一种噻吩‑咔唑‑噻吩衍生物及其制备方法与应用与流程
(一)技术领域
本发明涉及一种噻吩-咔唑-噻吩衍生物及其制备方法,以及将所述噻吩-咔唑-噻吩衍生物电聚合成膜并作为一种微孔结构材料在电致变色等领域的应用。
(二)
背景技术:
随着全球能源的急剧消耗和环境的不断恶化。节能环保材料吸引了人们的广泛关注。电致变色材料正是这样一种可以改变人类生产生活方式并且有助于合理利用能源的新型功能材料。该材料具有来源丰富、器件制备工艺简单及工作电压低等优点,且能广泛应用在智能窗、电子纸及显示器等领域。导电聚合物基电致变色材料因结构易修饰、着色效率较高、响应时间较短、光学对比度较高及较丰富的颜色变换而成为电致变色材料中最具有潜力的一类物质。
早期的研究工作主要集中在无机电致变色材料,如三氧化钨、二氧化铱等。无机电致变色材料具有良好的光化学稳定性和快速响应速度,但其加工成本较高、颜色种类单调、着色效率低下,限制了其在生产生活中的大规模应用。有机电致变色材料由于可以弥补无机变色材料的不足,已经成为人们研究的重点。相对于有机小分子变色材料的种类较少、分子结构不易修饰,导电聚合物电致变色(pec)材料则由于具有结构种类繁多、变色范围宽广、对比度高、加工性能好、能带可控以及响应速度快等优点而备受人们的关注,被认为是下一代ec材料的发展方向之一;而聚噻吩类导电聚合物,由于结构易修饰、合成相对简单等优点,是目前研究最广泛的一类电致变色材料。
聚噻吩类导电聚合物中,噻吩作为两侧基团,中间具有不同中心核的单体结构分子受到了研究者的广泛关注;本发明设计合成了一类新型的噻吩-咔唑-噻吩结构衍生物单体,将其电化学聚合成膜,系统地研究了该类化合物的电致变色性能。
(三)
技术实现要素:
本发明的目的是设计合成新型的基于噻吩-咔唑-噻吩结构的化合物,并将所述化合物电聚合成膜作为一种微孔结构材料应用于电致变色等领域。
本发明的技术方案如下:
如式(i-a)或式(i-b)所示的噻吩-咔唑-噻吩衍生物:
式(i-a)或式(i-b)中,o、p各自独立为:1、2或3。
本发明还提供了所述式(i-a)或式(i-b)所示的噻吩-咔唑-噻吩衍生物的制备方法。
a:所述式(i-a)所示的噻吩-咔唑-噻吩衍生物的制备方法为:
(1)氮气保护下,将3,6-二溴咔唑(iii)、2-噻吩硼酸(v-a)、四(三苯基磷)钯、碳酸钠溶解于四氢呋喃/甲苯体积比1:1~2混合溶剂中,于90~110℃下反应12~48h,之后反应液经后处理,得到中间化合物(vii-a);
所述3,6-二溴咔唑(iii)、2-噻吩硼酸(v-a)、四(三苯基磷)钯、碳酸钠的投料物质的量之比为1:2~6:0.05~0.1:1~4;
所述碳酸钠以2mol/l的水溶液形式进行投料;
所述四氢呋喃/甲苯混合溶剂的体积用量以3,6-二溴咔唑(iii)的质量计为30~50ml/g;
所述反应液后处理的方法为:反应结束后,待反应液冷却至室温(20~30℃,下同),加入水和二氯甲烷进行萃取,收集有机相,经无水硫酸镁干燥,减压浓缩,再进行柱层析纯化,以300~400目硅胶为固定相,二氯甲烷/石油醚体积比1:3~10混合液为流动相进行洗脱,收集含目标化合物的洗脱液,减压蒸除溶剂并干燥,得到中间化合物(vii-a);
(2)氮气保护下,将中间化合物(vii-a)、1,4-二溴苯衍生物(vi)、碘化亚铜、无水磷酸钾、反式1,2-环己二胺和甲苯混合,升温至100~110℃反应12~24h,之后反应液经后处理,得到产物(i-a);
式(vi)中,n为1、2或3;
所述中间化合物(vii-a)、1,4-二溴苯衍生物(vi)、碘化亚铜、无水磷酸钾、反式1,2-环己二胺的投料物质的量之比为1:0.3~0.5:0.04~0.05:1~1.1:1~1.6;
所述甲苯的体积用量以中间化合物(vii-a)的质量计为15~20ml/g;
所述反应液后处理的方法为:反应结束后,待反应液冷却至室温,用饱和碳酸铵水溶液淬灭,二氯甲烷萃取,收集有机相,经无水硫酸镁干燥,减压浓缩,再进行柱层析纯化,以300~400目硅胶为固定相,石油醚/二氯甲烷体积比1:1~3混合液为流动相进行洗脱,收集含目标化合物的洗脱液,减压蒸除溶剂并干燥,得到产物(i-a)。
b:将制备方法a中的2-噻吩硼酸(v-a)替换为2-(3,4-乙烯二氧)噻吩硼酸(v-b),其余不变,则步骤(1)得到中间化合物(viii-b),最终得到产物(ii-b)。
本发明通过核磁共振(nmr)、气质联用(gc-ms)表征了目标产物。
本发明所述的噻吩-咔唑-噻吩衍生物可作为单体用于电化学聚合成膜,所述膜可按如下方法进行制备:
将式(i-a)、式(i-b)所示的噻吩-咔唑-噻吩衍生物中的一种或两者任意比例的混合物作为单体,溶解于乙腈/二氯甲烷体积比1:7~9的混合溶剂中,加入四丁基高氯酸铵(tbap)作为电解质,采用1.2v恒电位或0~1.2v循环伏安,聚合扫速为100mv/s,聚合电量为0.02c,电聚合得到薄膜;
所述乙腈/二氯甲烷混合溶剂的体积用量以单体的质量计为1~3ml/mg;
所述四丁基高氯酸铵的用量以乙腈/二氯甲烷混合溶剂的体积计为0.1mol/l。
采用电化学工作站660e,紫外-可见吸收光谱表征了所得膜的电化学及光学性能。本发明制得的薄膜具有良好的氧化还原性质,在1100nm处的光学对比度为40%~60%,响应时间为0.5~3.5s,660nm处的光学对比度为30%~50%,响应时间为0.2~3s,同时该类材料具有良好的电化学循环稳定性。本发明制得的薄膜具有较大的比表面积,且具备电致变色性能,可应用于电致变色、超级电容器等方面。
本发明的有益效果在于:采用本发明方法合成噻吩-咔唑-噻吩衍生物,最终产物的产率提高了20%,并且由本发明噻吩-咔唑-噻吩衍生物聚合得到的薄膜可应用于电致变色领域,具有快速的响应时间,合理的光学对比度,良好的电化学稳定性等优异的性能,是潜在的电致变色材料。
(四)附图说明
图1:本发明实施例1~6中化合物的合成路线及化学结构;
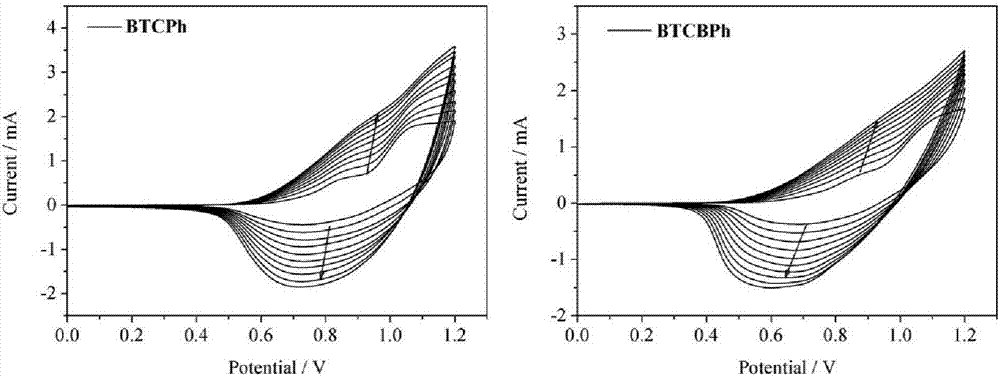
图2:本发明实施例7中薄膜pbtcph和pbtcbph的cv聚合曲线;
图3:本发明实施例7中薄膜pbtcph和pbtcbph不同电压下的紫外-可见吸收光谱;
图4:本发明实施例7中薄膜pbtcph的光学对比度;
图5:本发明实施例7中薄膜pbtcbph的光学对比度;
图6:本发明实施例7中薄膜pbtcbph和pbtcbph的响应时间。
(五)具体实施方式
下面以具体实施例对本发明的技术方案作进一步说明,但本发明的保护范围不限于此。
实施例14,4'-二(3,6-二噻吩-9氢-咔唑-9)-1,1'-联苯的合成
(1)在氮气保护下,将3,6-二溴咔唑(1.43g,3mmol),2-噻吩硼酸(0.896g,7mmol),四(三苯基磷)钯(0.2mmol)和碳酸钠(2.0m,5ml)溶解在四氢呋喃(20ml)和甲苯(30ml)混合溶剂中,体系回流反应48小时。待体系冷却后用去离子水和二氯甲烷萃取,所得有机相加入无水mgso4干燥后,减压浓缩,再用柱层析分离提纯,固定相为300-400目硅胶,流动相为二氯甲烷/石油醚(体积比1:3),最后得到白色固态的中间体产物3,6-二噻吩咔唑化合物0.68g,产率为68%。1hnmr(500mhz,cdcl3)δ:8.35(s,2h),8.12(s,h),7.73(m,2h),7.45(m,2h),7.38(m,2h),7.29(m,2h),7.14(t,2h).ms(ei)理论值(c20h13ns2)m/z:331.05,实测值:331.2。
(2)氮气保护下,将化合物3,6-二噻吩咔唑(331.05g/mol,5mmol,1.6552g)、1,4-二溴联苯(309.90g/mol,2mmol,0.6198g)、碘化亚铜(0.2mmol,0.038g)、无水磷酸钾(5mmol,1.061g)加入到100ml两口烧瓶中,加入反式1,2-环己二胺(840mg,7mmol),甲苯30ml,温度升至110℃,反应24h。反应结束后,待体系冷却至室温,用饱和碳酸铵水溶液淬灭反应,二氯甲烷萃取,合并有机相,以无水硫酸镁干燥过夜,抽滤除去干燥剂后,将有机相蒸出,重新加入二氯甲烷溶解,加入粗硅胶拌样,以层析法过柱,洗脱剂为石油醚与二氯甲烷混合溶液(体积比,1:1),最终得到白色目标产物0.64g,产率为40%。确认物质的表征结构如下:1hnmr(500mhz,cdcl3)δ8.44(d,j=1.4hz,4h),7.98(d,j=8.4hz,4h),7.80–7.72(m,8h),7.53(d,j=8.5hz,4h),7.42(dd,j=3.5,1.0hz,4h),7.32(dd,j=5.1,1.0hz,4h),7.16(dd,j=5.1,3.5hz,4h)。c52h32n2s4的ms(ei)数据表征实测为812.31。
实施例24,4'-二(3,6-二噻吩-9氢-咔唑-9)-1,1'-苯的合成
合成方法同实施例1,不同之处在于,步骤(2)中,将1,4-二溴联苯替换为1,4-二溴苯(235.90g/mol,2mmol,0.4718g),最终得到目标产物0.62g,产率为42%。
4,4'-二(3,6-二噻吩-9氢-咔唑-9)-1,1'-苯c46h28n2s4的ms(ei)表征实测为736.11。
实施例34,4'-二(3,6-二噻吩-9氢-咔唑-9)-1,1'-三联苯的合成
合成方法同实施例1,不同之处在于,步骤(2)中,将1,4-二溴联苯替换为1,4-二溴三联苯(388.10g/mol,2mmol,0.7762g),最终得到目标产物0.55g,产率为31%。
4,4'-二(3,6-二噻吩-9氢-咔唑-9)-1,1'-三联苯c58h36n2s4的ms(ei)表征实测为888.01。
实施例44,4'-二(3,6-二乙烯二氧噻吩-9氢-咔唑-9)-1,1'-联苯的合成
(1)在氮气保护下,将3,6-二溴咔唑(1.43g,3mmol),2-(3,4-乙烯二氧噻吩)硼酸(1.3g,7mmol),四(三苯基磷)钯(0.2mmol)和碳酸钠(2.0m,5ml)溶解在(20ml)和甲苯(30ml)混合溶剂中,体系回流反应48小时。待体系冷却后用去离子水和二氯甲烷萃取,所得有机相加入无水mgso4干燥后,减压浓缩,再用柱层析分离提纯,固定相为300-400目硅胶,流动相为二氯甲烷/石油醚(体积比1:3),最后得到白色固态的中间体产物3,6-二(3,4-乙烯二氧噻吩)咔唑化合物,产率为72%。1hnmr(500mhz,cdcl3)δ:10.1(s,1h),7.77(s,2h),7.46(dd,2h),7.30(dd,2h),5.9(s,2h),4.36(dd,4h).ms(ei):理论值(c20h13ns2)m/z:447.53,实测值:447.01。
(2)氮气保护下,将化合物3,6-二(3,4-乙烯二氧噻吩)咔唑(331.05g/mol,5mmol,1.6552g)、1,4-二溴联苯(309.90g/mol,2mmol,0.6198g)、碘化亚铜(0.2mmol,0.038g)、无水磷酸钾(5mmol,1.061g)加入到100ml两口烧瓶中,加入反式1,2-环己二胺(840mg,7mmol),甲苯30ml,温度升至110℃,反应24h。待体系冷却至室温,用饱和碳酸铵水溶液淬灭反应,二氯甲烷萃取,合并有机相,以无水硫酸镁干燥过夜,抽滤除去干燥剂后,将有机相蒸出,重新加入二氯甲烷溶解,加入粗硅胶拌样,以层析法过柱,洗脱剂为石油醚与二氯甲烷混合溶液(体积比,1:1),最终得到白色目标产物0.90g,产率为43%。确认物质的表征结构如下:1hnmr(500mhz,cdcl3)δ:7.77(s,2h),7.46(dd,2h),7.30(dd,2h),5.9(s,2h),4.36(dd,4h).ms(ei)表征实测为为1044.31。
实施例54,4'-二(3,6-二乙烯二氧噻吩-9氢-咔唑-9)-1,1'-苯的合成
合成方法同实施例4,不同之处在于,步骤(2)中,将1,4-二溴联苯替换为1,4-二溴苯(235.90g/mol,2mmol,0.4718g),最终得到目标产物0.89g,产率为46%。
4,4'-二(3,6-二乙烯二氧噻吩-9氢-咔唑-9)-1,1'-苯c54h36n2o8s4的ms(ei)表征实测为968.14。
实施例64,4'-二(3,6-二乙烯二氧噻吩-9氢-咔唑-9)-1,1'-三联苯的合成
合成方法同实施例4,不同之处在于,步骤(2)中,将1,4-二溴联苯替换为1,4-二溴三联苯(388.10g/mol,2mmol,0.7762g),最终得到目标产物0.76g,产率为34%。
4,4'-二(3,6-二乙烯二氧噻吩-9氢-咔唑-9)-1,1'-三联苯c66h44n2o8s4的ms(ei)表征实测为为1121.31。
实施例7
7.36mg4,4’-二(3,6-二噻吩-9氢-咔唑-9)-1,1’-苯(btcph)、8.13mg4,4’-二(3,6-二噻吩-9氢-咔唑-9)-1,1’-联苯(btcbph)各自溶解于二氯甲烷/乙腈溶液(体积比9:1)中,分别加入0.3419g四丁基高氯酸铵(tbap)作为电解质,并定容至10ml,超声3min,直至完全溶解,采用恒电位1.2v或循环伏安法0-1.2v电化学聚合成膜,在tbap/mecn中脱掺杂1min用于性能测试,所有电化学测试均在tbap/mecn中进行测试。
循环伏安、光谱电化学及电致变色测试阶跃电压为0v-1.2v,测试结果表明,聚合物薄膜均具有很好的氧化还原性能,在1100nm处的光学对比度为40%-60%,响应时间为0.3s-3.5s,660nm处的光学对比度为30%-50%,响应时间为0.2s-3s,同时该类材料具有良好的电化学循环稳定性,这些实验结果表明该类化合物是一类非常潜在的电致变色材料。
附图2~6为pbtcph膜和pbtcbph膜的cv曲线、不同电压下的紫外可见吸收光谱、光学对比度及响应时间。与线性的噻吩-咔唑-噻吩类聚合物(比如n-硝基苯取代二噻吩咔唑聚合物)电致变色材料相比,这类材料经电化学聚合形成了交联的聚合物薄膜,表现出快速的响应速度(最快可至毫秒级),合理的光学对比度,很好的聚合物结构稳定性及电化学稳定性。
- 还没有人留言评论。精彩留言会获得点赞!
- 用于生产化学品及其衍生物的组合物和方法与制造工艺
- 7‑OH‑8‑[(1‑胺基‑1‑苯基衍生物)‑次甲基]‑黄酮化合物及其制备方法及其应用与制造工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 含D型非天然氨基酸的抗菌肽类似物及其合成与应用的制造方法与工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 一种白杨素氨基酸衍生物的制备方法与制造工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 联苯胺衍生物、其制造方法和电子照相感光体与制造工艺
- 包含环糊精衍生物的可注射美法仑组合物及其制备和使用方法与制造工艺
- 一种黄芪甲苷衍生物及其制备方法和应用与制造工艺
- (Z,E)5‑OH‑3‑(苯基衍生物‑次甲基)‑二氢黄酮‑7‑O‑糖苷及其制备及其应用的制造方法与工艺
- 喹唑啉衍生物的制造方法与工艺
- 吡唑衍生物及其作为大麻素受体介体的用途的制造方法与工艺
- Harrisotone A衍生物的组合物在防治肝脏损伤药物中的应用的制造方法与工艺
- 水生态处理中淤泥及衍生物的再利用处理系统的制造方法与工艺
- Schiglautone A衍生物的组合物用于制备抗缺氧药物的制作方法
- Schiglautone A衍生物的组合物用于制备抗红细胞低下性贫血药物的制作方法