一种用于检测甲型肝炎病毒滴度的细胞系及其构建方法与应用与流程
本发明属于医学生物学检测
技术领域:
,具体涉及一种用于定性和定量检测甲型肝炎病毒滴度的细胞系及其构建方法与应用。
背景技术:
:甲型肝炎病毒(hepatitisavirus,hav)感染被认为是一个非常重要的公共卫生问题。在发展中或发达国家,hav是引起急性肝炎及部分急性肝功能衰竭的主要病原体之一。据统计,全世界每年大约有140万例甲型肝炎病毒感染。由于甲型肝炎减毒活疫苗与灭活疫苗的相继使用,目前,甲型肝炎病毒的传播已得到了有效的控制。甲型肝炎病毒是一种正链rna病毒,属于微小rna病毒科肝病毒属。hav感染细胞后不会产生细胞病变效应,这就为观察和检测hav感染带来了一定的难度。目前,一般采用免疫荧光、elisa、rt-pcr、组织培养半数感染量(ccid50)等方法对hav滴度及食品中的hav进行检测。elsa法主要检测hav的抗原,不能完全体现hav活病毒颗粒数,而且灵敏度也较差;组织培养半数感染量(ccid50)可以检测活病毒数,但检测周期较长需要21-28天;rt-pcr法通过对hav基因的目标片段进行扩增检测,具有简单和灵敏度高的特点,但是此种方法不能区分感染性与非感染性病毒颗粒数。因此如何克服现有技术的不足是目前医学检测
技术领域:
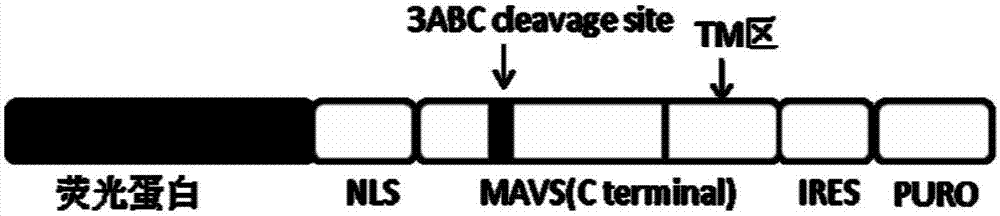
亟需解决的问题。技术实现要素:为解决甲型肝炎病毒感染性滴度检测耗时较长、操作复杂繁琐等问题,本发明的目的是提供一种快速、简单、准确的定性和定量检测甲型肝炎病毒滴度的细胞系及其构建方法与应用。为实现上述目的,本发明采用的技术方案如下:一种用于检测甲型肝炎病毒滴度的细胞系的构建方法,包括如下步骤:步骤(1),质粒构建:以huh7.0细胞或树鼩肝脏中的总rna为模板,采用rt-pcr扩增mavsc末端基因序列mavs,并对扩增产物和慢病毒表达载体进行双酶切,然后通过t4连接酶对双酶切后扩增产物和慢病毒表达载体通过t4连接酶进行连接,得到重组质粒lv-荧光蛋白-mavs(c-terminal)-ires-puro-wpre;所述的mavsc末端氨基酸序列如seqidno.1或seqidno.2所示;所述的慢病毒表达载体为lv-荧光蛋白-ires-puro-wpre载体;步骤(2),慢病毒包装:将步骤(1)得到的重组质粒与pmd2.g、spax2质粒一起采用脂质体的方法共转染293t细胞中,制得慢病毒;步骤(3),稳定细胞系构建:将步骤(2)制得的慢病毒感染huh7.0细胞,接着将表达荧光融合蛋白的huh7.0细胞系扩大培养,之后通过嘌呤霉素筛选表达荧光融合蛋白的huh7.0细胞系,得到用于检测甲型肝炎病毒的细胞系。进一步,优选的是,所述的荧光蛋白为egfp或mcherry。进一步,优选的是,以huh7.0细胞的总rna为模板,采用lv-egfp-ires-puro-wpre载体时,rt-pcr扩增所用引物如seqidno.3和seqidno.4所示;以huh7.0细胞的总rna为模板,采用lv-mcherry-ires-puro-wpre载体时,rt-pcr扩增所用引物如seqidno.4和seqidno.5所示;以树鼩肝脏中的总rna为模板,用lv-egfp-ires-puro-wpre载体时,rt-pcr扩增所用引物如seqidno.6和seqidno.7所示。进一步,优选的是,扩增程序为98℃预变性3min;再进行30个循环,循环所遵循条件为98℃变性10s,62℃退火30s,72℃延伸20s;最后采用72℃延伸5min。进一步,优选的是,步骤(2)中,步骤(1)得到的重组质粒与pmd2.g和、spax2质粒的质量比为5:2:3。作为替代方案,步骤(1)的质粒构建还可以为:合成恒河猴的mavsc末端基因序列,之后,将恒河猴的mavsc末端基因序列插入到慢病毒表达载体构建重组质粒,得到重组质粒lv-荧光蛋白-mavs(c-terminal)-ires-puro-wpre;所述的恒河猴的mavs末端基因序列如seqidno.9;所述的慢病毒表达载体为lv-荧光蛋白-ires-puro-wpre载体。本发明提供上述用于检测甲型肝炎病毒滴度的细胞系的构建方法构建的荧光融合蛋白。所述的荧光融合蛋白的组成:(1)、一段可定位于线粒体上的跨膜区(tm区),一段可被hav3abc蛋白酶特异性识别切割的序列,序列特征为:如seqidno.1、seqidno.2、seqidno.9所示。(2)、荧光蛋白。(3)、荧光蛋白与mavs之间加sv40核定位信号或不加sv40核定位信号本发明还提供上述用于检测甲型肝炎病毒滴度的细胞系的构建方法构建的细胞系。本发明同时提供上述用于检测甲型肝炎病毒滴度的细胞系在检测甲型肝炎病毒滴度中的应用。本发明提供一种检测甲型肝炎病毒滴度的方法,采用上述用于检测甲型肝炎病毒滴度的细胞系的构建方法构建的细胞系,包括如下步骤:将得到的细胞系接种于到细胞培养板中;所述的细胞培养板中含有含10%fbs的dmem;第二天,取hav,用含2%fbs的dmem按10倍梯度稀释为10-1~10-8,得到病毒稀释液;之后吸弃细胞培养板中的培养基,将病毒稀释液按100μl/孔加入细胞培养板中,摇匀后置于37℃、5%co2培养箱进行培养,每隔1h摇晃细胞培养板,4h后吸弃病毒稀释液,按250μl/孔加入含1%~2%甲基纤维素、2%fbs的dmem,或含0.4%~1%琼脂、2%fbs的dmem,于37℃、5%co2培养箱中培养3-5d;荧光显微镜下观察荧光噬斑的个数,之后通过如下公式计算hav滴度:hav滴度(ffu/ml)=荧光噬斑数/接种体积*稀释倍数。进一步,优选的是,细胞系接种于到细胞培养板时的接种量为5000个/孔。进一步,优选的是,将病毒稀释液按100μl/孔加入细胞培养板中时,每个稀释度设定3个复孔。本发明构思:1.质粒构建以huh7.0细胞或树鼩肝脏中的总rna为模板,采用rt-pcr进行扩增mavsc末端基因序列mavs(含有hav3abc蛋白酶的切割位点及定位于线粒体的跨膜区),经内切酶处理后,插入到经同样内切酶处理过的慢病毒表达载体lv-荧光蛋白-ires-puro-wpre载体中,构建lv-荧光蛋白-mavs(c-terminal)-ires-puro-wpre,表达的荧光蛋白-mavs(c-terminal)-ires-puro-wpre的结构如图1。以huh7.0细胞中的总rna为模板,采用rt-pcr进行扩增mavsc末端基因序列mavs(含有hav3abc蛋白酶的切割位点及跨膜区),其中引物中含有sv40核定位信号(nls)的核酸序列(seqidno.8),经内切酶处理后,插入到经同样内切酶处理过的慢病毒表达载体lv-荧光蛋白-ires-puro-wpre中,构建的荧光蛋白-nls-mavs(c-terminal)-ires-puro-wpre,其结构如图2。或者合成恒河猴的mavsc末端基因序列,构建重组质粒lv-荧光蛋白-mavs(c-terminal)-ires-puro-wpre。2.慢病毒包装将测序正确的重组质粒分别与包装质粒按比例混合(重组质粒:pmd2.g:pspax2=5:2:3),采用脂质体的方法进行转染,制备表达荧光蛋白-mavs(c-terminal)、荧光蛋白-nls-mavs(c-terminal)的慢病毒,分装后保存于-80℃冰箱。3.稳定细胞系构建及功能检测将制备的慢病毒感染huh7.0细胞,感染后次日将培养液更换成含10%fbs高糖dmem,继续培养48h,于荧光显微镜下观察绿色荧光蛋白或红色荧光蛋白的表达情况。将表达绿色荧光蛋白或红色荧光蛋白的huh7.0细胞系扩大培养,通过添加抗生素—puromycin筛选表达绿色荧光蛋白或红色荧光蛋白的huh7.0细胞系。取hav病毒液感染筛选得到的细胞系,显微镜观察荧光弥散情况或荧光入核情况。4.滴度检测将筛选得到的细胞系接种于48孔细胞培养板中,第二天,取10μlhav,用含2%fbs的dmem按10倍梯度稀释为10-1~10-8,吸弃48孔板中的培养基,将病毒稀释液按100μl/孔轻轻加入48孔板,每个稀释度设定3个复孔,摇匀后置于37℃、5%co2培养箱进行培养,每隔1h轻轻摇晃48孔板,4h后吸弃病毒稀释液,按250μl/孔加入含1%~2%甲基纤维素、2%fbs的dmem,或含0.4%~1%琼脂、2%fbs的dmem,于37℃、5%co2培养箱中培养1-4d。荧光显微镜下观察荧光噬斑的个数,从而计算hav的滴度。具体公式如下:hav滴度(ffu/ml)=荧光噬斑数/接种体积*稀释倍数。同时,将hav病毒做10倍系列稀释,取10-6、10-7、10-8稀释度的病毒液1ml,分别接种于人二倍体细胞-kmb17,每个稀释度做5个重复,置于35℃培养21天,收获后提取甲肝病毒,用酶联免疫法检测,按reed-muench法计算hav的滴度(ccid50)。(参照2015版《中华人民共和国药典》第三部)通过比较两种方法的滴度检测结果,得出两种方法测得滴度的比例关系。为了简化并准确检测hav感染的方法,本发明利用hav3abc与mavs相互作用的原理构建了一种直观、简单、快速的荧光蛋白定位系统来定性和定量检测hav病毒感染及其感染力。hav感染细胞的过程中,非结构蛋白3abc蛋白酶作用于细胞中定位于线粒体上的抗病毒信号蛋白—线粒体抗病毒信号蛋白(mitochondrialantiviralsignalingprotein,mavs),当mavs被3abc蛋白酶切割后,mavs蛋白的n端会从线粒体上脱落。mavs是一种定位于线粒体上的抗病毒信号蛋白,是hav3abc蛋白酶的作用底物。mavs的c端含有3abc蛋白酶的识别位点和线粒体定位信号,3abc蛋白酶作用位点是mavs蛋白c端,当被3abc蛋白酶切割后,mavs蛋白的n端会从线粒体上脱落。本发明利用3abc蛋白酶能与mavs的相互的性质,将mavsc端(含有hav3abc蛋白酶切割位点及定位于线粒体上的跨膜区)与荧光蛋白构建融合蛋白,并将其包装成慢病毒颗粒。慢病毒感染细胞后,表达的融合蛋白会锚定在细胞质中的线粒体上成点状分布,在抗生素-puromycin筛选下,可得到稳定表达融合蛋白的稳定细胞系,当此细胞系被hav感染后荧光蛋白会弥散在整个细胞或定位到细胞核中。为了快速、直观、简单地对hav滴度进行检测,以含有荧光蛋白报告系统的细胞系为基础,在感染的单层细胞上覆盖甲基纤维素等半固体营养培养基,由于甲基纤维素溶液有优良的增稠性,能很好地限制细胞中感染病毒颗粒的扩散,进而形成清晰的绿色荧光或红色荧光噬斑,通过观察荧光噬斑数可计算得到hav的滴度。与传统方法相比,利用荧光蛋白重定位系统检测hav滴度的方法仅需要1-4天,大大缩短了的疫苗生产周期,而且操作简单、灵敏度较高,可以直接观察到hav的感染过程。这种快速、简单、准确的检测hav滴度的方法的建立将为hav的基础研究及疫苗生产提供了一种实时有效的手段。本发明与现有技术相比,其有益效果为:与传统方法相比,利用荧光蛋白重定位系统检测hav滴度的方法是一种快速、简单、准确的定性及定量检测hav感染及其感染力的方法,此方法结果良好,重复性良好,完成甲肝减毒活疫苗滴度检测仅需要1-4天(传统方法需要21天-28天),而且大大缩短了的疫苗生产周期,保证了疫苗的质量,缩短了产品上市免疫接种时间,从而延长了疫苗的有效期。由于hav在体外培养周期长并且无明显的细胞病变,此方法还可以直接观察到hav的感染过程。此外此荧光蛋白重定位系统还可以用于hcv滴度及感染过程的检测。附图说明图1为荧光蛋白-mavs(c-terminal)-ires-puro结构图;图2为荧光蛋白-nls-mavs(c-terminal)-ires-puro结构图;图3为egfp-hmavs(c396-540)-ires-puro结构图;图4为荧光纤维镜观察huh7.0-egfp-hmavs图;图5为hav感染huh7.0-egfp-hmavs(c396–540)后荧光弥散图;图6为hav感染huh7.0-egfp-hmavs(c396–540)形成的荧光噬斑图;图7为mcherry-nls-hmavs(c396-540)-ires-puro结构图;图8为荧光纤维镜观察huh7.0-mcherry-nls-mavs(c396–540)图;图9为hav感染huh7.0-mcherry-nls-hmavs(c396–540)后荧光入核图;图10为hav感染huh7.0-mcherry-nls-hmavs(c396–540)形成的荧光噬斑图。图11为egfp-tmavs(c365-503)-ires-puro-wpre结构图;图12为荧光纤维镜观察huh7.0-egfp-tmavs(c365–503)图;图13为hav感染huh7.0-egfp-tmavs(c365–503)后荧光弥散图;图14为hav感染huh7.0-egfp-tmavs(c365–503)形成的荧光噬斑图。图15为egfp-rhmavs(c397-541)-ires-puro-wpre结构图;图16为荧光纤维镜观察huh7.0-egfp-rhmavs(c397-541)图;图17为hav感染huh7.0-egfp-rhmavs(c397-541)后荧光弥散图;图18为hav感染huh7.0-egfp-rhmavs(c397-541)形成的荧光噬斑图。具体实施方式下面结合实施例对本发明作进一步的详细描述。本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用材料或设备未注明生产厂商者,均为可以通过购买获得的常规产品。本发明中通常固体配制于液体中,百分号为质量百分浓度,比例为质量比;液体配制于液体中,百分号为体积百分浓度,比例为体积比,除非另有说明。实施例1步骤(1),质粒构建:以huh7.0细胞中的总rna为模板,以p1、p2为引物,采用rt-pcr进行扩增mavsc末端基因序列mavs(c396-540),bsrgi/mlui双酶切后,插入到用同样酶处理过的慢病毒表达载体lv-egfp-ires-puro-wpre载体中构建lv-egfp-mavs(c396–540)-ires-puro-wpre重组质粒,经测序分析,序列正确,大提保存于-20℃备用。具体方法如下:pcr扩增体系及扩增程序1、合成cdna采用takara的primescriptii1ststrandcdnasynthesiskit按说明书合成cdna:体系:体积(ul)huh7.0rna2引物p20.5dntp4rnasefreeddh2o6.5ultotal1365℃5min,之后迅速冰浴1min。向以上反应体系中加入5*fsbuffer:4ul,dtt:1ul,rnaseinhibitor:1ul,酶:1ul;55℃:1h,70℃:15min。2、pcrpcr扩增体系体积(ul)5xq5buffer45xq5enhancerbuffer4dntp1.6p10.4p20.4模板cdna1水10.2q5酶0.2pcr扩增程序:98℃for3min--(98℃for10s,62℃for30s,72℃for20s)×30cycle--72℃for5min,3、酶切酶切体系37℃,酶切过夜。4、连接连接体系16℃连接过夜。构建的egfp-hmavs(c396–540)-ires-puro的结构示意图如图3。步骤(2),慢病毒包装:转染前一天,消化293t细胞(用0.25%胰酶室温消化2min),以2.0×107个细胞量接种于t175培养瓶中(培养基为含10%fbs的dmem),转染当天细胞融合度达到40-50%时,进行转染。将重组质粒lv-egfp-hmavs(c396-540)-ires-puro-wpre与包装质粒(pmd2.g和pspax2)按比例混合(重组质粒:pmd2.g:pspax2=5:2:3),采用lipfectmin2000进行转染,其中,lipfectmin2000与重组质粒和包装质粒的总用量比为50ul:20ug,具体操作按说明书进行。转染后于37℃,5%co2培养箱中孵育12h;更换含10%(v/v)胎牛血清的dmem培养液,继续培养48h,荧光显微镜下观察荧光蛋白表达情况,收集含病毒颗粒的细胞上清液(用吸管取上清液),并经3000g、4℃离心5min,去除细胞碎片,收集病毒上清液,经24000rpm、4℃离心2h,去掉上清,加入200μl病毒冻存液,4℃溶解过夜,分装后,保存于-80℃冰箱。步骤(3),稳定细胞系构建:感染前一天,将huh7.0细胞传至6孔板中(培养基为含2ml10%fbs的dmem),放于37℃、5%co2细胞培养箱中培养,感染当天细胞融合度达到50%时,用制备的慢病毒进行感染。感染前,将细胞培养基更换成含2%fbs的dmem,加入收获的病毒液上清,其中,感染复数moi=0.1,于37℃培养过夜。次日将培养液更换成含10%fbs的高糖dmem,继续培养48h,于荧光显微镜下观察荧光蛋白的表达情况。将表达荧光蛋白的huh7.0细胞系用0.25%胰酶在室温消化5min,然后继续在t25培养瓶中培养,将培养基换成含5ug/mlpuromycin、10%fbs的dmem筛选表达荧光蛋白的huh7.0细胞系,命名为huh7.0-egfp-hmavs(c396-540)。感染前一天,将huh7.0-egfp-hmavs(c396-540)细胞传至48孔板中(培养基的体积为250ul),放于37℃、5%co2细胞培养箱中培养。将hav病毒液按moi=1接种于上述细胞中,显微镜观察荧光弥散情况。如果细胞被hav感染绿色荧光蛋白将会从细胞质弥散至整个细胞中。步骤(4),滴度检测:将筛选得到的细胞系接种于48孔细胞培养板中(培养基为含10%fbs的dmem,接种量为5000个/孔),第二天,取10μlhav,用2%(v/v)fbsdmem按10倍系列稀释为10-1~10-6,吸弃48孔板中的培养基,将病毒稀释液按100μl/孔轻轻加入48孔板,每个稀释度设定3个复孔,摇匀后置于37℃、5%co2培养箱进行培养,每隔1h轻轻摇晃48孔板,4h后吸弃病毒稀释液,按250μl/孔加入含1%甲基纤维素、2%fbs的dmem,于37℃、5%co2培养箱中培养3-4d。荧光显微镜下观察荧光噬斑的个数,从而计算hav的滴度。同时,将hav病毒做10倍系列稀释,取10-6、10-7、10-8稀释度的病毒液1ml,分别接种于人二倍体细胞-kmb17,每个稀释度做5个重复,置于35℃培养21天(培养基为含5%fbs的mem,培养过程每7天换液),收获后提取甲肝病毒,用酶联免疫法检测,按reed-muench法计算hav的滴度(ccid50)。(参照2015版《中华人民共和国药典》第三部)通过比较两种方法的滴度检测结果,得出两种方法测得滴度的比例关系。具体结果:1.获得了稳定表达egfp-hmavs(c396-540)的细胞系-huh7.0-egfp-hmavs(c396-540),而且荧光蛋白在细胞质中成点状分布如图4。2.hav感染huh7.0-egfp-hmavs(c396-540)后,荧光蛋白会弥散在细胞中(如图5)。3.hav感染huh7.0-egfp-hmavs(c396-540)后,荧光蛋白会弥散在细胞中,加入甲基纤维素后,会形成荧光“噬斑”点(如图6)。4.hav感染huh7.0-egfp-hmavs(c396-540)后,荧光蛋白会弥散在细胞中,加入甲基纤维素后,会形成荧光“噬斑”点,通过观察荧光“噬斑”的个数,计算hav的滴度。利用此方法检测3批hav的病毒滴度,结果良好,3批病毒的lgffu/ml分别是6.94、6.88、6.93,elisa法检测结果lgccid50/ml分别是7.67、7.35、7.53。与elisa法相比,利用荧光重定位系统检测的hav病毒滴度小于lgccid50/ml,约为lgccid50/ml的90%~95%,可用于甲肝减毒活疫苗的滴度检测。实施例2步骤(1),质粒构建:以huh7.0细胞中的总rna为模板,以p2、p3为引物,采用rt-pcr进行扩增mavsc末端基因序列hmavs(c396-540),bsrgi/mlui双酶切后,插入到用同样酶处理过的慢病毒表达载体lv-mcherry-ires-puro-wpre载体中构建lv-mcherry-nls-hmavs(c396-540)-ires-puro-wpre重组质粒,经测序分析,序列正确,大提保存于-20℃备用。构建的mcherry-nls-hmavs(c396-540)-ires-puro-wpre的结构见图7。1、合成cdna采用takara的primescriptii1ststrandcdnasynthesiskit按说明书合成cdna:体系:体积(ul)huh7.0rna2引物p20.5dntp4rnasefreeddh2o6.5ultotal1365℃5min,之后迅速冰浴1min。向以上反应体系中加入5*fsbuffer:4ul,dtt:1ul,rnaseinhibitor:1ul,酶:1ul;55℃:1h,70℃:15min。2、pcrpcr扩增体系体积(ul)5xq5buffer45xq5enhancerbuffer4dntp1.6p30.4p20.4模板cdna1水10.2q5酶0.2pcr扩增程序:98℃for3min--(98℃for10s,62℃for30s,72℃for20s)×30cycle--72℃for5min。3、酶切酶切体系37℃,酶切过夜。4、连接连接体系体积(ul)bsrgi/mlui酶切的pcr产物1.5bsrgi/mlui酶切的载体4.5t4连接酶1t4buffer1h2o216℃连接过夜。构建的mcherry-nls-hmavs(c396-540)-ires-puro的结构示意图如图7。步骤(2),慢病毒包装:转染前一天,消化293t细胞(用0.25%胰酶室温消化2min),以2.0×107个细胞量接种于t175培养瓶中(培养基为含10%fbs的dmem),转染当天细胞融合度达到40-50%时,进行转染。将重组质粒lv-mcherry-nls-hmavs(c396-540)-ires-puro-wpre与包装质粒(pmd2.g和pspax2)按比例混合(重组质粒:pmd2.g:pspax2=5:2:3),采用lipfectmin2000进行转染,其中,lipfectmin2000与重组质粒和包装质粒的总用量比为50ul:20ug,具体操作按说明书进行。转染后于37℃,5%co2培养箱中孵育12h;更换含10%(v/v)胎牛血清的dmem培养液,继续培养48h,荧光显微镜下观察荧光蛋白表达情况,收集含病毒颗粒的细胞上清液(用吸管取上清液),并经3000g、4℃离心5min,去除细胞碎片,收集病毒上清液,经24000rpm、4℃离心2h,去掉上清,加入200μl病毒冻存液,4℃溶解过夜,分装后,保存于-80℃冰箱。步骤(3),稳定细胞系构建:感染前一天,将huh7.0细胞传至6孔板中(培养基为含2ml10%fbs的dmem),放于37℃、5%co2细胞培养箱中培养,感染当天细胞融合度达到50%时,用制备的慢病毒进行感染。感染前,将细胞培养基更换成含2%fbs的dmem,加入收获的病毒液上清,其中,感染复数moi=0.1,于37℃培养过夜。次日将培养液更换成含10%fbs的高糖dmem,继续培养48h,于荧光显微镜下观察荧光蛋白的表达情况。将表达荧光蛋白的huh7.0细胞系用0.25%胰酶在室温消化5min,然后继续在t25培养瓶中培养,将培养基换成含5ug/mlpuromycin、10%fbs的dmem筛选表达荧光蛋白的huh7.0细胞系,命名为huh7.0-mcherry-nls-hmavs(c396-540)(如图8)。感染前一天,将huh7.0-mcherry-nls-hmavs(c396-540)细胞传至48孔板中(培养基的体积为250ul),放于37℃、5%co2细胞培养箱中培养。将hav病毒液按moi=1接种于上述细胞中,显微镜观察荧光荧光入核情况。如果细胞被hav感染,红色荧光蛋白将会从细胞质进入细胞核中(如图9)。步骤(4),滴度检测:将筛选得到的细胞系接种于48孔细胞培养板中(培养基为含10%fbs的dmem,接种量为5000个/孔),第二天,取10μlhav,用2%(v/v)fbsdmem按10倍系列稀释为10-1~10-6,吸弃48孔板中的培养基,将病毒稀释液按100μl/孔轻轻加入48孔板,每个稀释度设定3个复孔,摇匀后置于37℃、5%co2培养箱进行培养,每隔1h轻轻摇晃48孔板,4h后吸弃病毒稀释液,按250μl/孔加入含1%甲基纤维素、2%fbs的dmem,于37℃、5%co2培养箱中培养3-4d。荧光显微镜下观察荧光噬斑的个数,从而计算hav的滴度(如图10)。同时,将hav病毒做10倍系列稀释,取10-6、10-7、10-8稀释度的病毒液1ml,分别接种于人二倍体细胞-kmb17,每个稀释度做5个重复,置于35℃培养21天(培养基为含5%fbs的mem,培养过程每7天换液),收获后提取甲肝病毒,用酶联免疫法检测,按reed-muench法计算hav的滴度(ccid50)。(参照2015版《中华人民共和国药典》第三部)通过比较两种方法的滴度检测结果,得出两种方法测得滴度的比例关系。具体结果:1.获得了稳定表达mcherry-nls-hmavs(c396-540)的细胞系-huh7.0-mcherry-hmavs(c396-540),而且荧光蛋白在细胞质中成点状分布如图8。2.hav感染huh7.0-mcherry-nls-hmavs(c396-540)后,荧光蛋白会进入到细胞核中(如图9)。3.hav感染huh7.0-mcherry-nls-hmavs(c396-540)后,荧光蛋白会进入到在细胞核中,加入甲基纤维素后,会形成荧光“噬斑”点(如图10)。4.hav感染huh7.0-mcherry-nls-hmavs(c396-540)后,荧光蛋白会弥散在细胞中,加入甲基纤维素后,会形成荧光“噬斑”点,通过观察荧光“噬斑”的个数,计算hav的滴度。利用此方法检测3批hav的病毒滴度,结果良好,3批病毒的lgffu/ml分别是6.90、6.88、6.96,elisa法检测结果lgccid50/ml分别是7.67、7.35、7.53。与elisa法相比,利用荧光重定位系统检测的hav病毒滴度小于lgccid50/ml,约为lgccid50/ml的90%~95%,可用于甲肝减毒活疫苗的滴度检测。实施例3步骤(1),质粒构建:以树鼩肝脏的总rna为模板,以p4、p5为引物,采用rt-pcr进行扩增mavsc末端基因序列mavs(c365-503),bsrgi/mlui双酶切后,插入到用同样酶处理过的慢病毒表达载体lv-egfp-ires-puro-wpre载体中构建lv-egfp-tmavs(c365–503)-ires-puro-wpre重组质粒,经测序分析,序列正确,大提保存于-20℃备用。构建的egfp-tmavs(c365-503)-ires-puro-wpre的结构见图11。具体方法如下:1、合成cdna采用takara的primescriptii1ststrandcdnasynthesiskit按说明书合成cdna:体系:体积(ul)huh7.0rna2引物p20.5dntp4rnasefreeddh2o6.5ultotal1365℃5min,之后迅速冰浴1min。向以上反应体系中加入5*fsbuffer:4ul,dtt:1ul,rnaseinhibitor:1ul,酶:1ul;55℃:1h,70℃:15min。2、pcrpcr扩增体系体积(ul)5xq5buffer45xq5enhancerbuffer4dntp1.6p40.4p50.4模板cdna1水10.2q5酶0.2pcr扩增程序:98℃for3min--(98℃for10s,62℃for30s,72℃for20s)×30cycle--72℃for5min。3、酶切酶切体系37℃,酶切过夜。4、连接连接体系体积(ul)bsrgi/mlui酶切的pcr产物1.5bsrgi/mlui酶切的载体4.5t4连接酶1t4buffer1h2o216℃连接过夜。构建的egfp-tmavs(c365–503)-ires-puro的结构示意图如图11。步骤(2),慢病毒包装:转染前一天,消化293t细胞(用0.25%胰酶室温消化2min),以2.0×107个细胞量接种于t175培养瓶中(培养基为含10%fbs的dmem),转染当天细胞融合度达到40-50%时,进行转染。将重组质粒lv-egfp-tmavs(c365–503)-ires-puro-wpre与包装质粒(pmd2.g和pspax2)按比例混合(重组质粒:pmd2.g:pspax2=5:2:3),采用lipfectmin2000进行转染,其中,lipfectmin2000与重组质粒和包装质粒的的总用量比为50ul:20ug,具体操作按说明书进行。转染后于37℃,5%co2培养箱中孵育12h;更换含10%(v/v)胎牛血清的dmem培养液,继续培养48h,荧光显微镜下观察荧光蛋白表达情况,收集含病毒颗粒的细胞上清液(用吸管取上清液),并经3000g、4℃离心5min,去除细胞碎片,收集病毒上清液,经24000rpm、4℃离心2h,去掉上清,加入200μl病毒冻存液,4℃溶解过夜,分装后,保存于-80℃冰箱。步骤(3),稳定细胞系构建:感染前一天,将huh7.0细胞传至6孔板中(培养基为含2ml10%fbs的dmem),放于37℃、5%co2细胞培养箱中培养,感染当天细胞融合度达到50%时,用制备的慢病毒进行感染。感染前,将细胞培养基更换成含2%fbs的dmem,加入收获的病毒液上清,其中,感染复数moi=0.1,于37℃培养过夜。次日将培养液更换成含10%fbs的高糖dmem,继续培养48h,于荧光显微镜下观察荧光蛋白的表达情况。将表达荧光蛋白的huh7.0细胞系用0.25%胰酶在室温消化5min,然后继续在t25培养瓶中培养,将培养基换成含5ug/mlpuromycin、10%fbs的dmem筛选表达荧光蛋白的huh7.0细胞系,命名为huh7.0-egfp-tmavs(c365-503)(如图12)。感染前一天,将huh7.0-egfp-tmavs(c365–503)细胞传至48孔板中(培养基的体积为250ul),放于37℃、5%co2细胞培养箱中培养。将hav病毒液按moi=1接种于上述细胞中,显微镜观察荧光弥散情况。如果细胞被hav感染,绿色荧光蛋白将会从细胞质弥散至整个细胞中(如图13)。步骤(4),滴度检测:将筛选得到的细胞系接种于48孔细胞培养板中(培养基为含10%fbs的dmem,接种量为5000个/孔),第二天,取10μlhav,用2%(v/v)fbsdmem按10倍系列稀释为10-1~10-6,吸弃48孔板中的培养基,将病毒稀释液按100μl/孔轻轻加入48孔板,每个稀释度设定3个复孔,摇匀后置于37℃、5%co2培养箱进行培养,每隔1h轻轻摇晃48孔板,4h后吸弃病毒稀释液,按250μl/孔加入含1%甲基纤维素、2%fbs的dmem,于37℃、5%co2培养箱中培养3-4d。荧光显微镜下观察荧光噬斑的个数,从而计算hav的滴度(如图14)。同时,将hav病毒做10倍系列稀释,取10-6、10-7、10-8稀释度的病毒液1ml,分别接种于人二倍体细胞-kmb17,每个稀释度做5个重复,置于35℃培养21天(培养基为含5%fbs的mem,培养过程每7天换液),收获后提取甲肝病毒,用酶联免疫法检测,按reed-muench法计算hav的滴度(ccid50)。(参照2015版《中华人民共和国药典》第三部)通过比较两种方法的滴度检测结果,得出两种方法测得滴度的比例关系。具体结果:1.获得了稳定表达egfp-tmavs(c365–503)的细胞系-huh7.0-egfp-tmavs(c365–503),而且荧光蛋白在细胞质中成点状分布如图12。2.hav感染huh7.0-egfp-tmavs(c365–503)后,荧光蛋白会弥散到整个细胞中(如图13)。3.hav感染huh7.0-egfp-tmavs(c365–503)后,荧光蛋白会弥散到整个细胞中,加入甲基纤维素后,会形成荧光“噬斑”点(如图14)。4.hav感染huh7.0-egfp-tmavs(c365–503)后,荧光蛋白会弥散在细胞中,加入甲基纤维素后,会形成荧光“噬斑”点,通过观察荧光“噬斑”的个数,计算hav的滴度。利用此方法检测3批hav的病毒滴度,结果良好,3批病毒的lgffu/ml分别是7.02、6.93、6.98,elisa法检测结果lgccid50/ml分别是7.61、7.42、7.64。与elisa法相比,利用荧光重定位系统检测的hav病毒滴度小于lgccid50/ml,约为lgccid50/ml的90%~95%,可用于甲肝减毒活疫苗的滴度检测。实施例4步骤(1),合成重组质粒:合成恒河猴的mavsc末端基因序列rhmavs(c397-541),构建lv-egfp-rhmavs(c397-541)-ires-puro-wpre重组质粒,经测序分析,序列正确,大提保存于-20℃备用。构建的egfp-rhmavs(c397-541)-ires-puro-wpre的结构见图15。步骤(2),慢病毒包装:转染前一天,消化293t细胞(用0.25%胰酶室温消化2min),以2.0×107个细胞量接种于t175培养瓶中(培养基为含10%fbs的dmem),转染当天细胞融合度达到40-50%时,进行转染。将重组质粒lv-egfprhmavs(c397-541)-ires-puro-wpre与包装质粒(pmd2.g和pspax2)按比例混合(重组质粒:pmd2.g:pspax2=5:2:3),采用lipfectmin2000进行转染,其中,lipfectmin2000与重组质粒和包装质粒的的总用量比为50ul:20ug,具体操作按说明书进行。转染后于37℃,5%co2培养箱中孵育12h;更换含10%(v/v)胎牛血清的dmem培养液,继续培养48h,荧光显微镜下观察荧光蛋白表达情况,收集含病毒颗粒的细胞上清液(用吸管取上清液),并经3000g、4℃离心5min,去除细胞碎片,收集病毒上清液,经24000rpm、4℃离心2h,去掉上清,加入200μl病毒冻存液,4℃溶解过夜,分装后,保存于-80℃冰箱。步骤(3),稳定细胞系构建:感染前一天,将huh7.0细胞传至6孔板中(培养基为含2ml10%fbs的dmem),放于37℃、5%co2细胞培养箱中培养,感染当天细胞融合度达到50%时,用制备的慢病毒进行感染。感染前,将细胞培养基更换成含2%fbs的dmem,加入收获的病毒液上清,其中,感染复数moi=0.1,于37℃培养过夜。次日将培养液更换成含10%fbs的高糖dmem,继续培养48h,于荧光显微镜下观察荧光蛋白的表达情况。将表达荧光蛋白的huh7.0细胞系用0.25%胰酶在室温消化5min,然后继续在t25培养瓶中培养,将培养基换成含5ug/mlpuromycin、10%fbs的dmem筛选表达荧光蛋白的huh7.0细胞系,命名为huh7.0-egfp-rhmavs(c397-541)(如图16)。感染前一天,将huh7.0-egfp-rhmavs(c397-541)细胞传至48孔板中(培养基的体积为250ul),放于37℃、5%co2细胞培养箱中培养。将hav病毒液按moi=1接种于上述细胞中,显微镜观察荧光弥散情况。如果细胞被hav感染,绿色荧光蛋白将会从细胞质弥散至整个细胞中(如图17)。步骤(4),滴度检测:将筛选得到的细胞系接种于48孔细胞培养板中(培养基为含10%fbs的dmem,接种量为5000个/孔),第二天,取10μlhav,用2%(v/v)fbsdmem按10倍系列稀释为10-1~10-6,吸弃48孔板中的培养基,将病毒稀释液按100μl/孔轻轻加入48孔板,每个稀释度设定3个复孔,摇匀后置于37℃、5%co2培养箱进行培养,每隔1h轻轻摇晃48孔板,4h后吸弃病毒稀释液,按250μl/孔加入含1%甲基纤维素、2%fbs的dmem,于37℃、5%co2培养箱中培养1-2d。荧光显微镜下观察荧光噬斑的个数,从而计算hav的滴度(如图18)。同时,将hav病毒做10倍系列稀释,取10-6、10-7、10-8稀释度的病毒液1ml,分别接种于人二倍体细胞-kmb17,每个稀释度做5个重复,置于35℃培养21天(培养基为含5%fbs的mem,培养过程每7天换液),收获后提取甲肝病毒,用酶联免疫法检测,按reed-muench法计算hav的滴度(ccid50)。(参照2015版《中华人民共和国药典》第三部)通过比较两种方法的滴度检测结果,得出两种方法测得滴度的比例关系。具体结果:1.获得了稳定表达egfp-rhmavs(c397-541)的细胞系-huh7.0-egfp-rhmavs(c397-541),而且荧光蛋白在细胞质中成点状分布如图15。2.hav感染huh7.0-egfp-rhmavs(c397-541)后,荧光蛋白会弥散到整个细胞中(如图16)。3.hav感染huh7.0-egfp-rhmavs(c397-541)后,荧光蛋白会弥散到整个细胞中,加入甲基纤维素后,会形成荧光“噬斑”点(如图17)。4.hav感染huh7.0-egfp-rhmavs(c397-541)后,荧光蛋白会弥散在细胞中,加入甲基纤维素后,会形成荧光“噬斑”点,通过观察荧光“噬斑”的个数,计算hav的滴度。利用此方法检测3批hav的病毒滴度,结果良好,3批病毒的lgffu/ml分别是7.10、6.89、6.97,elisa法检测结果lgccid50/ml分别是7.43、7.51、7.69。与elisa法相比,利用荧光重定位系统检测的hav病毒滴度小于lgccid50/ml,约为lgccid50/ml的90%~95%,可用于甲肝减毒活疫苗的滴度检测。以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。部分序列表如下表所示。序列表<110>中国医学科学院医学生物学研究所<120>一种用于检测甲型肝炎病毒滴度的细胞系及其构建方法与应用<160>9<170>siposequencelisting1.0<210>1<211>145<212>prt<213>人工序列()<400>1glyalathrglyglyserseralatrpleuaspsersersergluasn151015argglyleuglysergluleuserlysproglyvalleualasergln202530valaspserpropheserglycysphegluaspleualaileserala354045serthrserleuglymetglyprocyshisglyproglugluasnglu505560tyrlyssergluglythrpheglyilehisvalalagluasnproser65707580ileglnleuleugluglyasnproglyproproalaaspproaspgly859095glyproargproglnalaasparglyspheglngluarggluvalpro100105110cyshisargproserproglyalaleutrpleuglnvalalavalthr115120125glyvalleuvalvalthrleuleuvalvalleutyrargargargleu130135140his145<210>2<211>119<212>prt<213>人工序列()<400>2glyalaleuthrglyserserprotrpprosersersersergluser151015trpglyprogluglngluleuserlysproservalvalcysserser202530aspleualailesertyrserasnserleuglyleuglyhisthrleu354045asnvalargserpheglyilehisvalglugluasnproserilepro505560leuleualaaspasnproglyproseralaproproleuglnglyglu65707580gluglugluglugluglnglugluprocysvalargvalsertrpleu859095glyvalalavalalaglyalaphevalalametleuleualaleuleu100105110tyrargargargproleugln115<210>3<211>31<212>dna<213>人工序列()<400>3agctgtacaagggcgccactggaggcagctc31<210>4<211>33<212>dna<213>人工序列()<400>4cagacgcgtctagtgcagacgccgccggtacag33<210>5<211>54<212>dna<213>人工序列()<400>5gctgtacaagccaaagaagaagcggaaggtcgccggcgccactggaggcagctc54<210>6<211>31<212>dna<213>人工序列()<400>6agctgtacaagggtgccctcaccggcagttc31<210>7<211>28<212>dna<213>人工序列()<400>7cagacgcgttcactggactgggcgccgc28<210>8<211>24<212>dna<213>人工序列()<400>8ccaaagaagaagcggaaggtcgcc24<210>9<211>145<212>prt<213>人工序列()<400>9glyalaalaglyglyargseralatrpleuaspsersersergluasn151015glyglypheglusergluleuserlysproglyileleuvalsergln202530alaaspserglnpheserglycyssergluaspleualaileserala354045serthrserleuglymetglyprocysargglyproglugluasnglu505560tyrlyssergluglythrpheglyilehisvalalagluasnproser65707580ileglnleumetgluglyasnproglyproproalaaspproglngly859095glyproargprohisileaspglnlyspheglngluarggluglypro100105110cyshisargproserproglyalaleutrpleuglnalaalavalala115120125glyvalleuvalvalthrleuleuvalalamettyrargargargleu130135140his145当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1