化学方法与流程
本申请是以下申请的分案申请:申请日:2013年8月14日;申请号:201380043392.3(pct/ep2013/067035);发明名称:同上。
发明领域
本发明涉及芜地溴铵(umeclidiniumbromide)的制备方法,以及用于制备芜地溴铵(umeclidiniumbromide)的中间体的制备方法。
发明背景
2005年4月27日提交的国际专利公开号wo2005/104745(glaxogrouplimited)公开了毒蕈碱乙酰胆碱受体拮抗剂。具体而言,wo2005/104745公开了式(i)的4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物,以及制备这种化合物的方法(实施例84):
4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物还可以称为芜地溴铵(umeclidiniumbromide)。
2010年9月10日提交的国际专利公开号wo2011/029896(glaxogrouplimited)公开了芜地溴铵(umeclidiniumbromide)的多步合成中的早期中间体1-氮杂双环[2.2.2]辛烷-4-甲酸乙酯的替代性制备方法。
存在对制备芜地溴铵(umeclidiniumbromide)的替代性方法的需要。具体而言,期望提供相对于先前公开在wo2005/104745和wo2011/029896中的那些的优点的方法。优点可以包括但不局限于:安全性、控制(即,对最终产物形式和物理特性的控制)、产率、可操作性、处理、规模可伸缩性和效率上的改善。
本发明概述
在第一个方面,本发明提供了制备芜地溴铵(umeclidiniumbromide)的方法,所述方法包括:
a)使式(ii)的((2-溴乙氧基)甲基)苯
与式(iii)的二苯基(奎宁环-4-基)甲醇
在沸点高于大约90℃的偶极非质子溶剂中或在沸点高于大约80℃的醇中反应;和任选地
b)将步骤(a)的产物重结晶。
本发明进一步涉及用于制备式(iii)的化合物的中间体,并由此涉及用于制备芜地溴铵(umeclidiniumbromide)的中间体。相比于wo2005/104745和wo2011/029896的现有技术方法,本文公开的方法提供了许多优点。
附图的简要说明
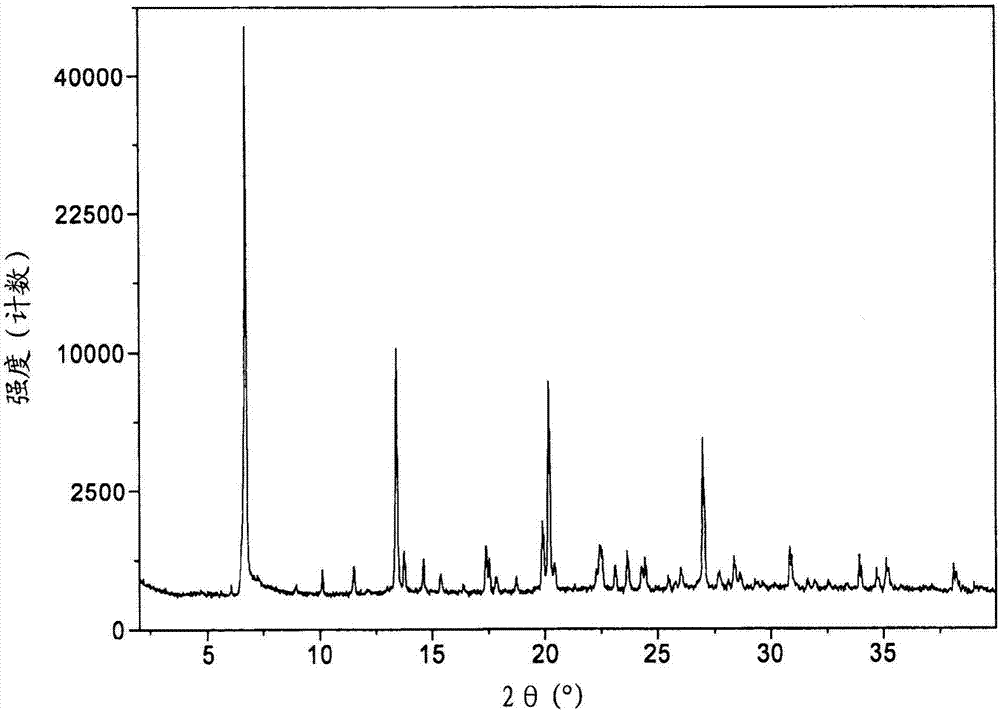
图1:实施例8制备的芜地溴铵(umeclidiniumbromide)的结晶形式1的xrpd数据。
图2:实施例7制备的芜地溴铵(umeclidiniumbromide)的结晶形式1的xrpd数据。
图3:实施例9制备的芜地溴铵(umeclidiniumbromide)的结晶形式2的xrpd数据。
图4:实施例10制备的芜地溴铵(umeclidiniumbromide)的结晶形式3的xrpd数据。
图5:实施例7至10制备的芜地溴铵(umeclidiniumbromide)的结晶形式1(有和没有加入晶种)、2和3的xrpd数据的叠加图。
图6:实施例8制备的芜地溴铵(umeclidiniumbromide)的结晶形式1的dsc热谱图。
图7:实施例9制备的芜地溴铵(umeclidiniumbromide)的结晶形式2的dsc热谱图。
图8:实施例10制备的芜地溴铵(umeclidiniumbromide)的结晶形式3的dsc热谱图。
本发明的详细说明
在第一个方面,本发明提供了制备芜地溴铵(umeclidiniumbromide)的方法,所述方法包括:
c)使式(ii)的((2-溴乙氧基)甲基)苯
与式(iii)的二苯基(奎宁环-4-基)甲醇
在沸点高于大约90℃的偶极非质子溶剂中或在沸点高于大约80℃的醇中反应;和任选地
d)将步骤(a)的产物重结晶。
在一方面,用于制备芜地溴铵(umeclidiniumbromide)的醇可以是,例如,丙醇或丁醇的任何异构体,例如正丙醇。或者,该反应可以在沸点高于90℃的偶极非质子溶剂,包括但不局限于:dmf、dma、dmso或nmp的存在下进行。
进行步骤(a)时的温度范围可以根据在所选择的溶剂中式(iii)的化合物的溶解度和所述溶剂的沸点加以确定。例如,在正丙醇中的反应可以在大约60℃和97℃之间进行。
芜地溴铵(umeclidiniumbromide)正作为用于治疗呼吸系统疾病,例如哮喘和copd的未溶剂化形式被开发。因此,需要制备这种形式的有效的、商业上可行的方法,而提供所述方法已经被证实是具有挑战性的,因为该化合物容易形成溶剂化物。迄今为止,已经鉴定了芜地溴铵(umeclidiniumbromide)的许多溶剂化物,包括甲醇、乙醇、异丙醇、异丁醇、氯苯和对二甲苯溶剂化物。已经意外地发现,使式(ii)和(iii)的化合物在正丙醇中反应,最小化了溶剂化物形成的风险,并由此免除了对先前需要的在乙酸乙酯、甲醇和水中重新形成浆液(wo2005/104745,实施例84的方法b)的需要。
与wo2005/104745的现有技术方法相比较,在正丙醇中进行该反应还导致了显著更高的转化率(43.3%对实施例5的~90%),并且由于从所述方法(wo2005/104745的实施例84)中排除了氯仿和乙腈,从而是更安全的。此外,还显著地减少了反应时间(由16小时减少到在正丙醇中的3小时)。
使用本领域中已知的标准方法,例如,冷却结晶或加入反溶剂进行结晶,可以将步骤(a)的产物重结晶(步骤(b))。在冷却结晶过程中,使含有溶解的步骤(a)的产物的反应混合物慢慢地冷却,并任选地加入晶种,导致芜地溴铵(umeclidiniumbromide)的晶体的形成,所述晶体将从溶液中分离出来。
意外的是,使用含水的正丙醇溶剂混合物进行重结晶(通过冷却结晶),能够稳定的控制芜地溴铵(umeclidiniumbromide)的最终未溶剂化的形式和物理特性。在本发明的进一步方面,在正丙醇水溶液中进行重结晶,其中水与正丙醇的比例是2:1。
结晶之后,可以通过过滤分离晶体,使用合适的溶剂如冷的正丙醇洗涤,并真空干燥。
在进一步方面,本发明提供了制备式(iii)的化合物的方法,所述方法包括:使在二丁醚中的苯基锂与式(iv)的化合物在合适的溶剂中反应
其中r1是c1-6烷基、芳基或苄基。
在进一步方面,本发明提供了制备芜地溴铵(umeclidiniumbromide)的方法,所述方法包括:制备式(iii)的化合物,其包括使在二丁醚中的苯基锂与式(iv)的化合物在合适的溶剂中反应的步骤,
其中r1是c1-6烷基、芳基或苄基,然后将式(iii)的化合物转化为芜地溴铵(umeclidiniumbromide)。
在一方面,r1是乙基。
本文使用的术语“烷基”是指具有具体数目的成员原子的饱和烃链。例如,c1-6烷基是指具有1至6个成员原子的烷基。烷基可以是直链的或支链的。代表性的支链烷基具有一个、两个或三个支链。烷基包括甲基、乙基、丙基(正丙基和异丙基)、丁基(正丁基、异丁基和叔丁基)、戊基(正戊基、异戊基和新戊基)和己基。
本文使用的术语“芳基”指的是苯基或萘基。芳基可以任选被一个或多个取代基取代,所述取代基例如卤素、c1-6烷基和c1-6烷氧基。
苯基锂和式(iv)的化合物之间的反应是在合适的溶剂中进行的,所述溶剂例如非质子溶剂,例如甲苯、thf、methf、tbme或烷烃,例如己烷和环己烷。在本发明的进一步方面,该反应是在作为溶剂的甲苯中和/或在0℃的温度下进行的。
由于使用的特定试剂和溶剂系统,wo2005/104745中概述的式(iii)的化合物的合成不得不在低温学温度(例如-30℃)下操作。因此,与本领域的已知方法相比,本文公开的方法有利地提供了改善的可操作性,并由此更适合于大规模的、商业化的生产。此外,反应时间被显著地减少了(例如,在0℃下在甲苯中由16小时减少至1小时)。
在制备式(iii)的化合物期间产生的反应副产物可以通过水性后处理来除去。例如,氢氧化锂可以通过下列方法除去:向反应混合物中加入水和极性高沸点的与水不相混溶的溶剂,例如,丁醇和戊醇的异构体,加热至适宜的温度,然后进行液-液萃取。在一方面,溶剂是正丁醇,并且温度可以为从79至85℃。认识到使用极性高沸点的与水不相混溶的溶剂(例如,正丁醇)可以除去无机副产物,这为该方法的下一个阶段提供了进一步的益处:在所述下一个阶段中意想不到地发现这些杂质的存在使反应减慢。由此,充分除去无机副产物,改善了该方法的效率。
在进一步方面,本发明提供了制备式(iv)的化合物的方法,所述方法包括:使式(v)的化合物与选自khmds、lihmds和nahmds的合适的碱在合适的溶剂中反应
其中r1是c1-6烷基、芳基或苄基,并且y是离去基团。
在进一步方面,本发明提供了制备芜地溴铵(umeclidiniumbromide)的方法,所述方法包括:制备式(iv)的化合物,其包括使式(v)的化合物与选自khmds、lihmds和nahmds的合适的碱在合适的溶剂中反应的步骤,
其中r1是c1-6烷基、芳基或苄基,并且y是离去基团,
然后将式(iv)的化合物转化为芜地溴铵(umeclidiniumbromide)。
在一方面,r1是乙基。
在一方面,碱是khmds。
y的示例性离去基团包括但不局限于:-ots、-oms、-otf、cl或br。在一方面,y是cl。
制备式(iv)的化合物的合适的溶剂包括但不局限于:非质子溶剂,例如甲苯、thf、methf、tbme或烷烃,例如己烷和环己烷。在一方面,溶剂是甲苯。
wo2005/104745中概述的式(iv)的化合物的制备,涉及式(v)的化合物与lda(碱)在thf中的反应,且这种方法需要再一次使用低温学温度(-50℃)。本文所描述的方法条件意外地允许反应在大约50℃下充分进行。用例如khmds替代lda,以及将溶剂由thf更换为例如甲苯,也会有利地导致反应时间的减少(在紧接着的上面概述的条件下由16小时减少至大约1小时)。
在进一步方面,本发明提供了制备式(v)的化合物的方法,所述方法包括:
a)在合适的碱的存在下,使式(vi)的化合物
其中r1是c1-6烷基、芳基或苄基,
与式(vii)的化合物反应,
其中x是离去基团;和
b)用合适的试剂将步骤(a)的产物转化为式(v)的化合物。
在进一步方面,本发明提供了制备芜地溴铵(umeclidiniumbromide)的方法,所述方法包括:制备式(v)的化合物,其包括:
a)在合适的碱的存在下,使式(vi)的化合物,
其中r1是c1-6烷基、芳基或苄基,
与式(vii)的化合物反应
其中x是离去基团,和
b)用合适的试剂将步骤(a)的产物转化为式(v)的化合物;然后将式(v)的化合物转化为芜地溴铵(umeclidiniumbromide)。
在一方面,x是cl或br和/或r1是乙基。
反应的步骤(a)是在合适的碱的存在下进行的,所述碱包括但不局限于碳酸钾和dbu。在本发明的进一步方面,提供了氯乙醇(x是cl)在dbu的存在下的组合,其提供了可接受的反应速率和极好的产率。
步骤(b)的转化采用合适的试剂进行,所述试剂例如选自亚硫酰氯、磺酰卤如磺酰氯、磺酰基酸酐(sulfonylanhydrides)和卤化磷的试剂。
在一方面,步骤(b)中的试剂选自:亚硫酰氯、对甲苯磺酰氯、甲磺酰氯、甲磺酸酐、对甲苯磺酸酐、三氟甲磺酸酐、磷酰氯、三溴化磷和五氯化磷。
在进一步方面,该试剂是亚硫酰氯。
与wo2005/104745中概述的方法相比,这种两步方法可以以显著更高的产率有利地提供式(v)的化合物。例如,使用溴乙醇和碳酸钾作为试剂,本文公开的方法提供大约80%的转化(参见实施例1)。与此相反,wo2005/104745的方法提供38.6%的产率(实施例1:1-(2-氯乙基)-4-哌啶甲酸乙酯)。
wo2005/104745中概述的低产率主要是二聚体杂质1,1'-(乙烷-l,2-二基)双(哌啶-4-甲酸二乙酯)形成的结果,随后必须通过色谱法从感兴趣的化合物中分离出这种杂质,从而增加了对高毒性的式(v)的化合物的暴露。本发明的方法排除了这种主要二聚体杂质的形成,避免了对高暴露分离阶段的需要,并由此提供了安全得多的替代方案。
wo2011/029896概述了经由4-(2-氯乙基)-哌啶-4-甲酸乙酯(参考实施例9)来制备奎宁环-4-甲酸乙酯(式(iv)的化合物)的替代方法,这种方法避免了形成高毒性的中间体(式(v)的化合物)。然而,这种替代性的制备也导致了非常低的产率(即,前体向4-(2-氯乙基)-哌啶-4-甲酸乙酯的产率为1.71-45.56%,参考实施例1-7,wo2011/029896)。
在制备式(v)的化合物中的两个步骤都在合适的溶剂(例如,甲苯)的存在下方便地进行。该反应的每个步骤都可以使用不同的溶剂,或溶剂混合物。
从式(vi)的化合物起始来制备式(iv)的化合物可以在一系列单独的反应中进行(由此分离每个中间体),或可以作为套叠式合成方法(telescopicsynthesis)来进行。
芜地溴铵(umeclidiniumbromide)以许多结晶形式存在,可以使用许多常规分析技术来表征和区分它们,所述分析技术包括但不限于:x射线粉末衍射(xrpd)、红外光谱(ir)、拉曼光谱、示差扫描量热法(dsc)、热重分析法(tga)和固态核磁共振(ssnmr)。
在一方面,本发明提供了结晶固态形式的芜地溴铵(umeclidiniumbromide)。
在进一步方面,提供了芜地溴铵(umeclidiniumbromide)的结晶固态形式,其特征在于基本上如图1、2、3或4所示的x射线粉末衍射(xrpd)图,和/或具有在表1中所示的2θ值处的显著的衍射峰。
在进一步方面,本发明涉及芜地溴铵(umeclidiniumbromide)的结晶固态形式,其特征在于具有在6.7、8.9、10.1、11.5、13.4、13.8、14.6、15.4、17.5、17.9、18.7、19.9、20.2、22.6、23.1、24.3、24.4和/或27.0的2θ值(±0.10°2θ实验误差)处的衍射峰的x射线粉末衍射图(1型)。
在进一步方面,本发明涉及芜地溴铵(umeclidiniumbromide)的结晶固态形式,其特征在于具有在6.6、7.8、10.7、13.4、14.0、14.9、16.4、19.7、20.1、20.7、20.9、21.4和/或25.6的2θ值(±0.10°2θ实验误差)处的衍射峰的x射线粉末衍射图(2型)。
在进一步方面,本发明涉及芜地溴铵(umeclidiniumbromide)的结晶固态形式,其特征在于具有在6.9、9.3、12.5、12.9、16.1、16.7、17.9、18.5、19.4、20.1、20.9、23.3和/或25.1的2θ值(±0.10°2θ实验误差)处的衍射峰的x射线粉末衍射图(3型)。
通过示差扫描量热法(dsc)图,进一步表征芜地溴铵(umeclidiniumbromide)的结晶形式(1型、2型和3型)。在进一步方面,本发明涉及芜地溴铵(umeclidiniumbromide)的结晶固态形式,其特征在于具有大约236℃(1型,没有加入晶种)、232℃(2型)和232℃(3型)的出峰温度的dsc图。
实验部分
缩写
dmf:二甲基甲酰胺
dma:二甲基乙酰胺
dmso:二甲亚砜
nmp:n-甲基-2-吡咯烷酮
khmds:双(三甲基甲硅烷基)氨基钾
lda:二异丙基氨基锂
lihmds-双(三甲基甲硅烷基)氨基锂
nahmds-双(三甲基甲硅烷基)氨基钠
dbu-1,8-二氮杂双环(5.4.0)十一-7-烯;
thf-四氢呋喃
methf-甲基四氢呋喃
tbme-甲基叔丁基醚
在下述实施例中举例说明本发明。
实施例1:1-(2-氯乙基)-4-哌啶甲酸乙酯的制备
将在甲苯(4000ml)中的异哌啶酸乙酯(400.1g)、碳酸钾粉末(448.7g)和2-溴乙醇(256ml)加热至回流(大约110℃),并搅拌160分钟。将该反应混合物冷却至60℃,并加入水(1200ml),然后冷却至20℃。搅拌之后,分离水层,并用甲苯(2000ml)萃取。通过真空蒸馏,将合并的有机层浓缩至4200ml。将该反应混合物的一部分(4050ml)加热至50℃,并加入亚硫酰氯(193ml)。搅拌1小时之后,在40℃加入乙酸乙酯(4600ml),然后加入1-(2-氯乙基)-4-哌啶甲酸乙酯晶种(0.4g)。将该浆液老化35分钟,然后冷却至20℃,并老化40分钟。过滤出产物,用乙酸乙酯(1500ml)洗涤,在45℃下真空干燥,得到白色固体(502.7g,80%)。ei-msm/z220(m+h+)rt(2.1min)。
1hnmr(400mhz;dmso-d6):4.14-4.01(4h,m),3.57-3.42(4h,m),3.01(2h,m),2.59(1h,m),2.05(2h,m),1.88(2h,m),1.19(3h,t)。
实施例2:1-氮杂双环[2.2.2]辛-4-基(二苯基)甲醇的制备
将1-(2-氯乙基)-4-哌啶甲酸乙酯(199g,例如,按照实施例1制备)(在水(800ml)中)加入到甲苯(2000ml)和碳酸钾(118g)(在水(800ml)中)中,并用水(400ml)冲洗。搅拌该混合物,直到获得双相溶液为止。分离水层,并用甲苯(3000ml)萃取。通过真空蒸馏,将合并的有机层浓缩至4000ml。在40℃,将此溶液加入到0.5m在甲苯(1700ml)中的六甲基二甲硅烷基氨基钾和甲苯(2000ml)中。加入乙酸(178ml),通过真空蒸馏,将该混合物浓缩至4000ml,并加入到18%w/w碳酸钾水溶液(2432g)中。分离各层,通过真空蒸馏,将有机层浓缩至3000ml,并加入甲苯(1000ml)。将该溶液冷却至-15℃,并加入2m苯基锂(在二丁醚(800ml)中)。加入水(2000ml)和正丁醇(700ml),并将该混合物加热至75℃。除去水层,并将有机层用水(1000ml)洗涤。加入甲苯(1000ml),蒸馏该混合物,直到除去3000ml溶剂为止,然后冷却至20℃,并搅拌过夜。过滤出产物,用甲苯(2×200ml)洗涤,并在40℃下真空干燥,得到白色固体(131g,57%)。ei-msm/z294(m+h+)rt(3.6min)。
1hnmr(400mhz;meod):7.55(4h,m),7.27(4h,m),7.18(2h,m),2.84(6h,m),1.83(6h,m)。
实施例3:1-氮杂双环[2.2.2]辛-4-基(二苯基)甲醇的制备
将在甲苯(2700ml)中的异哌啶酸乙酯(300g)、碳酸钾粉末(330g)和2-溴乙醇(150ml)在dean&stark条件下回流4小时。将该反应混合物冷却至20℃,并加入水(900ml)。搅拌之后,分离水层,并用甲苯(1500ml)萃取。通过真空蒸馏,将合并的有机层浓缩至2700ml。将该反应混合物加热至60℃,并加入亚硫酰氯(150ml)。搅拌90分钟之后,将该混合物冷却至20℃,搅拌30分钟,并加入甲苯(1800ml)。加入水(900ml)和26%w/w碳酸钾水溶液(2028g)。分离各层,并将水层用甲苯(7500ml)萃取。将合并的有机层用水(300ml)洗涤,通过加入甲苯(3000ml)和浓缩至4800ml(通过真空蒸馏)进行干燥。在40℃,加入0.5m在甲苯(4200ml)中的六甲基二甲硅烷基氨基钾,并将该混合物搅拌1小时。加入乙醇(192ml)和乙酸(426ml),并将该混合物在40℃下搅拌2小时。加入26%w/w碳酸钾水溶液(4038g),并分离各层。用甲苯(2500ml)萃取水层。将合并的有机层用水(300ml)洗涤,并通过真空蒸馏浓缩至4500ml。将该溶液冷却至0-5℃,并加入2m在二丁醚(1920ml)中的苯基锂。一个小时之后,加入水(1500ml)和正丁醇(1680ml),并将该混合物加热至78℃。除去水层,并将有机层用水(1500ml)洗涤。通过在加入甲苯的情况下蒸馏,将有机相浓缩至6000ml,然后冷却至20℃,并搅拌过夜。过滤出产物,用甲苯(2×600ml)洗涤,并真空干燥,得到白色固体(300g,53%)。ei-msm/z294(m+h+)rt(3.7min)。
1hnmr(400mhz;meod):7.54(4h,m),7.26(4h,m),7.17(2h,m),2.83(6h,m),1.82(6h,m)。
实施例4:1-氮杂双环[2.2.2]辛-4-基(二苯基)甲醇的制备
将异哌啶酸乙酯(600g)和dbu(600ml)溶于甲苯(3000ml)中,并加热到100℃。用2小时加入2-氯乙醇(330ml),并将该混合物在109℃下进一步搅拌4.5小时。将温度调节至55-65℃,并加入亚硫酰氯(378ml)。搅拌1小时之后,将该混合物冷却至30℃,并加入水(1800ml)和40%w/w碳酸钾水溶液(3350g)。分离各层,并将水层用甲苯(3000ml)萃取。将合并的有机层用水(600ml)洗涤,通过加入甲苯(2000ml)和浓缩至6000ml(通过真空蒸馏)进行干燥。溶液测定显示从异哌啶酸乙酯的94%的转化。在45-50℃,加入0.5m在甲苯(8400ml)中的六甲基二甲硅烷基氨基钾,并将该混合物搅拌2小时。进一步加入0.5m在甲苯(1260ml)中的六甲基二甲硅烷基氨基钾,并将该混合物搅拌30分钟。加入乙醇(390ml),并通过真空蒸馏将该混合物浓缩至8400ml。加入乙酸(850ml),并将该混合物在45℃下搅拌15小时。加入26%w/w碳酸钾水溶液(8110g),并分离各层。用甲苯(4800ml)萃取水层。将合并的有机层分为两个半份,使用助滤剂(38gcelite或harborlite)过滤,重新合并,并通过真空蒸馏浓缩至6000ml。将该溶液冷却至0-5℃,并加入2m在二丁醚(3840ml)中的苯基锂。一个小时之后,加入水(3000ml)和正丁醇(3960ml),并将该混合物加热至83℃。除去水层,并将有机层用水(3000ml)洗涤。通过在加入甲苯的情况下蒸馏,将有机相浓缩至12000ml,然后冷却至20℃,并搅拌过夜。过滤出产物,用甲苯(2×1200ml)洗涤,并在70℃下真空干燥,得到白色固体(561g,50%)。ei-msm/z294(m+h+)rt(3.7min)。
1hnmr(400mhz;dmso-d6):7.51(4h,m),7.25(4h,m),7.15(2h,m),2.65(6h,m),1.60(6h,m)。
实施例5:4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物(中间体等级-没有加入晶种的步骤)的制备
将1-氮杂双环[2.2.2]辛-4-基(二苯基)甲醇(31.7kg)和苄基2-溴乙基醚(25.7kg)在正丙醇(257.5kg)中的溶液回流13小时。用不少于一个小时将该溶液冷却至50-55℃,搅拌40分钟,以诱导结晶。用不少于1小时将该浆液冷却至17-23℃,并搅拌60分钟。然后用不少于1小时将该浆液冷却至0-5℃,并老化2小时。过滤出产物,用正丙醇洗涤两次(34.8kg和33.8kg)。在50℃下真空干燥,得到白色固体(47.95kg,87%)。
实施例6:4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物(中间体等级)的制备
将1-氮杂双环[2.2.2]辛-4-基(二苯基)甲醇(445.6g,例如,按照实施例4制备)和苄基2-溴乙基醚(360.9g)在正丙醇(4456ml)中的溶液回流3小时。将该溶液冷却至87℃,加入芜地溴铵(umeclidiniumbromide)晶种(1型)(0.44g),进一步冷却至82℃,并老化1小时。用2.5小时将该浆液冷却至0-5℃,并老化1小时。过滤出产物,并用正丙醇(2×900ml)洗涤。在50℃下真空干燥,得到白色固体(690g,89%)。ei-msm/z428(m+)rt(4.7min)。
1hnmr(400mhz;dmso-d6):7.57(4h,d),7.40-7.30(9h,m),7.26(2h,t),5.94(1h,s),4.52(2h,s),3.84(2h,m),3.49(6h,t),3.38(2h,m),2.02(6h,t)。
实施例7:4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物的1型晶体的制备
在80℃,将4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物(165g)溶于正丙醇(495ml)和水(990ml)中。将得到的溶液冷却至50℃,加入1型晶种(0.825g)(在正丙醇(2.8ml)中),并进一步用正丙醇(5.5ml)冲洗。在50℃老化一小时之后,用80分钟将该浆液冷却至40℃,然后用105分钟进一步冷却至0-5℃。将浆液样品在0-5℃老化3小时,过滤,并将产物用正丙醇(2×330ml)洗涤。在60℃下真空干燥,得到白色固体(146g,88%)。通过xrpd进行表征(参见图2和表1)。
实施例8:4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物的1型晶体的制备(实施例6的替代方案,没有加入晶种的步骤)
在80℃,将4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物(20g)溶于正丙醇(60ml)和水(120ml)中,并进行澄清。将得到的溶液冷却至45℃,并老化2小时。形成稠浆液,用3小时将其冷却至0-5℃。将浆液样品在0-5℃下老化1小时,过滤,并将产物用正丙醇(2×40ml)洗涤。在50℃下真空干燥,得到白色固体(16g,80%)。通过xrpd(参见图1和表1)和dsc(参见图6)进行表征。
实施例9:4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物的2型晶体的制备
在21℃,将硝基甲烷(105ml)和正丙醇(45ml)加入到4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物(6g)中。将得到的混合物在室温(大约21℃)下搅拌30.5小时。使用玻璃漏斗和滤纸,在重力下,过滤该浑浊的混合物。在真空条件下,将该澄清溶液放置在旋转蒸发器上(6-7mbar)达15分钟,并将所得到的白色固体在50℃下真空干燥。产率:5.8g(97%)。通过xrpd(参见图3和表1)和dsc(参见图7)进行表征。
实施例10:4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物的3型晶体的制备
在21℃,将二氯甲烷(105ml)和1-戊醇(45ml)加入到4-[羟基(二苯基)甲基]-1-{2-[(苯基甲基)氧基]乙基}-1-氮鎓双环[2.2.2]辛烷溴化物(6g)中。将得到的混合物在室温(大约21℃)下搅拌30小时。使用玻璃漏斗和滤纸,在重力下,过滤该浑浊的混合物。在真空条件下,将该澄清溶液放置在旋转蒸发器上(6-7mbar)达20分钟,直到获得混悬液为止。过滤收集白色固体,并在50℃下真空干燥。产率:5.1g(85%)。通过xrpd(参见图4和表1)和dsc(参见图8)进行表征。
仪器参数
lc-ms实验条件
柱:5cm×2.1mm,3μm,luna(c18)
移动相:水/乙腈+0.05%v/vtfa。
0%至95%乙腈,历时8分钟。
总运行时间:10分钟。
流速:1.0ml/min
柱温:40℃
质量范围:100至1000da
波长范围:205至400nm
1hnmr
在meod或dmso-d6中,在brukerdpx400,400mhz仪器上记录1hnmr谱。
x射线粉末衍射(xrpd)
在配备有x'celerator检测器的panalyticalx'pertpro粉末衍射仪上获取xrpd数据。获取条件是:辐射:cukα,发电机电压:40kv,发电机电流:45ma,起始角度:2.0°2θ,终止角度:40.0°2θ,步长:0.0167°2θ。每个步长的时间是31.750s。如下制备样品:将几毫克样品固定在零背景si薄片上,得到粉末的薄层。特征峰位置和相应的d-间距汇总在表1中。使用panalyticalhighscore软件,由原始数据计算这些数据。峰位置的实验误差为大约±0.10°2θ。相对峰强度将由于优先取向而改变,并因此没有对其记录。
表1:芜地溴铵(umeclidiniumbromide)的三个固态形式的特征性的xrpd峰位置。突出显示的峰是每种形式的独特的峰。
示差扫描量热法(dsc)
使用tainstrumentsq2000量热计,获得dsc热谱图。将样品称到铝盘中;将盘盖放在顶部,并轻轻地压褶而并非密封该盘。使用10℃min-1的加热速率,进行该实验。
- 还没有人留言评论。精彩留言会获得点赞!