氢二钠):12.8g,KH2PO4(磷酸二氢钾)3g,NaCl(氯化钠):0.5g,NH4Cl(氯化铵)2g,MgSO4·7H2O(七水合硫酸镁)0.25g,Yeast Extract(酵母提取物)2g,Glycerol(甘油):20g。
将所得基因工程菌在5mL含有卡那霉素50 μg/ml,氨苄青霉素100 μg/ml,链霉素50 μg/ml的LB培养基中37℃,220rpm/min培养12h,转入M9培养基,M9培养基在使用前加入卡那霉素,氨苄青霉素,链霉素(卡那霉素50 μg/ml,氨苄青霉素100 μg/ml,链霉素50 μg/ml),37℃,培养月3h左右,加入IPTG(IPTG 0.2mM),并转入25℃培养,培养约2h后,加入乳糖,继续培养4h后,取样。该方法的具体步骤如下:
1)分别制备含有磷酸甘露糖酶基因(manB),磷酸鸟嘌呤转移酶基因(manC),GDP-甘露糖- 4,6-脱水酶基因(gmd),GDP-fucose合成酶基因(fcl),甘露糖-6-磷酸异构酶基因(manA),GDP-fucose的合成的一个正向调控因子(rcsA),β-半乳糖苷透性酶基因(lacY),α-1,2-岩藻糖转移酶基因 (futC)的重组质粒,获得构建代谢途径的质粒;
2)将质粒pSim导入转化到宿主菌E.coli BL21(DE3)中,获得携带质粒的宿主菌;
3)分别扩增带有β-半乳糖苷酶基因lacZ,UDP-葡萄糖脂质载体转移酶基因wcaJ,ATP依赖的蛋白酶基因lon同源臂的抗性敲除片段;
4)先向步骤2所得的携带质粒pSim的宿主菌中转化一个基因带有同源臂的抗性消除片段,获得缺失一个基因的重组菌;
5)将步骤4)所获得的缺失一个基因的重组菌进行溶源化处理,利用pCP20质粒进行抗性消除;
6)以步骤5)所得的缺失一个基因的重组菌为宿主菌,重复步骤4)和步骤5)的操作,获得缺失两个基因的重组菌,再重复步骤4)和步骤5)的操作,每次操作均以上一次操作获得的重组菌为宿主菌,直至将步骤3)所述的基因全部敲除,获得缺失3个基因的重组大肠杆菌;
7)将步骤6)中基因敲除的大肠杆菌进行溶源化处理,再将步骤1)所得的代谢途径构建质粒转化到溶源菌中,获得能够利用乳糖合成2'-岩藻乳糖的重组大肠杆菌;
8)利用步骤7)所得的重组大肠杆菌合成2'-岩藻乳糖;
所述任一大肠杆菌工程菌在生产2'-岩藻乳糖中的应用,也在本发明的保护范围之内。所述应用的步骤如下:
1) 活化大肠杆菌基因工程菌,获得种子液;
2) 将步骤1)获得的种子液与含有氯霉素、氨苄青霉素和链霉素的培养基,按照种子液:培养基=1:100的比例接种至新鲜的培养基,35~37℃,180rpm~220rpm/min,培养至OD600=0.6~0.8,并加入诱导剂IPTG至终浓度为0.2mM~0.3mM, 然后转入20~25℃,180rpm~220rpm/min,补加乳糖继续培养4~6h;发酵生产2'-岩藻乳糖过程中,大肠杆菌基因工程菌所用的培养基包括各种适于所选用的宿主细胞(大肠杆菌)生长的培养基,碳源优选为低成本的葡萄糖。
本发明公开的构建重组大肠杆菌生物合成2'-岩藻乳糖的方法与现有技术相比所具有的积极效果在于:
(1)本发明所构建的大肠杆菌基因工程菌具有以葡萄糖为碳源发酵合成2'-岩藻乳糖的特点,发酵4h,可以得到5~8g/L的2'-岩藻乳糖。
(2)本发明建立了一条2'-岩藻乳糖的合成途径,本发明同时克服了现有的技术难题,获得了一个高效的基因敲除方案;原始菌株在通过基因工程改造以后,除了生产2'-岩藻乳糖外,没有改变菌株的其它特性,不影响发酵生产;该菌株采用的质粒为成熟的大肠杆菌质粒,因此在代谢过程中不影响细菌生长和正常代谢。
附图说明
图1是质粒pACYCDuet-1-rcsA-manA的图谱,用来过表达2'-岩藻乳糖合成途径中的rcsA、manA基因;
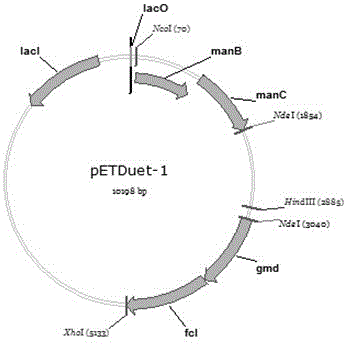
图2是质粒pET-Duet1-manB-manC-gmd-fcl的图谱,用来过表达2'-岩藻乳糖合成途径中的manB、manC、gmd、fcl基因;
图3是质粒pCDF-Duet-1-futC的图谱,用来过表达2'-岩藻乳糖合成途径中的futC基因;
图4是敲除wcaj基因后的PCR验证产物的电泳图;
(图中,M:Marker;泳道1-2分别为:阴性对照,wcaj
图5是敲除lon基因后的PCR验证产物的电泳图;
(图中,M:Marker;泳道1-2分别为:阴性对照lon
图6是敲除lacZ基因后的PCR验证产物的电泳图;
(图中,M:Marker;泳道1-2分别为:阴性对照 lacZ
图7是本发明中构建的合成2'-岩藻乳糖的代谢途径;
图8是ESI-MS检测重组菌株发酵液,其中487.03为目的产物2'-岩藻乳糖。
具体实施方式
下面通过具体的实施方案叙述本发明。除非特别说明,本发明中所用的技术手段均为本领域技术人员所公知的方法。另外,实施方案应理解为说明性的,而非限制本发明的范围,本发明的实质和范围仅由权利要求书所限定。对于本领域技术人员而言,在不背离本发明实质和范围的前提下,对这些实施方案中的物料成分和用量进行的各种改变或改动也属于本发明的保护范围。本发明所用原料及试剂均有市售。以下实施例中所用的材料、试剂、仪器和方法未经特殊说明均为本领域中的常规材料、试剂、仪器和方法,均可通过商业渠道获得;例如Tryptone(胰蛋白胨),Yeast Extract(酵母提取物),Agar(琼脂);卡那霉素,氨苄青霉素,链霉素,E.coli DH5α等等。
本发明中质粒提取采用生工生物工程(上海)有限公司的SanPrep柱式质粒DNA小量抽提试剂盒(Catalog NO.:B518191),切胶回收是采用生工生物工程(上海)有限公司的SanPrep柱式胶回收试剂盒(Catalog NO.:B518131),DNA片段的连接是使用fermentas公司的T4 DNA Ligase(Catalog NO.:EL0014),DNA片段的扩增使用fermentas公司的pfu DNA polymerase(Catalog NO.:EP0571),PCR质粒模板的消化使用fermentas公司的Falst Digelst KpnI(Catalog NO.:FD0524),BamHI(Catalog NO.:FD0054) NcoI(Catalog NO.:FD0574)BglII(Catalog NO.:FD0083)E.coli电击转化实验使用Bio-Rad的电转仪(Catalog NO.:165-2100)。细菌基因组提取使用北京康为世纪生化科技有限公司的细菌基因组提取试剂盒(Catalog NO.:CW0552S)。
实施例1
基因的获取:
本实施例中,获取来源于大肠杆菌MG1655的磷酸甘露糖酶基因manB(基因登录号 GI:946574),磷酸鸟嘌呤转移酶基因manC(基因登录号 GI:946580),GDP-甘露糖- 4,6-脱水酶基因gmd(基因登录号 GI:946562),GDP-fucose合成酶基因fcl(基因登录号 GI:946563),甘露糖-6-磷酸异构酶基因manA(基因登录号 GI:944840),GDP-fucose的合成的一个正向调控因子rcsA(基因登录号 GI:946467),获取来源于大肠杆菌BL21的β-半乳糖苷透性酶基因lacY(基因登录号 GI:949083),获取来源于幽门螺旋杆菌的α-1,2-岩藻糖转移酶基因 futC(基因登录号 GI:CP010436)。
实施例2
重组质粒的制备
使用设计的引物F1,R1对实施例1中获得的来源于大肠杆菌MG1655的磷酸甘露糖酶基因manB,磷酸鸟嘌呤转移酶基因manC进行PCR扩增,扩增后的片段进行切胶纯化,并用NcoI和HindIII进行双酶切,酶切后的片段与同样经过NcoI和HindIII双酶切的质粒pETDuet-1质粒进行连接,将载体:目的片段按摩尔比为1:3的比例混合,加入T4 DNA Ligase后在22℃下酶连5h,连接产物转化E.coli DH5α,并在氨苄青霉素板上进行筛选,获得重组质粒pETDuet-1-manB-manC。
使用设计的引物F2,R2对实施例1中获得的来源于大肠杆菌MG1655的GDP-甘露糖- 4,6-脱水酶基因gmd,GDP-fucose合成酶基因fcl进行PCR扩增,扩增后的片段进行切胶纯化,并用NdeI和XhoI进行双酶切,酶切后的片段与同样经过NdeI和XhoI双酶切的质粒pETDuet-1-manB-manC质粒进行连接,将载体:目的片段按摩尔比为1:3的比例混合,加入T4 DNA Ligase后在22℃下酶连5h,连接产物转化E.coli DH5α,并在链霉素板上进行筛选,获得重组质粒pETDuet-1-manB-manC-gmd-fcl。
使用设计的引物F3,R3对实施例1中获得的来源于大肠杆菌BL21的β-半乳糖苷透性酶基因(lacY),来源于幽门螺旋杆菌的α-1,2-岩藻糖转移酶基因 futC进行PCR扩增,扩增后的片段进行切胶纯化,并用NcoI和HindIII进行双酶切,酶切后的片段与同样经过NcoI和HindIII双酶切的质粒pCDFDuet-1质粒进行连接,将载体:目的片段按摩尔比为1:3的比例混合,加入T4 DNA Ligase后在22℃下酶连5h,连接产物转化E.coli DH5α,并在链霉素板上进行筛选,获得重组质粒pCDFDuet-1-lacY-futC。
使用设计的引物F4,R4对实施例1中获得的来源于大肠杆菌MG1655的甘露糖-6-磷酸异构酶基因manA,GDP-fucose的合成的一个正向调控因子rcsA进行PCR扩增,扩增后的片段进行切胶纯化,并用NcoI和HindIII进行双酶切,酶切后的片段与同样经过NcoI和HindIII双酶切的质粒pACYCDuet-1质粒进行连接,将载体:目的片段按摩尔比为1:3的比例混合,加入T4 DNA Ligase后在22℃下酶连5h,连接产物转化E.coli DH5α,并在链霉素板上进行筛选,获得重组质粒pACYCDuet-1-manA-rcsA。
实施例3
基因的敲除
本实施例使用λRed重组系统敲除E.coli BL21(DE3)的多个基因,该方法每敲除一个基因都进行抗性的消除。下面以wcaj基因为例,详细阐述基因敲除的步骤,其余2个基因的敲除与此相同。
在NCBI中查找E.coli BL21(DE3) wcaj基因的核苷酸序列,设计wcaj基因的缺失引物和鉴定引物。wcaj基因的缺失引物和鉴定引物的核苷酸序列如SEQ ID NO.5-SEQ ID NO.8所示。
实施例4
缺失型E.coli BL21(DE3) ∆3的构建
4.1质粒pSim的转化
挑取-80℃冻存的野生型E.coli BL21(DE3)在无抗性的LB平板上划线,37℃过夜培养。第二天挑取单克隆,接种至5mL LB培养基中,37℃,220rpm/min,过夜培养。第二天按1%的接种量,转接至200ml的LB培养基中。37℃,220rpm/min,培养至OD600约为0.6~0.8,冰浴20min,5500rpm,5min,收集菌体于已灭菌的50ml离心管中,4℃,5500rpm,离心5min,弃上清,用50ml冰浴过的无菌的10%甘油重悬菌体,4℃,5500rpm,再离心5min,重复上述操作3次,最后一次,利用弃上清时的残余液体重悬菌体,取80μL于至新的无菌EP管中。-80℃冻存。
将-80℃冻存的感受态细胞置于冰中融化10min,加入1μL的pSim质粒,混匀后加入电击杯中,冰浴2min,1.8KV电击转化,电击后立即加入1ml的LB培养基,30℃,220rpm/min复苏20min,取适量菌液涂布于灭瘟素平板上(灭瘟素浓度200μg/ml),30℃倒置过夜培养,第二天长出的单克隆即是携带pSim质粒的E.coli BL21(DE3)。
4.2 目的基因的敲除
4.2.1同源重组片段的制备
以pKD3质粒为模板,使用引物SEQ ID NO.5-SEQ ID NO.6进行PCR扩增,切胶纯化回收,获得两端含有wcaj同源臂的敲除片段wcaj-Fragment I。
4.2.2 第一步同源重组(wcaj-Fragment I的转化)
挑取携带pSim质粒的E.coli BL21(DE3)单克隆接种至5ml LB培养基中,37℃,220rpm/min,过夜培养。第二天按1%的接种量,转接至200ml的LB培养基中。37℃,220rpm/min,培养至OD600约为0.6~0.8,将菌液转至42℃水浴摇床中,150rpm/min,20min,然后冰浴20min,4℃,5500rpm/min,离心5min,收集菌体于已灭菌的50ml离心管中,4℃,5500rpm,离心5min,弃上清,用50ml冰浴过的无菌的10%甘油重悬菌体,4℃,5500rpm,再离心5min,重复上述操作3次,最后一次,利用弃上清时的残余液体重悬菌体,取80μL于至新的无菌EP管中,加入4μLlacZ-Fragment I片段,混匀后加入电击杯中,冰浴2min,1.8KV电击转化,电击后立即加入1ml的LB培养基,37℃,180rpm/min,培养2h,取适量菌液涂布于抗性平板上,37℃过夜培养。第二天挑取单克隆,PCR鉴定出wcaj基因被wcaj-Fragment I替换的正确克隆,即E.coli BL21(DE3) 。
基因缺失后抗性的消除
4.3.1E.coli BL21 (DE3) ∆wcaj菌株感受态细胞的制备
挑取E.coli BL21 (DE3) ∆wcaj单克隆接种至5ml的LB培养基中,按照上面步骤制备感受态,电转pCP20质粒,电击后立即加入1ml的LB培养基,30℃,180rpm/min,培养20min,取适量菌液涂布于氨苄青霉素平板上(氨苄青霉素浓度100 μg/ml),30℃过夜培养。第二天挑取单克隆至5ml的LB培养基中(氯霉素浓度25μg/ml),30℃,180rpm/min, 培养10h,转移至新的液体培养基,不加抗生素,42℃,培养6h后,稀释涂板,37℃,倒置过夜培养,第二天挑单克隆,分别在无抗板和氯霉素板上进行影印。氯霉素板上未长,但是无抗板对应位置上长出的单克隆,即为抗性消除成功的阳性克隆,进一步通过PCR验证。
4.4 剩余2个基因的敲除
剩余2个基因的敲除原理和步骤与wcaj相同,以敲除了wcaj基因的菌株为基础,通过重复实验步骤中的4.2和4.3,可依次将全部3个基因敲除,最终构建出缺失的E.coli BL21 (DE3) ∆3。
图4为野生型E.coli BL21(DE3)和E.coli BL21 (DE3) ∆3的PCR验证结果,两组结果使用相同的鉴定引物,图4中泳道2的条带泳道1的条带相比,出现了显著的减小,表明相应目的基因已经被成功敲除。pSim质粒为温度敏感型质粒,培养温度高于30℃的条件下,质粒将会丢失,因此,在将pSim质粒转入E.coli BL21(DE3)后,菌株将一直在30℃条件下培养,以防止pSim质粒的丢失。
实施例5
产2'-岩藻乳糖E.coli BL21 (DE3) ∆3菌株的构建
以敲除3个基因的E.coli BL21 (DE3)∆3菌株为基础,制备感受态细胞(方法参照实验步骤4.1),将pCDFDuet-1-lacY,pETDuet-1-manB-manC-gmd-fcl,pACYCDuet-1-rcsA-manA转入,在LB平板(氯霉素50 μg/ml,氨苄青霉素100 μg/ml,链霉素50 μg/ml)上筛选正确的克隆。经双酶切验证得到携带整个2'-岩藻乳糖合成途径的菌株E.coli BL21(DE3) ∆3/2'-FL。得到5~8g/L的2'-岩藻乳糖,图7为本发明中合成2'-岩藻乳糖的代谢途径。
实施例6
E.coli BL21 ∆3/2'-FL菌株2'-FL合成的验证
将菌株接种至5mL的LB培养基中(氯霉素50 μg/ml,氨苄青霉素100μg/ml,链霉素50μg/ml),37℃,220rpm/min,过夜培养。第二天按1%的接种量转接至优化的M9培养基中,OD600约为0.6时诱导(IPTG浓度0.2mM),转至25℃,诱导2h后,加入2mL乳糖,约4h后取样,4℃,7000rpm/min,离心10min,将上清与沉淀分开。上清过0.22μm滤膜。通过ESI-MS和高效液相色谱检测。
沉淀首先用灭菌水悬浮,4℃,7000rpm/min,离心10min,重复一次,然后采取反复冻融来破碎细胞,破碎后,4℃,5100rpm/min,离心25min,
离心,将上清转移至10mL的离心管中,过0.22μm滤膜,通过ESI-MS检测。
2'-岩藻乳糖的检测:,
仪器:Finnigan LCQ Advantage MAX ion trap mass spectrometer (Thermo Electron,CA)
离子化模式:电喷雾负离子模式;
电喷雾范围:400-700m/z;
干燥器温度:220℃;
喷雾压力:45psi;
毛细管电压:4500V;
进样量:0.2mL/min;
碰撞气体为氮气,辅助气体为氦气;
高效液相检测
色谱柱:Venusil C18柱(5μm particle size,4.6 by 250mm);
流动相:10% 乙腈, 90% 三乙胺冰醋酸(pH 6.0);
流速:0.6mL/min;
进样量5μL。
以上实施例仅用以说明本发明的技术方案,而非对其进行限制,尽管参照上述实施例对本发明进行了详细的说明,对于本领域的普通技术人员来说,依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。而这些修改或替换,并不使相应技术方案的本质脱离本发明所要求保护的技术方案的范围和精神。
SEQUENCE LISTING
<110> 南开大学
<120> 一种构建重组大肠杆菌生物合成2'-岩藻乳糖的方法
<160> 12
<170> PatentIn version 3.5
<210> 1
<211> 79
<212> DNA
<213> 人工序列
<400> 1
atgaccatga ttacggattc actggccgtc gttttacaac gtcgtgactg ggaaaaccgt 60
gtaggctgga gctgcttcg 79
<210> 2
<211> 79
<212> DNA
<213> 人工序列
<400> 2
ttatttttga caccagacca actggtaatg gtagcgaccg gcgctcagct ggaattccgc 60
atatgaatat cctccttag 79
<210> 3
<211> 18
<212> DNA
<213> 人工序列
<400> 3
ctggcacgac aggtttcc 18
<210> 4
<211> 19
<212> DNA
<213> 人工序列
<400> 4
tgtagccaaa tcgggaaaa 19
<210> 5
<211> 79
<212> DNA
<213> 人工序列
<400> 5
gttacgctta aagatgccta atccgccaac ggcttacatt ttacttattg aggtgaatag 60
tcttgagcga ttgtgtagg 79
<210> 6
<211> 79
<212> DNA
<213> 人工序列
<400> 6
ctgttttacg agatcaacat tacctatctg agcttgtccg cctggtgtca tactttctcc 60
ttaacggctg acatgggaa 79
<210> 7
<211> 20
<212> DNA
<213> 人工序列
<400> 7
aatcaggtcg gattgacgcc 20
<210> 8
<211> 20
<212> DNA
<213> 人工序列
<400> 8
caatcaggcg ataaaccgcc 20
<210> 9
<211> 59
<212> DNA
<213> 人工序列
<400> 9
tctgattacc tggcggaaat taaactaaga gagagctctg tcttgagcga ttgtgtagg 59
<210> 10
<211> 59
<212> DNA
<213> 人工序列
<400> 10
gccagccctg tttttattag tgcattttgc gcgaggtcac ttaacggctg acatgggaa 59
<210> 11
<211> 20
<212> DNA
<213> 人工序列
<400> 11
gggaaacatc cccatatact 20
<210> 12
<211> 20
<212> DNA
<213> 人工序列
<400> 12
gggaaacatccccatatact 20
- 还没有人留言评论。精彩留言会获得点赞!