一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法与流程
本发明涉及一种利用表面羧基修饰磁球固定化酶筛选芳香化酶抑制剂的方法,具体利用的技术为利用固定化芳香化酶,筛选潜在的芳香化酶抑制剂,属于酶学及酶工程领域。
背景技术:
芳香化酶(aromatase,又名cyp19)是体内雌激素合成的关键酶,属于细胞色素p450酶系中的一种,它在烟酰胺腺嘌呤二核苷酸磷酸(nadph)的帮助下,可以将雄烯二酮与睾酮的a环芳香化,分别转化为雌酮与雌二醇;在体内,雌激素能够促进乳腺癌细胞的增殖,芳香化酶是雌激素依赖型乳腺癌治疗的一个重要靶点,尤其是对于绝经后妇女,其体内的雌激素主要由周围组织的雄激素转化而来,使用芳香化酶抑制剂已经成为治疗的重要手段,一些药物已逐渐上升为一线药物,如:依西美坦,来曲唑等;但是其在带来治疗作用的同时,会对骨骼系统产生如骨丢失等副作用;因此,寻找高效、低毒的潜在芳香化酶抑制剂已经成为近年来的研究热点。
对已有芳香化酶抑制剂进行结构改造或者从天然产物中寻找潜在的活性化合物是芳香化酶抑制剂筛选的一种重要手段;最早的体外筛选体系中主要采用胎盘微粒体作为酶源,以雄烯二酮或者睾酮作为底物,通过直接或者间接的检测产物雌激素的量的变化来达到筛选目的。存在诸多问题:比如胎盘来源存在医学伦理问题;胎盘微粒体又是多种酶的混合物,存在着底物竞争问题,易造成假阳性结果。检测手段主要有薄层层析法、3h2o法和酶联免疫吸附法(elisa),也会存在一些问题,如薄层层析法操作复杂,3h2o法的放射性会给操作人员带来危害,elisa法成本太高。后来研究者们逐渐采用昆虫细胞表达的人源芳香化酶作为酶源,以dibenzylfluorescein(dbf)为底物,通过酶标仪来检测产物的荧光强度,进而获知抑制情况。近年来专利cn103149186b发明了一种利用均相时间分辨荧光技术实现对芳香化酶抑制活性进行高通量筛选,但是相对成本也很高。
上述以游离芳香化酶为酶源建立体外酶抑制剂筛选体系已经成为了科研工作的瓶颈,而固定化酶技术的使用相比直接使用游离酶来说具有诸多优势:比如极易与底物、产物分离;可以反复使用,降低了酶的成本;能够提高酶的稳定性等。酶的固定化方法有吸附法、共价键合法、交联法和包埋法。专利201610268351.x公开了一种利用环氧基修饰的多孔硅胶为载体采用吸附法固定化芳香化酶,实现芳香化酶的首次固定化。而采用共价键合的方式来固定化芳香化酶的研究至今未有人涉足;磁球作为新兴的载体应用于固定化酶中可以很方便的借助磁铁来回收固定化酶,具有方便、高效等优势,本发明采用表面羧基修饰的磁球利用共价键合法来固定化芳香化酶,利用亲和色谱原理来进行酶潜在抑制剂筛选,用阳性药物与阴性药物对筛选模型做了验证,后续采用该模型筛选雷公藤系列混合标准品溶液中潜在抑制剂,筛选出雷公藤红素与芳香化酶具有亲和作用,并用游离酶体外温孵体系测试出其半数抑制浓度(ic50)值。该模型可为新药开发提供可选择的工具。
技术实现要素:
本发明的目的是提供一种简便有效的方法固定芳香化酶,同时基于亲和色谱机理筛选潜在的抑制剂。本发明的方法是通过表面羧基修饰的磁球利用共价固定法使芳香化酶固定化及基于亲和作用去筛选潜在抑制剂。
一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法,方法包括如下步骤:
步骤一:合成载体的母体四氧化三铁微球,进行表面功能化修饰得表面羧基修饰磁球。
步骤二:利用步骤一中的表面羧基修饰磁球作为载体,固定化芳香化酶。
步骤三:利用高效液相色谱法检测温孵反应体系中的固定化芳香化酶酶活力。
步骤四:建立以液质联用法为检测手段的固定化芳香化酶抑制剂筛选模型。
步骤五:利用步骤四中的筛选模型,筛选芳香化酶抑制剂。
步骤六:抑制剂抑制效果的评价。
本发明的一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法,步骤一中利用溶剂热法合成四氧化三铁微球母体。母体表面包覆二氧化硅层的方法为stober法。硅烷偶联剂为3-氨丙基三乙氧基硅烷(aptes)或3-氨丙基三甲氧基硅烷(aptms)。羧基修饰剂为丁二酸酐或戊二酸酐。
本发明的一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法,步骤二中,固定化方法为碳二亚胺(edc)/n-羟基琥珀酰亚胺(nhs)共价固定法。羧基活化剂为edc·hcl和nhs或者edc·hcl和sulfo-nhs。edc与nhs的摩尔浓度比值为1∶2-3∶8。所用芳香化酶为原核细胞或者真核细胞表达系统表达所得,浓度为1-6mg/ml。磁球用量:酶量=1∶5-4∶5(mg∶μl),固定化温度为4℃-37℃,固定化时间2h-24h。
本发明的一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法,步骤三中,雄烯二酮浓度为8.75-35μmol/l;辅酶nadph终浓度为0.1-1mmol/l;固定化酶用量为0.5-4mg;恒温摇床温度为37-45℃;反应时间为20-60min。
本发明的一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法,步骤四中,采用液质联用法进行检测,以依西美坦和来曲唑为阳性对照药物,以雷公藤甲素为阴性对照药物,验证筛选模型。
本发明的一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法,步骤五中,所述筛选模型是应用于对小分子单体、天然产物单体、天然产物的混合物、天然植物提取物、多肽单体、多肽混合物进行广泛的筛选,获得对芳香化酶有效抑制的药物筛选应用。
本发明的一种利用表面羧基修饰磁球制备固定化酶筛选芳香化酶抑制剂的方法,雷公藤红素作为活性成分在抑制芳香化酶药物中的用途。
本发明的固定化酶的制备方法中,芳香化酶以共价键合的形式固定于表面羧基修饰磁球表面,这种共价键为酰胺键。
本发明的另一个目的是提供用上述方法制备的固定化芳香化酶并且运用于潜在抑制剂筛选,通过该方法所制备的固定化酶在37℃下的热稳定性以及在4℃下的保存稳定性较游离酶有很大提高,且磁球的引入使得酶在参加反应后通过借助磁场可以快速方便的回收再利用。随后利用固定化酶对雷公藤植物中各化合物单体进行了潜在抑制剂筛选,在所建筛选体系条件下测试发现雷公藤红素与芳香化酶具有亲和结合能力,其ic50达到23.625μg/ml。
本发明的有益效果:
①本发明的固定化酶的制备方法为共价固定法,简单有效,采用的载体为表面羧基修饰的磁球,为首次运用共价固定法将芳香化酶固定于磁球的表面,具有很高的潜在研究价值。
②本发明所制备的固定化酶在37℃下的热稳定性以及在4℃下的保存稳定性较游离酶有很大提高,且磁球的引入使得酶在参加反应后通过借助磁场可以快速方便的回收再利用。
③利用固定化酶对雷公藤植物中各化合物单体进行了潜在抑制剂筛选,在所建筛选体系条件下测试发现雷公藤红素与芳香化酶具有亲和结合能力,其ic50达到23.625μg/ml。
附图说明
附图1为fe3o4磁球(a)与fe3o4@sio2(b)的透射电镜(tem)对比图。
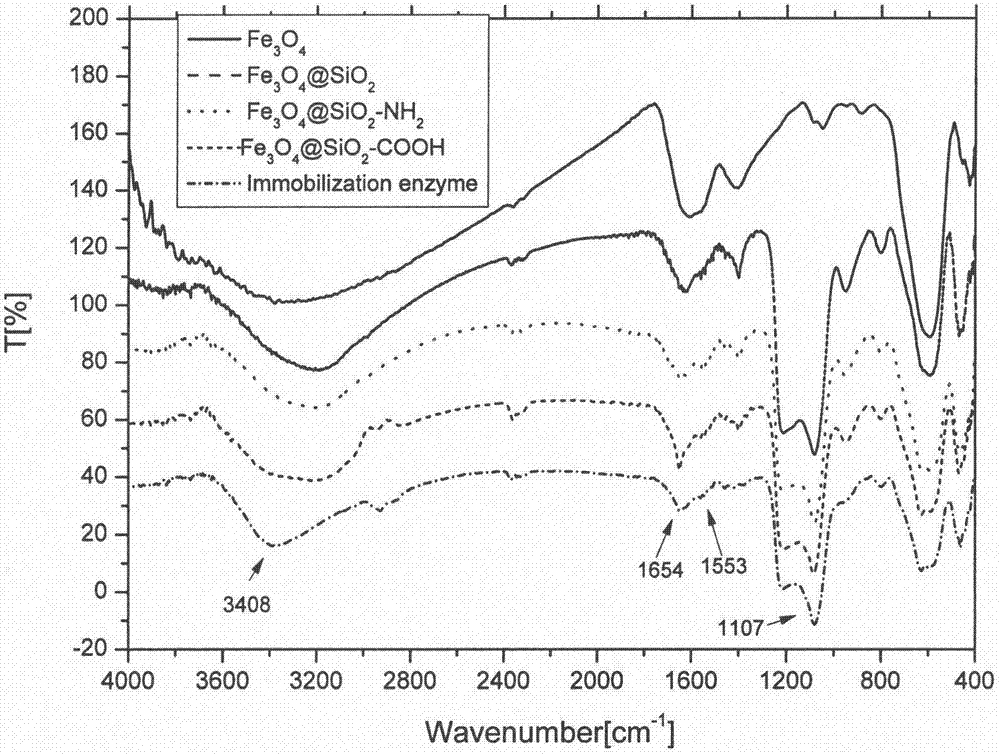
附图2为各合成阶段磁球以及固定化酶的红外光谱图对比。
附图3为各合成阶段磁球以及固定化酶的磁力回滞曲线对比图。
附图4为固定化酶催化反应完成后上清液中产物(①雌酮)与底物(②雄烯二酮)分离的高效液相色谱图。
附图5为固定化酶与游离酶在37℃下稳定性对比图。
附图6为固定化酶与游离酶在4℃下储存稳定性对比图。
附图7为加入雷公藤红素(25μg/ml)与不加抑制剂对照所得酶活力差异对比图。
附图8为加入雷公藤红素的半数抑制浓度(ic50)图。
具体实施方式:
下述非限制性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。
实施例1表面羧基修饰磁球载体的制备
(1)fe3o4磁核的制备:称取一定量的氯化铁(fecl3·6h2o)、柠檬酸三钠和乙酸铵置于干燥的圆底烧瓶中。加入一定量的乙二醇,搅拌使充分溶解。于170-200℃油浴锅加热搅拌反应1-2h。待溶液稍微冷却后,将黑色的反应液倒入带有聚四氟乙烯的高压反应釜中,于200℃的烘箱中密闭反应8-16h。随后取出反应釜,自然冷却至室温,将反应结束的磁球溶液倒入烧杯中进行磁力沉降,待出现明显的固液分层后,弃去上清液,固体磁球用无水乙醇与去离子水分别洗涤三遍。之后放入50℃的真空干燥箱中干燥,密封备用。
(2)fe3o4@sio2的制备:准确称取0.5g步骤(1)所得到的fe3o4磁核溶于250ml0.1mol/lhcl溶液中,超声20min进行活化,使其充分暴露表面的羟基。随后通过磁力沉降作用,用去离子水洗涤3次。加入380ml乙醇,100ml去离子水,5ml28%氨水溶液,室温搅拌10min后,将1.625mlteos溶解于20ml无水乙醇中,利用恒流泵以21.7ml/h的速度泵入上述反应体系中,于室温下搅拌反应6h。借助磁力沉降弃去上清液,用乙醇充分洗涤除去未反应的teos,随后用去离子水洗涤3次后于50℃条件下真空干燥收集得fe3o4@sio2。
(3)fe3o4@sio2-nh2的制备:称取50mg步骤(2)所得的fe3o4@sio2,加入25ml50%乙醇的水溶液(去离子水配制,用ch3cooh或者hcl调节体系的ph为3),将0.5ml的aptes溶解于2ml的无水乙醇中,利用恒流泵在搅拌条件下以5ml/h的速度泵入上述反应体系中,然后在50℃条件下搅拌反应5h。借助磁力沉降弃去上清液,用乙醇充分洗涤除去未反应的aptes,随后用去离子水洗涤3次后于50℃条件下真空干燥收集得fe3o4@sio2-nh2。
(4)fe3o4@sio2-cooh的制备:准确称10mg步骤(3)所得的fe3o4@sio2-nh2置于2mlep管中,加入0.6ml10%的丁二酸酐-dmf溶液,超声分散后在n2保护下,置于试管旋转混匀器20rpm,室温下反应24h。反应停止后借助磁力沉降,用去离子水洗涤3次后,40℃真空干燥收集,得到干燥的羧基化磁性fe3o4微球,置于4℃保存备用。
(5)对fe3o4与fe3o4@sio2进行tem表征,附图1所示,从图(a1)(a2)(a3)可看出fe3o4成球体,表面粗糙,分散性好;并且粒径比较均一,为500nm左右。图(b1)(b2)(b3)为fe3o4@sio2的tem扫描图,显示磁核外部包裹上了一层无孔二氧化硅,呈现典型的核壳型结构,且包裹均匀,厚度大约为30nm,该二氧化硅层能很好地保护磁核,避免其被氧化,延长磁球的寿命,而且表面暴露的硅羟基非常利于后续的官能团修饰偶联酶分子。
实施例2表面羧基修饰磁球固定化芳香化酶的制备
(1)按照实施例1制备表面羧基修饰磁球(fe3o4@sio2-cooh)。
(2)磷酸钾缓冲液(100mmol/l,ph7.4)的配制:称取4.56gk2hpo4溶于200ml去离子水为a液,称取2.72gkh2po4溶于200ml去离子水为b液,a与b以80.2∶19.8的比例混合即得。
(3)0.1mol/l2-(n-吗啉)乙磺酸一水合物(mes)缓冲液的配制:称取1.9524gmes溶于100ml去离子水,用50%naoh调节ph至4.0-7.0,置于4℃保存备用。
(4)称取7.68mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc·hcl)和11.5mgn-羟基琥珀酰亚胺(nhs)于5ml的ep管中,加入2ml的0.1mol/lmes缓冲液,涡旋混匀备用,作为活化液。
(5)称取4mg步骤(1)制备的fe3o4@sio2-cooh置于一2ml的ep管中,用去离子水清洗3次后,再用0.1mol/lmes缓冲液清洗一次。再加入1ml步骤(4)所得活化液,涡旋混匀后,室温下旋转震摇1h,随后磁力去除上清液,立即用去离子水清洗三次,去除上清液后,再用磷酸钾缓冲液(100mmol/l,ph7.4)清洗一次后,立即加入10μl的芳香化酶(美国,康宁)和190μl磷酸钾缓冲液(100mmol/l,ph7.4),于4℃下旋转震摇20h,然后磁力吸去上清液,沉淀用磷酸钾缓冲液(100mmol/l,ph7.4)清洗3次,最终的沉淀用200μl的含0.1mmol/l的edta、dtt,0.5mmol/l的蔗糖,0.5μmol/l的雄烯二酮和20%的甘油的磷酸钾缓冲液(100mmol/l,ph7.4)分散保存于-20℃即得固定化酶。
(6)附图2为该固定化条件下固定化酶与表面羧基修饰磁球的红外光谱对比图;比较fe3o4与fe3o4@sio2的红外图可知,在1107cm-1处的红外吸收,此处为si-o键的伸缩振动,说明sio2的包裹成功。固定化酶在1654cm-1与1553cm-1特有的吸收峰,为酰胺键(-conh-)的典型振动峰。在3400em-1处的吸收峰,为o-h以及n-h的伸缩振动。根据红外结果可以确证,芳香化酶被成功固定于羧基磁球表面。附图3为该固定化条件下固定化酶与表面羧基修饰磁球的磁力回滞曲线对比图。fe3o4磁核外层包裹二氧化硅层、嫁接氨基、羧基以及偶联固定化酶后会影响其磁响应能力,实验通过测定室温磁力回滞曲线(vsm)观察磁球的磁性变化发现,随着后续每一步合成反应步骤的完成,磁球的磁响应能力逐渐减弱,但是仍能够有效的对外加磁场产生响应,实现快速的固液分离。
实施例3表面羧基修饰磁球固定化酶酶活力的测定
(2)烟酰胺腺嘌呤二核苷酸磷酸(nadph)工作液的配制:称取8.33mg的nadph(biosharp公司)加入1ml容量瓶中,用磷酸钾缓冲液(100mmol/l,ph7.4)稀释至刻度,混匀,保存于4℃冰箱中即得。
(3)固定化酶的活力测定方法为:吸取20μl实施例2步骤(5)所制得固定化酶于2mlep管中,磁力吸去上清液后,加入175μl磷酸钾缓冲液(100mmol/l,ph7.4),5μl雄烯二酮工作液,混匀,37℃恒温震荡5min后,加入20μlnadph工作液启动反应,反应20min,随后磁力收集上清液,上清液取50μl注入高效液相色谱分析。沉淀用200μl的磷酸钾缓冲液(100mmol/l,ph7.4)洗涤3次后分散在200μl的含0.1mmol/l的edta、dtt,0.5mmol/l的蔗糖,0.5μmol/l的雄烯二酮和20%的甘油的磷酸钾缓冲液(100mmol/l,ph7.4)中并保存于-20℃。固定化酶酶活力(u)的表示方法为每μg的固定化酶每分钟催化雄烯二酮所产生的雌酮的量(μg)。
(4)高效液相色谱分析条件:采用十八烷基硅烷键合硅胶(250mm×4.6mm,5μm)作为固定相,乙腈∶水(55∶45)作为流动相,柱温为40℃,流速为1.0ml/min,进样量为50μl,检测波长为205am,分析时间为20min。所得色谱图(附图4)中产物雌酮的出峰时间为7.880min,底物雄烯二酮的出峰时间为8.680min。用步骤(4)所述的固定化酶活力测定方法测定此固定化条件下固定化酶的酶活力:固定化芳香化酶的酶活力为u,固定化率定义为固定化成功的酶量与参与固定化的游离酶量的比值,此条件下固定化率可达到80.35%。
实施例4固定化酶的热稳定性研究
(1)按照实施例2制备固定化酶。按照实施例3测定固定化酶酶活力。
(2)游离芳香化酶酶活力测定:2mlep管中依次加入170μl磷酸钾缓冲液(100mmol/l,ph7.4,5μl的雄烯二酮工作液,2μl的芳香化酶,涡旋混合均匀后,37℃恒温震摇5min后,加入20μlnadph启动反应,反应20min后,随后立即加入100μl冰乙腈终止反应,4℃高速离心(12000rpm×5min)后,上清液取50μl注入高效液相色谱分析。游离芳香化酶酶活力(u)的表示方法为每μg的芳香化酶每分钟催化雄烯二酮所产生的雌酮的量(μg)。
(3)将固定化酶与游离酶置于37℃恒温箱中,放置1、2、12h后,分别测定残余的酶活力,并作比较,如附图5。由附图5得知芳香化酶经固定化以后得到了保护,热稳定性较游离酶有明显提高。
实施例5固定化酶储存稳定性实验
(1)按照实施例2制备固定化酶。按照实施例3步骤(3)测定固定化酶酶活力。
(2)将固定化酶与游离酶置于4℃冰箱中,放置4天,每天分别测定残余的酶活力,并作比较,如附图6。由附图6得知芳香化酶经固定化以后得到了保护,放置4天后,游离的芳香化酶活力仅为初始的15.36%,而固定化酶还剩下88.8%的活力,由此可见酶经过固定化以后储存稳定性得到了明显的提高。
实施例6固定化酶重复使用性实验
(1)按照实施例2制备固定化酶。
(2)用实施例3步骤(3)所述的固定化酶活力测定方法测定固定化芳香化酶的酶活力。活力测定完成后用200μl的磷酸钾缓冲液(100mmol/l,ph7.4)洗涤3次后,再次测定酶活力,此步骤重复进行直至固定化酶失活,实验发现固定化酶至少可重复利用4次。
实施例7筛选模型的验证-结合时间的考察
(1)来曲唑与依西美坦混合对照品溶液的配制:精密称取依西美坦和来曲唑适量,用dmso分别稀释成浓度为1mg/ml的储备液,分别取储备液适量,用磷酸钾缓冲液(100mmol/l,ph7.4)稀释成浓度为依西美坦(2.5ng/ml),来曲唑(1ng/ml)的混合对照品溶液。
(2)取2mlep管6个并分别标号,1-5号分别加入100μl按实施例2步骤(5)制备的固定化酶,6号加入2mg空白载体。磁力沉降吸去1-5号上清液,固定化酶和空白载体分别用200μl的磷酸钾缓冲液(100mmol/l,ph7.4)进行清洗,随后吸去上清液,再在各个试管中分别加入200μl的步骤(1)的混合对照品溶液,混合均匀后,在37℃恒温震荡仪中预温孵5min后,加入10μl2μm的napdh溶液混合均匀后,置于37℃恒温震荡仪中反应分别反应0.5h、1h、2h、4h、8h后,空白管反应8h,之后采用磁力沉降的方式吸取上清液,同时对反应后的固定化酶用磷酸钾缓冲液(100mmol/l,ph7.4)洗三次,上清液以及洗液注入lc-tof/ms以及lc-ms/ms分析。结合率按公式1计算:
结合率binding(%)=100-[100×(a1+-a1-)/(a0+-a0-)](公式1)
公式中,a1+表示固定化酶与混合单体化合物反应后的化合物的质谱图峰面积,a1-代表不加化合物的样品管阴性对照;a0+表示相应的空白载体与混合单体化合物反应后的化合物的质谱图峰面积。a0-表示不加化合物载体管的阴性对照。
(3)lc-tof/ms条件:来曲唑,流动相为甲醇∶水=50∶50,色谱柱为菲罗门c18(50mm×2mn,5μm),流速0.5ml/min。esi电喷雾负离子模式,干燥气温度350℃;干燥器流量10l/min;nebulizer,35psig;vcap,3500v;fragmentor,135v;skimmer,65v;质量扫描范围,m/z150-1000.提取离子色谱eic284.0592为[m-h]-。
(4)lc-ms/ms条件:依西美坦,流动相为甲醇∶水=65∶35,色谱柱为菲罗门c18(50mm×2mm,5μm),流速1ml/min。esi正离子检测模式:正离子化喷雾电压3500v;毛细管温度350℃;二级质谱扫描子离子模式:碰撞气氩气压力1.3mtorr。质谱扫描范围m/z50-500,mrm模式,依西美坦m/z297.2→m/z121.0。
(5)与空白载体对照管相比,0.5h后芳香化酶与依西美坦和来曲唑的结合已经基本完成,清洗后的上清液中也未检测到相关化合物,为了使得后续实验结合完全,把结合反应时间决定为1h。
实施例8筛选模型的验证-结合浓度的考察
(1)依西美坦、来曲唑和雷公藤甲素系列混合标准溶液的配制:精密称取依西美坦、来曲唑和雷公藤甲素适量,依西美坦和来曲唑用dmso分别稀释成浓度为1mg/ml的储备液,雷公藤甲素用80%的甲醇配置成浓度为50μg/ml的储备液,分别取各储备液适量,用磷酸钾缓冲液(100mmol/l,ph7.4)稀释成各成分浓度为5,10,20,40ng/ml的混合对照品工作液。
(2)采用游离酶温孵体系对来曲唑(1μg/ml),依西美坦(1μg/ml),雷公藤甲素(25μg/ml)进行抑制剂验证(见表1),通过抑制率表明来曲唑和依西美坦能显著抑制芳香化酶的活性,而雷公藤甲素却几乎无抑制作用,上述结合实验也表明其与固定化酶无结合能力,因此实验选用其为阴性对照药物。
抑制率按公式2计算:
inhibition(%)=100-[(ai+-ai-/a0+-a0-)×100](公式2)
公式中,ai+表示加入抑制剂后产物雌酮的峰面积,ai-表示加入抑制剂但不加入辅酶时雌酮的峰面积;a0+表示不加入抑制剂时雌酮的峰面积,a0-表示不加入抑制剂且不加入辅酶时雌酮的峰面积。
表1各化合物对芳香化酶的的抑制情况
(3)取2mlep管8个并分别标号,1-4号分别加入100μl按实施例2步骤(5)制备的固定化酶,5-8号分别加入2mg空白载体。磁力沉降吸去1-4号上清液,1-8号固定化酶和空白载体分别用200μl的磷酸钾缓冲液(100mmol/l,ph7.4)进行清洗,随后吸去上清液,在1-4号试管中分别加入200μl的浓度分别为5,10,20,40ng/ml的依西美坦、来曲唑和雷公藤甲素系列混合对照品工作液,5-8号空白载体对照管也相应分别加入上述混合对照品工作液,各管在37℃恒温震荡仪中预温孵5min后,加入10μl2μm的napdh溶液混合均匀后,置于37℃恒温震荡仪中反应1h,之后采用磁力沉降的方式吸取各管上清液,同时对反应后的固定化酶用磷酸钾缓冲液(100mmol/l,ph7.4)清洗三次,上清液以及洗液注入lc-tof/ms以及lc-ms/ms分析。
(4)lc-tof/ms条件:来曲唑按照实施例7步骤(3)的条件实施。
雷公藤甲素,流动相为甲醇∶水=50∶50,色谱柱为菲罗门c18(50mm×2mm,5μm),流速1ml/min。esi电喷雾正离子模式,干燥气温度350℃;干燥器流量10l/min;nebulizer,35psig;vcap,3500v;fragmentor,135v;skimmer,65v;质量扫描范围,m/z150-1000.eic399.1204为[m+k]+。
(5)lc-ms/ms条件:依西美坦按照实施例7步骤(4)的条件实施。
(6)通过对不同浓度来曲唑、依西美坦和雷公藤甲素的混合溶液进行结合反应,雷公藤甲素作为对照药物,结果发现(表2和表3),结合反应在抑制剂浓度为5、10、20ng/ml呈现明显的结合。当抑制剂浓度达到40ng/ml的时候,样品与对照峰面积已基本相当,说明此时抑制剂浓度已经达到过饱和。高浓度的抑制剂不利于筛选反应的进行。而雷公藤甲素在样品与对照中的峰面积并无明显变化,说明与芳香化酶无结合(表4),综合以上实验,我们可以得出该结合反应具有特异性,可以用于复杂药物成分中抑制剂的筛选。为了使筛选结果更灵敏,推荐化合物浓度在20ng/ml以下,而且每一次筛选只会消耗100μl的固定化酶(相当于游离酶5μl,约28μg)的酶量即可进行高通量筛选,进一步说明该筛选模型成本低廉,具有很强的实用性。
表2固定化酶对各浓度依西美坦的结合率情况
表3固定化酶对各浓度来曲唑的结合率情况
表4固定化酶与空白载体对雷公藤甲素的作用情况(阴性对照)
实施例9芳香化酶抑制剂的筛选
(1)雷公藤系列混合标准溶液的配制:精密称取雷公藤甲素,雷公藤内酯酮,雷公藤定碱,雷公藤红素适量,用80%的甲醇分别配置成各组分浓度为50μg/ml的储备液,分别取各储备液适量,用磷酸钾缓冲液(100mmol/l,ph7.4)稀释成各成分浓度为10,20,40,80ng/ml的混合对照品工作液。
(2)取2mlep8个并标号,样品管分别加入100μl按实施例2步骤(5)制备的固定化酶,对照管分别加入2mg空白载体。磁力沉降吸去固定化酶上清液,然后和空白载体分别用200μl的磷酸钾缓冲液(100mmol/l,ph7.4)进行清洗,随后吸去上清液,在样品管中分别加入200μl的浓度分别为10,20,40,80ng/ml的混合对照品工作液,空白载体对照管也同法加入上述工作液,各管在37℃恒温震荡仪中预温孵5min后,加入10μl2μm的napdh溶液混合均匀后,置于37℃恒温震荡仪中反应1h,之后采用磁力沉降的方式吸取各管上清液,同时对反应后的固定化酶用磷酸钾缓冲液(100mmol/l,ph7.4)洗三次,上清液以及洗液注入lc-tof/ms以及lc-ms/ms分析。
(3)对于雷公藤甲素和雷公藤内酯酮采用lc-tof/ms分析,条件为:流动相为甲醇∶水=50∶50,色谱柱为菲罗门c18(50mm×2mm,5μm),流速1ml/min。esi电喷雾正离子模式,干燥气温度350℃;干燥器流量10l/min;nebulizer,35psig;vcap,3500v;fragmentor,135v;skimmer,65v;质量扫描范围,m/z150-1000.雷公藤甲素eic399.1204为[m+k]+,雷公藤内酯酮eic397.1048为[m+k]+。
(4)对于雷公藤红素,采用lc-ms/ms分析,条件为:流动相为甲醇∶水=85∶15,色谱柱为菲罗门c18(50mm×2mm,5μm),流速1ml/min。esi正离子检测模式:正离子化喷雾电压3500v;毛细管温度350℃;二级质谱扫描子离子模式:碰撞气氩气压力1.3mtorr。质谱扫描范围m/z50-m/z500,mrm模式,雷公藤红素m/z452.1→m/z201.0。
对于雷公藤定碱,采用lc-ms/ms分析,条件为:流动相为甲醇∶水=85∶15,色谱柱为菲罗门c18(50mm×2mm,5μm),流速1ml/min。esi正离子检测模式:正离子化喷雾电压3500v;毛细管温度350℃;二级质谱扫描子离子模式:碰撞气氩气压力1.3mtorr。质谱扫描范围m/z50-m/z500,mrm模式,雷公藤定碱m/z833.4→m/z856.1。
(5)通过对不同浓度雷公藤甲素,雷公藤内酯酮,雷公藤红素的混合溶液进行结合反应研究,结果发现,结合反应在雷公藤红素浓度为10、20、40、80ng/ml时呈现明显的结合(表5和表6)。实验发现,反应后的固定化酶在清洗一次后洗液中也并未检测出与芳香化酶结合的雷公藤红素,说明雷公藤红素与芳香化酶为基于亲和作用结合。在而固定化酶对雷公藤甲素、雷公藤内酯酮和雷公藤定碱在化合物浓度为10ng/ml时几乎无亲和作用。
表5固定化酶对各浓度雷公藤红素的结合率情况(进样量20μl)
表6固定化酶对各浓度雷公藤红素的结合率情况(进样量增大至50μl)
实施例10雷公藤红素抑制效果评价
(1)各浓度雷公藤红素母液配制:称取4mg雷公藤红素对照品(含量>98%)于1ml的容量瓶中,用dmso溶解并稀释至刻度为储备液,然后该4mg/ml的储备液用dmso分别稀释成0μg/ml,0.125μg/ml,0.5μg/ml,0.25μg/ml,0.125μg/ml备用。
(2)雷公藤红素对芳香化酶的抑制效果评价:分别于2mlep管中,加入168μl磷酸钾缓冲液(100mmol/l,ph7.4),2μl的游离酶,5μlad工作液,5μl的各浓度雷公藤红素溶液,震摇均匀后,置于37℃的恒温震荡仪中预温孵5min后,在各管中加入20μl的nadph,之后反应20min,反应结束后各管中加入100μl冰乙腈,混匀。离心(12000r/min×5min)除去蛋白,取上清液注入hplc分析;不加抑制剂的空白对照(须将5μl的各浓度雷公藤红素溶液换为5μl的磷酸钾缓冲液(100mmol/l,ph7.4)),然后同法操作。
按公式2计算出各浓度雷公藤红素下的抑制率。
(1)附图7为雷公藤红素(25μg/ml)对芳香化酶的抑制作用色谱图。(a图为含雷公藤红素25μg/ml;b图为不加抑制剂对照)。通过表6和附图8,计算抑制率表明雷公藤红素在25μg/ml时能显著抑制芳香化酶的活性,抑制率达到71.99%。
本发明已以将较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权力要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!