HANP-含FC分子缀合物的制作方法
本发明涉及包含负载在充当载体的含fc分子上的hanp肽的缀合物(由此hanp肽在皮下施用后逐渐迁移至血液中且急剧延长hanp的药理作用的持续时间)、含有所述缀合物作为活性成分的药物、用于产生所述缀合物的方法等。
背景技术:
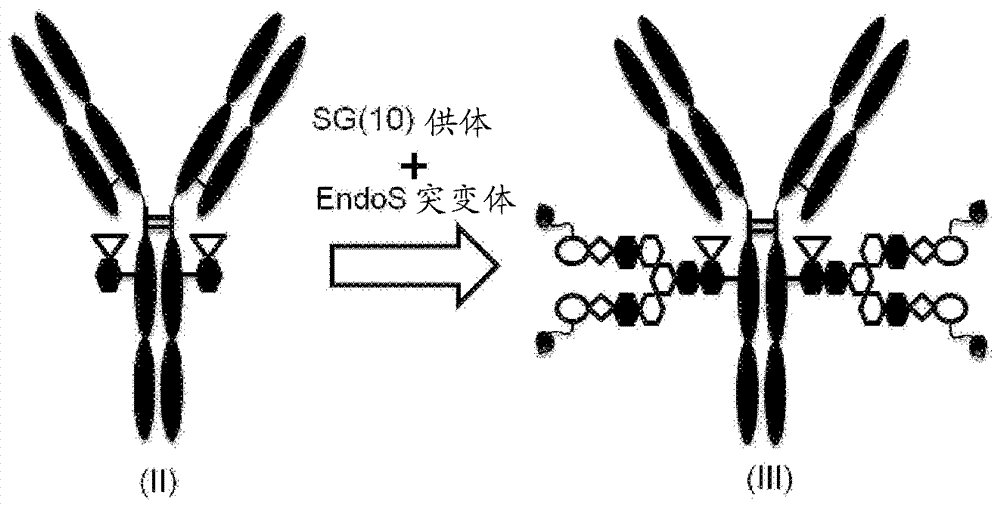
:人心房利钠肽(hanp)是具有血管舒张作用、利尿作用、细胞生长抑制作用、静脉回流降低作用和交感神经活动抑制作用的生物活性肽。天然hanp,例如,通过血液中的中性内肽酶(nep)的切割,迅速失去其在血液中的活性。在日本,hanp临床上用作急性心力衰竭的治疗药物,但需要在血压监测下经由静脉内输注等连续施用,其目的是避免施用后血压的急剧下降。内源性生物活性肽诸如hanp对其特异性受体具有非常高的选择性,且因此可以预期其具有高效力和安全性。另一方面,已知此类生物活性肽在血液中具有非常短的半衰期,因为生物活性肽在全身循环期间被各种代谢酶快速代谢或通过肾脏中的肾小球过滤迅速排泄。因此,已经尝试通过向其赋予代谢酶抗性或避免肾脏排泄来延长此类肽在血液中的半衰期。其实例包括各种方法,诸如糖基化肽(专利文献1)、融合多肽(专利文献2)、白蛋白融合肽(专利文献3和非专利文献1)、免疫球蛋白fc融合肽(专利文献4和非专利文献2)和聚乙二醇(peg)修饰的肽(非专利文献3和4)。通过由新生儿fc受体(fcrn)介导的再循环机制,与肽药物或蛋白药物相比,抗体药物在血液中具有非常长的半衰期(非专利文献5)。因此,预期产生类似再循环效果的融合肽也已被认为是延长hanp的半衰期的手段(专利文献3和4以及非专利文献1和2)。然而,已经提出,当在血液中快速代谢的hanp直接加载至载体蛋白上时,载体上的hanp部分在循环期间经历代谢。因此,“hanp-蛋白融合体”难以以与血液中保留载体蛋白部分等效的水平保留在血液中(非专利文献2)。近年中,已经积极地进行抗体-药物缀合物的技术开发,并且已经报道各种合成方法(非专利文献6、7和8)。然而,如果采用降低载体蛋白与待加载于其上的药物部分的相容性的缀合方法,则载体部分或药物部分可能去稳定化,使得不延长血液中的半衰期或增加凝集。因此,选择最佳的缀合方法或接头是非常重要的。通常,已知生物药物通过皮下施用而不是静脉内施用逐渐迁移至血液中(非专利文献9)。然而,难以预测化学修饰抗体的药代动力学。因此,需要开发具有以下所有的hanp制剂:逐渐迁移至血液中,在血液中足够的保留时间,以及维持药理作用所必需的活性。引文列表专利文献专利文献1:pct国际专利申请公开号wo2014/115797a1专利文献2:美国专利申请公开号us2006-036227专利文献3:美国专利申请公开号us2007-0162986a1专利文献4:pct国际专利申请公开号wo2008/154226a1非专利文献非专利文献1:regulatorypeptides,2012,175,7-10非专利文献2:bioconjugatechem.,2012,23,518-526非专利文献3:proc.natl.acad.sci.usa1994,91,12544-12548非专利文献4:bioconjugatechem.,2008,19,342-348非专利文献5:chem.soc.rev.,2012,41,2686-2695非专利文献6:bioconjugatechem.,2015,26,176-192非专利文献7:j.am.chem.soc.,2012,134,12308-12318非专利文献8:angew.chem.int.ed.,2016,55,2361-2367非专利文献9:drugmetab.dispos.,2014,42,1881-1889。发明概述技术问题本发明的一个目的是发现一种hanp的改进形式,其在皮下施用中具有药效和安全性。问题的解决方案本发明人已经对在皮下施用中具有药效和安全性的hanp的改进形式进行了勤奋的研究,并且因此发现:包含经由特定peg接头连接至附接至对应于igg重链的位置297的asn残基的聚糖的hanp肽的缀合物使表达gc-a受体的细胞中的细胞内cgmp浓度升高,当施用于大鼠时延长hanp肽在血液中的持续时间,并且甚至在施用缀合物后168小时或更晚可持续地升高血液中的cgmp浓度;采用糖基化hanp作为hanp肽的缀合物具有特别有利的物理特性;等。本发明人已经进行了进一步研究,由此完成了本发明。本发明提供了以下内容:(1)包含经由聚乙二醇接头(l(peg))与附接至含fc分子的asn297的聚糖(n297聚糖)键合的hanp肽的缀合物,或其药学上可接受的盐,其中:所述hanp肽任选地缺乏由seqidno:1代表的氨基酸序列中的n末端起连续1至5个氨基酸和/或一个c末端氨基酸,并且任选地在其n末端和c末端的任一个或两个处糖基化;所述l(peg)是包含10至35个乙二醇结构且任选地包含额外的结合结构和/或修饰结构的接头结构;所述含fc分子是具有对应于人iggfc区的氨基酸序列且不具有特异性结合人生物分子的能力的分子;且所述n297聚糖是聚糖n297-(fuc)sg、n297-(fuc)msg1或n297-(fuc)msg2,其具有由下式代表的结构:其中[l(peg)]代表l(peg)结合键合至β-man的1-3和1-6支链两者的非还原末端处的唾液酸残基的2-位的羰基,其中[l(peg)]代表l(peg)结合键合至β-man的1-3支链的非还原末端处的唾液酸残基的2-位的羰基,且其中[l(peg)]代表l(peg)结合键合至β-man的1-6支链的非还原末端处的唾液酸残基的2-位的羰基。(2)根据(1)所述的缀合物或其药学上可接受的盐,其中所述hanp肽是hanp(1-28)、hanp(2-28)、hanp(3-28)、hanp(1-27)、hanp(2-27)或hanp(3-27)。(3)根据(1)或(2)所述的缀合物或其药学上可接受的盐,其中所述hanp肽是糖基化肽,其中其侧链通过n-糖苷键附接至由下式代表的聚糖ag5、ag7、ag9和sg中任一者的天冬酰胺或谷氨酰胺键合至所述肽的n末端或c末端:其中"-(n/q)"代表通过n-糖苷键与天冬酰胺或谷氨酰胺的侧链的结合。(4)根据(3)所述的缀合物或其药学上可接受的盐,其中所述hanp肽是hanp(1-28),其中其侧链通过n-糖苷键附接至聚糖sg的天冬酰胺键合至所述肽的n末端。(5)根据(1)所述的缀合物或其药学上可接受的盐,其中所述l(peg)是包含25至30个乙二醇结构且包含酰胺键和1,2,3-三唑环作为结合结构的接头结构。(6)根据(1)所述的缀合物或其药理学上可接受的盐,其中所述l(peg)是由下式中任一者代表的接头结构:其中“hanp”代表与所述hanp肽的n末端结合;“n297gly”代表与n297-附接的聚糖的非还原末端的结合;每个接头结构具有在1,2,3-三唑环位点处形成键期间形成的几何异构结构,如所述式中的结构l(peg)-x1和l(peg)-x2(这些统称为l(peg)-x(x为a、b、c、d、e、f、g或h))中所示;并且,在缀合物的一个分子中的接头中仅存在所述结构中任一者,或者两个结构作为接头共存于缀合物的一个分子中。(7)根据(1)所述的缀合物或其药学上可接受的盐,其中所述含fc分子是针对作为抗原的除了人生物物质以外的物质的人igg单克隆抗体,或具有人iggfc区且缺乏可变区的人igg的片段或工程改造形式。(8)根据(7)所述的缀合物或其药学上可接受的盐,其中所述含fc分子是衍生自人igg的fc或由人igg恒定区组成的clch。(9)根据(7)所述的缀合物或其药学上可接受的盐,其中所述含fc分子是由重链和轻链的组合组成的抗体(mab-a),所述重链由来自seqidno:3的氨基酸位置20至474的氨基酸序列组成,且所述轻链由来自seqidno:5的氨基酸位置21至234的氨基酸序列组成;由重链和轻链的组合组成的clch(clch-a),所述重链由来自seqidno:7的氨基酸位置20至349的氨基酸序列组成,且所述轻链由来自seqidno:9的氨基酸位置21至125的氨基酸序列组成;由重链和轻链的组合组成的clch(clch-b),所述重链由来自seqidno:11的氨基酸位置20至349的氨基酸序列组成,且所述轻链由来自seqidno:9的氨基酸位置21至125的氨基酸序列组成;由来自seqidno:15的氨基酸位置21至243的氨基酸序列组成的fc片段(fc-b);或由来自seqidno:17的氨基酸位置21至247的氨基酸序列组成的fc片段(fc-a);或任何这种抗体的工程改造形式。(10)根据(1)所述的缀合物或其药学上可接受的盐,其中所述hanp肽是hanp(1-28)或(sg-)asn-hanp(1-28),所述peg接头是l(peg)-a、l(peg)-b、l(peg)-c、l(peg)-d、l(peg)-e、l(peg)-f、l(peg)-g或l(peg)-h,所述含fc分子是mab-a、clch-a、clch-b、fc-a或fc-b,且所述n297聚糖是n297-(fuc)sg。(11)根据(1)所述的缀合物或其药学上可接受的盐,其中所述hanp肽是(sg-)asn-hanp(1-28),所述peg接头是l(peg)-b,所述含fc分子是fc-a或fc-b,且所述n297聚糖是n297-(fuc)sg。(12)药物,其包含根据(1)至(11)中任一项所述的缀合物或其药学上可接受的盐作为活性成分。(13)根据(12)所述的药物,其中所述药物是用于可通过活化gc-a治疗的疾病的治疗剂或预防剂。(14)根据(12)所述的药物,其中所述药物是用于高血压、急性心力衰竭、慢性心力衰竭、急性肾衰竭或慢性肾衰竭的治疗剂或预防剂。(15)用于产生根据(1)至(11)中任一项所述的缀合物的方法,其包括以下步骤:步骤a:用水解酶处理动物细胞中产生的含fc分子以产生(fucα1,6)glcnac-含fc分子的步骤;步骤b1:在糖基转移酶存在的情况下使聚糖供体分子与(fucα1,6)glcnac-含fc分子反应以合成具有引入唾液酸中的叠氮化物基团的sg型聚糖重建的含fc分子的步骤,其中所述聚糖供体含有sg(10)或msg(9),其具有引入唾液酸中的叠氮化物基团且在还原末端处具有噁唑啉,或步骤b2:在两种内切糖苷酶存在的情况下使聚糖供体分子与(fucα1,6)glcnac-含fc分子反应以合成具有引入唾液酸中的叠氮化物基团的sg型聚糖重建的含fc分子的步骤,所述聚糖供体分子是具有引入其唾液酸中的叠氮化物基团的(sg-)asn或(msg-)asn;和步骤c:使在一侧具有hanp肽且在另一侧具有dbco的接头分子与步骤b中制备的具有引入唾液酸中的叠氮化物基团的sg型聚糖重建的含fc分子反应以合成根据(1)所述的缀合物的步骤。(16)根据(15)所述的产生方法,其进一步包括通过涉及使用羟基磷灰石柱纯化的步骤从步骤a的反应溶液纯化所述(fucα1,6)glcnac-含fc分子的步骤。(17)用于治疗或预防可通过活化gc-a治疗的疾病的方法,其包括将作为活性成分的根据(1)至(11)中任一项所述的缀合物或其药学上可接受的盐施用于需要所述施用的受试者。(18)根据(1)至(11)中任一项所述的缀合物或其药学上可接受的盐,其用于治疗或预防可通过活化gc-a治疗的疾病。(19)根据(1)至(11)中任一项所述的缀合物或其药学上可接受的盐用于产生可通过活化gc-a治疗的疾病的治疗剂或预防剂的用途。在方面(17)至(19)中,可通过活化gc-a治疗的疾病优选为高血压、急性心力衰竭、慢性心力衰竭、急性肾衰竭或慢性肾衰竭。本发明的有利效果本发明的缀合物具有以下所有:当皮下施用时,逐渐迁移至血液中,血液中的长期半衰期,以及长期维持药理作用。因此,本发明的缀合物在临床上适用于迄今已知的hanp制剂没有治疗效果的疾病,并且使得能够开发为患者提供改善的便利性的药物。此外,利用糖基化的hanp肽作为hanp肽的本发明的缀合物表现出急剧抑制的凝集和有利的物理特性。因此,预期其应用于一系列形式的制剂。附图简述[图1]图1示意性显示本发明的缀合物(分子(i))。部分(a)描绘hanp肽,部分(b)描绘peg接头,且部分(c)描绘n297聚糖(其中空心椭圆描绘neuac(sia),空心六边形描绘man,实心六边形描绘glcnac,空心长菱形描绘gal,且空心倒三角形描绘fuc)。y形状描绘含fc分子,为方便起见,其被举例说明为含有fab的全长igg。然而,本发明的缀合物中的含fc分子可以是具有与n297聚糖键合的fc区的任何分子。在该示意图中,为方便起见,n297聚糖表示为n297-(fuc)sg,并且以peg接头与所有支链的非还原末端处的唾液酸残基键合的形式显示。然而,作为n297聚糖采用的n297-(fuc)msg可以在两个支链中的任一个中具有与peg接头键合的唾液酸,并且在另一支链的非还原末端处不具有唾液酸。在整个本说明书中应用这种符号,除非另有指明。[图2]图2是显示(fucα1,6)glcnac-含fc分子(图2a的分子(ii))和sg型聚糖重建的含fc分子(图2b的分子(iii))(其为用于产生本发明的缀合物的中间体)的结构的示意图。在两个图中,y形状描绘与图1中相同的含fc分子。在图2a中,部分(d)描绘仅由在fuc的1-和6-位具有α-糖苷键的glcnac组成的n297聚糖。在图2b中,部分(c)描绘与图1中相同的n297聚糖,并且部分(e)描绘作为peg接头的部分结构的末端官能团(本文例举叠氮化物基团,尽管官能团不限于此),并进行与另一接头分子结合。peg接头的结合模式如图1中所述。[图3a]图3a和3b各自是从动物细胞中产生的含fc分子产生sg型聚糖重建的含fc分子的步骤的示意图。在图中,分子(ii)和(iii)分别描绘(fucα1,6)glcnac-含fc分子和sg型聚糖重建的含fc分子,如图2中所示。分子(iv)是在动物细胞中产生的含fc分子,并且是具有异质n297聚糖的分子的混合物。图3a显示用水解酶诸如endos处理分子(iv)中的异质n297聚糖以制备均质(fucα1,6)glcnac-含fc分子(ii)的步骤。本文使用的sg型聚糖供体分子在sg(10)、msg1(9)或msg2(9)的非还原末端处具有peg接头修饰的唾液酸。在制备的sg型n297聚糖重建的含fc分子中,非还原末端处的唾液酸也被类似地修饰,如图2b中所述。在图中,为方便起见,供体分子以使用sg(10)的形式表示。然而,通过使用msg1(9)或msg2(9)作为聚糖供体,合成重建的含fc分子(其中在n297聚糖的两个非还原末端中的任一个处具有官能团的接头分子与重建的抗体键合)作为分子(iii)。[图3b]图3a和3b各自是从动物细胞中产生的含fc分子产生sg型聚糖重建的含fc分子的步骤的示意图。在图中,分子(ii)和(iii)分别描绘(fucα1,6)glcnac-含fc分子和sg型聚糖重建的含fc分子,如图2中所示。分子(iv)是在动物细胞中产生的含fc分子,并且是具有异质n297聚糖的分子的混合物。图3b显示通过使用糖基转移酶诸如endosd233q突变体将sg型聚糖供体分子的聚糖转糖基化至含fc分子(ii)中的n297聚糖的glcnac以制备sg型聚糖重建的含fc分子(iii)的步骤。本文使用的sg型聚糖供体分子在sg(10)、msg1(9)或msg2(9)的非还原末端处具有peg接头修饰的唾液酸。在制备的sg型n297聚糖重建的含fc分子中,非还原末端处的唾液酸也被类似地修饰,如图2b中所述。在图中,为方便起见,供体分子以使用sg(10)的形式表示。然而,通过使用msg1(9)或msg2(9)作为聚糖供体,合成重建的含fc分子(其中在n297聚糖的两个非还原末端中的任一个处具有官能团的接头分子与重建的抗体键合)作为分子(iii)。[图4]图4显示化合物1-10([n3-peg(3)]2-sg(10)-ox)的nmr图。[图5]图5显示化合物1-11([n3-peg(3)]-msg1(9)-ox)的nmr图。[图6]图6是显示给予本发明的缀合物的皮下施用的大鼠的血浆cgmp浓度的时间依赖性变化的图。横坐标显示施用后经过的时间(h),且纵坐标显示cgmp浓度(pmol/ml)。每个数据通过平均值±标准偏差(n=2至4)表示。具有实心圆圈的实线显示化合物3-1的结果,具有空心圆圈的实线显示化合物3-2的结果,具有实心正方形的实线显示化合物3-3的结果,具有空心正方形的实线显示化合物3-4的结果,具有空心圆圈的虚线显示化合物3-5的结果,且具有空心正方形的虚线显示化合物3-6的结果。[图7]图7是显示给予本发明的缀合物的皮下施用的大鼠的血浆中检测到的所有含人fc分子和缀合物的量的时间依赖性变化的图。横坐标显示施用后经过的时间(h),且纵坐标显示检测到的物质浓度(μg/ml)。每个数据通过平均值±标准偏差(n=3至4)表示。实心圆圈显示化合物3-1的结果,空心圆圈显示化合物3-2的结果,实心正方形显示化合物3-3的结果,且空心正方形显示化合物3-4的结果。对于每种化合物,实线描绘检测到的缀合物的量,且虚线描绘检测到的含人fc分子的量。对于每种化合物,实线和虚线之间的重合显示缀合物在血液中维持而不比含fc分子更快速地降解单独的hanp(1-28)部分或不从含fc分子解离hanp(1-28)。[图8]图8是显示给予本发明的缀合物的皮下施用的大鼠的血浆cgmp浓度的时间依赖性变化的图。横坐标显示施用后经过的时间(h),且纵坐标显示cgmp浓度(pmol/ml)。每个数据通过平均值±标准偏差(n=2至4)表示。具有实心长菱形的实线显示化合物3-7的结果,具有空心长菱形的实线显示化合物3-8的结果,具有x-标记的实线显示化合物3-9的结果,具有空心三角形的实线显示化合物3-10的结果,具有实心三角形的实线显示化合物3-11的结果,具有实心长菱形的虚线显示化合物3-12的结果,具有x-标记的虚线显示化合物3-13的结果,且具有实心三角形的虚线显示化合物3-14的结果。[图9]图9是显示给予本发明的缀合物的皮下施用的猴的血浆中检测到的所有含人fc分子和缀合物的量的时间依赖性变化的图。横坐标显示施用后经过的时间(h),且纵坐标显示检测到的物质浓度(μg/ml)。每个数据通过平均值±标准偏差(n=3至4)表示。实心圆圈显示化合物3-2的结果,空心圆圈显示化合物3-4的结果,实心正方形显示化合物3-15的结果,空心正方形显示化合物3-16的结果,且实心长菱形显示化合物3-17的结果。对于每种化合物,实线描绘检测到的缀合物的量,且虚线描绘检测到的含人fc分子的量,如图7中所示。实施方案的描述其后,将详细描述本发明。本发明提供了包含经由peg接头连接至附接至含fc分子中保守的天冬酰胺(asn297)的n-连接的聚糖(n297聚糖)的hanp肽的缀合物,或其药学上可接受的盐。本发明的缀合物是大分子,其中连接多个结构单元诸如hanp肽、peg接头、含fc分子和构成它们的部分结构。该结构的示意图显示于图1中。在本发明中,当描述多个结构单元时,术语“连接”意味着这些结构单元通过共价键直接键合或经由接头间接键合,使得结构单元存在于一个分子中。连接结构单元的化学结构没有特别限制。构成本发明的缀合物的结构单元具有非常大的分子量和复杂的结构,且因此,为了方便起见,还可以通过使用符号以简化的形式描述。在这种符号描述中,hanp肽被称为“hanp”,peg接头被称为“l(peg)”或“peg(n)”(“n”是其中含有的连续的乙二醇单元(结构单元(-ch2-ch2-o-))的数量),含fc分子中含有的n297聚糖被称为“n297gly”,且含fc分子被称为"mab"、"clch"或"fc"。如下所述,聚糖也用“sg”等表示。结构单元之间的每个键可以从一般结合模式的符号描述中省略,而特征性结构或官能团可以通过有机合成化学领域中通常使用的表述来指示。在本发明的缀合物或其部分结构的符号中,氨基酸或肽的n末端(氨基)和c末端(羧基)分别在左侧和右侧表示,除非另有规定。在右侧具有符号“*”的氨基酸或肽(例如,gln*)代表与该规则相反,c末端和n末端分别在左侧和右侧表示。在氨基酸的符号中,直接键合至中心碳原子(α碳)的氨基和羧基(其为氨基酸必需的结构)分别被称为“α氨基”和“α羧基”。在本说明书中的缀合物或其部分结构的符号中,当氨基酸或肽在其n末端氨基连接至另一个接头时,代表待连接的结构单元的符号用连字符表示,并且在代表该肽或氨基酸的符号的左侧没有括号。在该情况下,连字符代表在肽或氨基酸的氨基和由接头结构携带的羧基之间形成的酰胺键。例如,其中sg与asn的氨基连接的结构被称为“sg-asn”。相反,当氨基酸或肽在其c末端羧基连接至本说明书中的另一个结构单元时,代表待连接的结构单元的符号用连字符表示,并且在代表该氨基酸或肽的符号的右侧没有括号。在该情况下,连字符代表在肽或氨基酸的c末端羧基和由接头结构携带的氨基(或叠氮化物基团)之间形成的酰胺键。例如,sg与tyr的羧基连接的结构被称为“tyr-sg”。当氨基酸在其侧链与本说明书中的聚糖连接时,该部分结构被称为例如“(sg-)asn”,其中侧链部分包括在括号中。在本发明中,“聚糖”意指通过糖苷键与彼此键合的两个或更多个单糖的结构单元。特定的单糖或聚糖也用缩写(例如“glcnac-”或“sg-”)表示。当聚糖由具有这些缩写的结构式代表时,在代表聚糖的缩写中不包括属于在还原末端处具有另一个结构单元的糖苷键的氧原子或氮原子,除非另有定义。在本说明书中,充当聚糖的基本单元的单糖以其环结构表示,其中为方便起见,与构成环的氧原子键合且与羟基(或属于糖苷键的氧原子)直接键合的碳原子被定义为1-位(仅对于唾液酸为2-位),除非另有指定。实施例的化合物根据其整个化学结构命名,使得该规则不一定适用于此。聚糖中含有的单糖没有特别限制,只要单糖具有糖的基本结构。可以使用各种单糖诸如6-元和5-元糖。单糖可以是天然存在的糖或可以是人工合成的糖。天然存在的糖是优选的。单糖的实例可以包括葡萄糖(glu)、果糖(fru)、甘露糖(man)、半乳糖(gal)、葡糖胺(glc)、n-乙酰葡糖胺(glcnac)、葡糖醛酸(gluca)、神经氨酸(neu)、唾液酸/n-乙酰神经氨酸(sia/neunac/neu5ac)、半乳糖胺,n-乙酰半乳糖胺(galnac)、木糖(xyl)、艾杜糖醛酸(idoa)、岩藻糖(fuc)、丙醛糖、甘油醛、丁醛糖、赤藓糖、苏阿糖、戊醛糖、核糖、来苏糖、阿拉伯糖、己醛糖、阿洛糖、塔罗糖、古洛糖、醛糖、艾杜糖、酮丙糖、二羟基丙酮、丁酮糖、赤藓酮糖、戊酮糖、木酮糖、核酮糖、己酮糖、阿洛酮糖、山梨糖和塔格糖。当聚糖在本说明书中由符号(例如,gly、sg或glcnac)表示时,该符号还包括在还原末端处的碳并且不包括属于n-或o-糖苷键的n或o,除非另有定义。同样地,当hanp肽由符号(例如,hanp或hanp(1-28))表示时,该符号通常还包括n末端-nh和c末端c=o。n末端和c末端分别在左侧和右侧表示,除非另有指明。具体地,未修饰的hanp肽被称为h-hanp-oh。<hanp肽>在本发明中,“hanp肽”意指由包含至少人心房利钠肽((seqidno:1;下文中,称为天然hanp或hanp(1-28)),其是由28个氨基酸组成的生物活性肽)的氨基酸序列中的7-至27-位的氨基酸的氨基酸序列组成的肽。天然hanp通过结合在细胞表面上表达的gc-a受体(chinkersm,等人,nature338;78-83,1989))、活化受体的细胞内结构域中存在的鸟苷酸环化酶并升高细胞内cgmp浓度而发挥其生物活性。关于天然hanp,biochem.biophys.res.commun.,vol.118,p.131,1984中描述的α-hanp已经在日本被批准以“卡培立肽”的通用名称制造和销售,并且是商业可得的(商品名:hanp)。α-hanp通常也被称为人pro-anp[99-126]。天然hanp具有由seqidno:1的7-位和23-位的cys残基通过二硫键形成的分子内环结构。已知该环结构和直至27-位的arg残基的c末端氨基酸对于hanp活化gc-a受体是重要的(silver,ma,curr.opin.nephrol.hypertens.(2006),15,p.14-21;和a.calderone,minervaendocrinol.(2004),29,p.113-127)。因此,由该环结构组成的hanp(7-27)被认为是用于活化gc-a的最小单位。本发明的hanp肽是由可以缺乏seqidno:1中的n末端起连续1至6个氨基酸和/或28-位的氨基酸的氨基酸序列组成的肽,并且是优选地,是可以缺乏seqidno:1的1-位、1-和2-位和28-位的氨基酸位点中的至少一个的肽,更优选地,是由可以缺乏seqidno:1的1-位的氨基酸或1-和2-位的氨基酸的氨基酸序列组成的肽(hanp(2-28),hanp(3-28),等),最优选地,是由seqidno:1的氨基酸序列组成的肽(hanp(1-28))。在本发明中,例如,seqidno:1中所示的肽被称为“hanp(1-28)”,缺乏seqidno:1的1-位的氨基酸的肽被称为“hanp(2-28)”,并且缺乏seqidno:1的1-和2-位的氨基酸的肽被称为“hanp(3-28)”。缺乏一部分氨基酸的其它肽也以对应于上文所示的方式提及。本发明的缀合物的一个分子中的hanp分子的数量根据与含fc分子或peg接头键合的n297聚糖的结构而不同,且通常为2或4。在本发明的缀合物中,peg接头可以通过例如与n末端α-氨基或c末端α-羧基的酰胺键与hanp肽键合。优选地,peg接头与hanp肽的n末端键合。在本发明的缀合物中,hanp肽可以是糖基化的。专利文献1中描述了hanp肽的糖基化的各种形式。通过使用这些糖基化形式,可以将多种糖基化肽应用于本发明。结构上类似于存在于人体内的聚糖的聚糖被适当地用作用于修饰hanp肽的聚糖。天然糖蛋白中含有的聚糖被广泛分类为与糖蛋白的天冬酰胺附接的n-连接的聚糖和与其丝氨酸或苏氨酸附接的o-连接的聚糖,其二者都具有其特征性的基本结构。自然地,n-连接的聚糖通过n-糖苷键与蛋白的氨基酸侧链键合,而o-连接的聚糖通过o-糖苷键与其键合。人工聚糖可以通过任何糖苷键与其它化合物键合。因此,糖苷键的类型不受这种聚糖结构的限制。例如,聚糖在其还原末端进行叠氮化,并且该叠氮化的聚糖可以在三苯基膦存在的情况下与具有羧基的化合物反应,以通过n-糖苷键将具有所需结构的化合物键合至聚糖。或者,聚糖可以与具有羟基的化合物(诸如醇)反应,以通过o-糖苷键将聚糖键合至所需化合物。n-连接的聚糖的基本结构由以下结构式和序列代表。具有该聚糖结构的聚糖被称为ag5。其中"-(n/q)"代表通过n-糖苷键与asn或gln的侧链的结合。大多数n-连接的聚糖具有该基本结构。其非还原末端或支链糖可以进一步与另一糖键合。人类聚糖或人类相容的聚糖是已知在人体内不表现出抗原性的聚糖。例如,n-连接的聚糖的高甘露糖、复杂和复合形式是已知的。高甘露糖形式是具有富含甘露糖的结构的聚糖,其由在n-连接的基本结构的非还原末端处的多个连续的甘露糖残基构成。复杂形式是在n-连接的基本结构的非还原末端处具有galβ1-4glcnac基序结构的聚糖。复合聚糖是在n-连接的基本结构的非还原末端处具有galβ1-4glcnac基序结构并且还具有由多个甘露糖残基组成的富含甘露糖的结构的聚糖。n-连接的复杂聚糖通常是在鸡蛋黄中所含的唾液酸化糖肽(下文中称为“sgp”)中所含的聚糖。其实例可以包括具有由以下结构式和序列代表的结构的唾液酸化聚糖(下文中,称为“sg”):其中"-(n/q)"代表通过n-糖苷键与asn或gln的侧链的结合。可以根据常规方法、例如wo2011/0278681中描述的方法从鸡蛋黄分离和纯化sgp。或者,sgp的纯化产物是商业可得的(tokyochemicalindustryco.,ltd.或fushimipharmaceuticalco.,ltd.)并且可以购买。此外,例如,仅由在sg的聚糖部分的还原末端处缺乏一个glcnac的聚糖(下文中,该聚糖被称为“sg(10)”)组成的二唾液酸八糖(由tokyochemicalindustryco.,ltd.制造)是商业可得的。在本说明书中,仅sg(10)β-man的两个支链中的任一个的非还原末端处缺乏唾液酸的聚糖结构被称为msg(9),仅在聚糖的1-3支链中具有唾液酸的聚糖结构被称为msg1(9),且仅在聚糖的1-6支链中具有唾液酸的聚糖结构被称为msg2(9)。使用二唾液酸糖,通过使用已知的转糖基化反应,可以制备通过用另一糖替代sg的还原末端处的glcnac而在还原末端处工程改造的聚糖。通过用glc替代在sg的还原末端处的glcnac而在还原末端处工程改造的聚糖被称为sg(glc)。通过用man替代在sg的还原末端处的glcnac而在还原末端处工程改造的聚糖被称为sg(man)。可用作本发明的聚糖的工程改造的聚糖的具体实例可以包括ag9(下面给出ag9的结构式和序列),其由于sg的神经氨酸酶处理而在两个非还原末端处缺乏neu5ac残基;ag(7)(下面给出ag7的结构式和序列),其由于ag9的半乳糖苷酶处理而在两个非还原末端处缺乏gal残基;和ag5(具有n-连接的基本结构的聚糖,如上所述),其由于用n-乙酰氨基葡糖苷酶进一步处理ag7在两个非还原末端处缺乏glcnac残基。此外,在ag(9)、ag(7)和ag(5)的还原末端处工程改造的聚糖(例如,ag(9)的还原末端处的glcnac被glc替代的ag(9-glc),和ag(9)的还原末端处的glcnac被man替代的ag(9-man))可以通过与上述相同的处理使用在sg的还原末端处工程改造的聚糖(例如,sg(glc)或sg(man))代替sg来获得,并且可以用于本发明的糖基化。其中"-(n/q)"代表通过n-糖苷键与asn或gln的侧链的结合。其中"-(n/q)"代表通过n-糖苷键与asn或gln的侧链的结合。在本发明的缀合物中,与hanp肽键合的聚糖数没有上限。聚糖的数目是例如3或更少,优选2或1。附接至hanp肽的聚糖可以具有相同的结构或可以是结构不同的聚糖的混合物。优选地,所有这些聚糖都具有相同的聚糖结构。本发明的糖基化hanp肽的一个实例可以包括hanp肽,其包含n-连接的聚糖,如上所提及,其通过n-糖苷键附接至asn或gln的侧链("(gly-)asn"或"(gly-)gln"。在这种肽中,seqidno:1的氨基酸序列的1-至5-和28-位的任何一个或多个氨基酸可以用(gly-)asn或(gly-)gln替代。或者,(gly-)asn或(gly-)gln可以通过肽键键合至由包含seqidno:1的6-至27-位的连续氨基酸序列组成的肽的n-末端和c-末端中任一个或两个。这种肽优选为具有通过肽键键合至hanp(1-28)、hanp(2-28)、hanp(3-28)、hanp(4-28)、hanp(5-28)或hanp(6-28)的n末端和/或c末端的(gly-)asn或(gly-)gln的肽,更优选地具有通过肽键键合至hanp(1-28)、hanp(2-28)或hanp(3-28)的n末端的(sg-)asn或(sg-)gln的肽,甚至更优选地(sg-)asn-hanp(1-28)。在本发明中,在使用天然存在的糖蛋白-或糖脂-衍生的聚糖作为用于修饰hanp肽的聚糖的情况下,根据例如专利文献1中描述的方法,可以在通过使用水解酶切割或分离或通过使用糖基转移酶的转糖基作用转移至所需化合物(受体化合物)后使用该聚糖。例如,可以根据已知方法通过经由与水解酶(endom等)反应来水解切割sgp或通过使用内切糖苷酶(endomn175q突变体等)转移至所需化合物来分离sg。此外,sgp用肽酶诸如肌动蛋白酶处理,且由此降解成(sg-)asn或含有(sg-)asn的肽片段,并且聚糖键合的级分可以通过已知的分离方法纯化以获得聚糖附接的氨基酸。因此获得的(sg-)asn、(sg-)gln或含有其的肽片段可以通过肽键通过使用保护基团的常规反应键合至hanp肽的n末端和/或c末端,以产生糖基化的hanp肽。<含fc分子>在本发明的缀合物中,“含fc分子”经由igg重链fc区域中良好保守的天冬酰胺(被称为“asn297”;其经历通过n-连接聚糖的修饰)的侧链的聚糖(该聚糖被称为“n297聚糖”)连接至hanp肽,并且作为载体蛋白发挥功能,用于将hanp肽在血液中维持长时段。因此,所述含fc分子需要具有对应于人iggfc区的氨基酸序列且不具有特异性结合人生物分子的能力。对应于iggfc区的氨基酸序列的实例可以包括来自seqidno:7的氨基酸位置128至349的氨基酸序列,来自seqidno:11的氨基酸位置128至349的氨基酸序列,来自seqidno:15的氨基酸位置22至243的氨基酸序列,来自seqidno:17的氨基酸位置26至247的氨基酸序列,和由这些序列工程改造的氨基酸序列。igg由重链和轻链组成。重链和轻链通过二硫键连接,并且重链在铰链区通过二硫键进一步彼此连接以形成同二聚体。igg还具有结构域结构,其中包含可变区且具有结合抗原的能力的fab结构域经由铰链区连接至结合fc受体的fc结构域。本发明的含fc分子没有特别限制,只要含fc分子包含fc区。例如,可以使用全长igg重链、含有fc区的抗体片段或通过部分工程改造衍生自任何其氨基酸序列的工程改造形式。本发明的含fc分子可以仅由重链组成,或者可以进一步具有适合于重链的结构的轻链。充当含fc分子的起源的igg的亚类没有特别限制,并且可以选择任何亚类。亚类优选为igg1、igg2、igg3或igg4,更优选为igg1。igg恒定区的氨基酸序列是非常保守的,并且每个氨基酸由edelman等人(biochemistry,(1969)vol.63,pp.78-85)提供的eu索引定义。例如,在fc区中附接至n-连接的聚糖的asn297对应于基于eu索引的297-位。即使实际氨基酸位置由于分子片段化或区域缺陷而不同,也根据eu索引通过显示明确地标识氨基酸。在采用全长igg作为含fc分子的情况下,全长igg没有特别限制,只要igg不具有特异性结合通常存在于人体内的物质的能力。如果有的话,针对作为抗原的非人动物蛋白的igg可能表现出与相应或相关的人分子的交叉反应性。因此,优选的是选择针对不含相应或相关人分子的抗原的单克隆抗体。甚至可以采用可能结合人体内的物质的抗体作为本发明的含fc分子,只要该分子已被修饰,因此其由于通过基因工程改造方法向其可变区中引入突变而缺乏针对人体物质的结合活性。用作本发明的含fc分子的全长igg更优选为针对作为抗原的非哺乳动物生物体衍生分子的igg,甚至更优选地针对作为抗原的微生物衍生分子的igg,进一步优选地针对作为抗原的脂多糖(lps)的igg。这种单克隆抗体描述于例如wo2015/046505中。其具体实例包括mab-a,其由重链和轻链的组合组成,所述重链由来自seqidno:3的氨基酸位置20至474的氨基酸序列组成(氨基酸位置1至19对应于信号肽,且编码重链的核苷酸序列显示于seqidno:2),且所述轻链由来自seqidno:5的氨基酸位置21至234的氨基酸序列组成(氨基酸位置1至20对应于信号肽,且编码轻链的核苷酸序列显示于seqidno:4)。在采用抗体片段或工程改造形式作为本发明的含fc分子的情况下,抗体片段或工程改造形式没有特别限制,只要抗体片段或工程改造形式包含允许二聚化的形式的fc区域。可以采用各种序列。可以采用维持可变区的一部分或全部的工程改造形式,只要充当其来源的抗体不具有如上所提及的特异性结合人体物质的能力。缺乏可变区的片段或工程改造形式是优选的。这种工程改造形式的实例可以包括缺乏人igg重链可变区且由人igg重链恒定区组成的ch。在采用ch作为本发明的含fc分子的情况下,充当其来源的igg的亚类没有特别限制。衍生自人igg1的ch的氨基酸序列(该ch被称为“ch-a”)显示于seqidno:7的氨基酸位置20至349(n末端1-至19-位对应于信号肽,且编码ch-a的核苷酸序列显示于seqidno:6)。在该序列中,铰链区是从118-位的glu至132-位的pro的epkscdkthtcppcp,fc区是从133-位的ala至349-位的lys,且199-位的asn对应于asn297(对于seqidno:11也是如此)。在选择ch作为含fc分子的情况下,可以仅采用ch,或者可以采用具有ch与仅由轻链恒定区组成的cl的组合的clch。igg1轻链cl的氨基酸序列(被称为“cl-a”)显示于seqidno:9的氨基酸位置21至125(1-至20-位的n末端氨基酸对应于信号肽,且编码cl-a的核苷酸序列显示于seqidno:8)。本发明的含fc分子的优选实例可以包括clch。更优选组合具有ch-a和cl-a的clch-a。在采用主要由fc区组成的片段或工程改造形式作为本发明的含fc分子的情况下,例如,可以采用由在其n末端附接至氨基酸序列cppc(铰链区的一部分)的iggfc区的氨基酸序列组成的抗体片段(igg1的序列的实例是来自seqidno:7的128-至349-位的序列)。铰链区是增强含fc分子的结构自由度且不影响如本发明中所预期的其作为载体蛋白的功能的区域。因此,可以采用其长度适当调整的铰链区,只要铰链区包含cppc。主要由fc构成的这种抗体片段的实例包括igg1衍生的含fc分子(其由来自seqidno:7或seqidno:11的118-位的glu至349-位的lys的氨基酸序列组成,其可以从所述氨基酸序列的n末端连续缺乏1至10个氨基酸),以及由其氨基酸序列工程改造的工程改造形式。主要由fc组成的抗体片段优选为含fc分子,其由来自seqidno:7的123-位的asp至349-位的lys、127-位的thr至349-位的lys或126-位的his至349-位的lys的氨基酸序列组成。与fc-a和fc-b相关的分子的实例可以包括由以下组成的含fc分子:来自seqidno:15的氨基酸位置22至243的氨基酸序列或来自seqidno:17的氨基酸位置26至247的氨基酸序列,或其在其n末端附接至氨基酸残基t、ht、tht、ktht或dktht的氨基酸序列,以及由这些氨基酸序列工程改造的工程改造形式。该分子优选为由以下组成的含fc分子:来自seqidno:15的氨基酸位置21至243的氨基酸序列,来自seqidno:17的氨基酸位置25至247的氨基酸序列,或来自seqidno:17的氨基酸位置21至247的氨基酸序列。这些fc片段中的asn297是seqidno:15的93-位的asn或seqidno:17的97-位的asn。在设计抗体片段或工程改造形式作为本发明的含fc分子的情况下,可以采用在1至数个位点(优选5个或更少位点,更优选地3、2或1个位点)具有1至数个(每个工程改造位点,优选地20个或更少,更优选地15个或更少,甚至更优选地10个或更少,进一步优选地7、6、5、4、3、2或1个)氨基酸的取代、缺失、插入和/或添加的工程改造形式,而不损害作为载体蛋白的功能,只要维持用于在铰链区中二聚化的cys,有助于fc区内的分子内二硫键的cys,用于附接n297聚糖的asn297,和其邻近的氨基酸。对于氨基酸序列中的工程改造位点,进行n末端和/或c末端氨基酸的取代、缺失和/或添加等。具体地,n末端氨基酸可以通过生物工程改造方法影响含fc分子的产生,并且可以被工程改造成适合于所需生产系统的氨基酸序列。基于eu索引的氨基酸leu234和leu235已知为通过抗体与fc受体结合而通过t细胞活化影响效应子活性的展示的位点。在该区域中含有的leu可以用ala替代(所得突变体被称为“lala形式”),使得可以消除该效应子活性,以便降低不良反应的风险(美国专利号us5885573)。如果必要,可以进行这种工程改造。在本发明的实施例中,分别制备clch-b和fc-a作为clch-a的llca形式和fc-b的lala形式,并证实其作为本发明的缀合物中适当地作为载体分子发挥功能。为了改进目标分子的表达或纯化效率的目的,可以向其中添加信号序列、肽酶识别序列或标签序列诸如gst。用于本发明中的含fc分子具有n297聚糖,并且在使用动物细胞的生产过程中通过翻译后修饰将n297聚糖重建为来自最初附接至含fc分子的asn297的异源聚糖的具有以下给出的任何结构的sg型聚糖(sg型n297聚糖)。通常,在从动物细胞生产期间,聚糖附接至二聚体的两个单体中的asn297残基,以产生每个二聚的含fc分子具有两个n297聚糖部分的正常形式。然而,取决于含fc分子的生产条件或结构,可以以给定速率产生其中n297聚糖附接至仅单体(每个二聚的含fc分子具有一个n297聚糖部分的分子)之一的聚糖缺失突变体。甚至可以在本发明中使用包含聚糖缺失突变体的这种含fc分子。其中[l(peg)]代表与键合至β-man的1-3和1-6支链两者的非还原末端处的唾液酸残基的2-位的羰基结合的l(peg)。其中[l(peg)]代表与键合至β-man的1-3支链的非还原末端处的唾液酸残基的2-位的羰基结合的l(peg)。其中[l(peg)]代表与键合至β-man的1-6支链的非还原末端处的唾液酸残基的2-位的羰基结合的l(peg)。当本发明的缀合物中的n297聚糖是n297-(fuc)msg1或n297-(fuc)msg2时,缀合物是具有两个peg接头部分和两个与其键合的hanp肽部分的分子(二价hanp肽),因为含fc分子是通常在两个单体中具有n297聚糖的二聚体(当含fc分子是聚糖缺失突变体时,n297-(fuc)msg1或n297-(fuc)msg2仅附接至单体之一以形成单价hanp肽)。另一方面,当n297聚糖是n297-(fuc)sg时,缀合物是具有四个peg接头部分和四个与其键合的hanp肽部分的分子(四价hanp肽),因为含fc分子是二聚体(当含fc分子是聚糖缺失突变体时,n297-(fuc)sg仅附接至所述单体之一以形成具有二价hanp肽的缀合物)。本发明的“缀合物”可以代表具有这些多种类型的n297聚糖的分子,或者作为这些正常形式的分子以及作为聚糖缺失突变体的分子的混合物(为方便起见,在本发明中,这种混合物被表示为正常形式的缀合物),或者可以是具有具备任何一种结构的n297聚糖的分子。本发明的缀合物优选为具有具备任何一种结构的sg型n297聚糖的分子,更优选地具有n297-(fuc)sg作为n297聚糖且具有四价hanp肽(或用于聚糖缺失突变体的二价hanp肽)的缀合物。在这些聚糖结构中,在还原末端处的岩藻糖基化glcnac(fucα1,6)glcnac)衍生自动物细胞中产生的含fc分子,由此将非还原末端侧的聚糖重建为类似于上面提及的sg的聚糖结构的聚糖结构。在任何情况下,通过使用与在非还原末端处的唾液酸的2-位键合的羧酸,所述聚糖与peg接头键合。具有sg型n297聚糖的这种含fc分子可以通过如图3中所示的方法根据例如wo2013/120066中描述的方法来产生。当根据已知方法使用动物细胞产生含fc分子作为重组蛋白时,n297聚糖具有岩藻糖基化的n-连接的聚糖结构作为基本结构。在这种情况下,含fc分子被获得为具有具备各种结构的聚糖的抗体或其片段的混合物,其中非还原末端处的结构或组成糖被不同地修饰(图3a的分子(iv))。在动物细胞中产生的这种含fc分子用水解酶诸如endos处理,使得在还原末端处的壳二糖结构的glcnacβ1和4glcnac之间的糖苷键被水解,以获得具有具备仅(fucα1,6)glcnac作为n297聚糖的单一聚糖结构的含fc分子(该含fc分子被称为“(fucα1,6)glcnac-含fc分子”;参见图2a)(图3a)。例如,endos或其维持水解活性的突变型酶可用作用于n297聚糖的水解反应中的这种酶。可以通过使用内切糖苷酶诸如endosd233q突变体使通过水解反应获得的(fucα1,6)glcnac-含fc分子作为聚糖受体分子与sg型聚糖供体分子反应以获得具有具备上面提及的结构的sg型n297聚糖的含fc分子(参见图2b)(图3b)。当目标化合物是具有四价hanp肽的缀合物时,具有sg(10)作为聚糖的聚糖供体分子用于其转糖基化反应中。使用的这种sg(10)聚糖可以通过水解等从例如sgp获得,或者可以是单独的sg(10)聚糖,诸如商业可得的二唾液酸八糖(tokyochemicalindustryco.,ltd.)。当目标化合物是具有二价hanp肽的缀合物时,采用具有msg1(9)或msg2(9)作为聚糖的聚糖供体分子。这种聚糖可以根据实施例1-11中描述的方法使用商业可得的单唾液酸-asn游离形式(1s2g/1g2s-10nc-asn,glytech,inc.)作为起始材料通过分离来使用,或者可以在没有分离的情况下用作混合物。供体分子中含有的sg型聚糖的还原末端处的glcnac优选在通过用2-氯-1,3-二甲基-1h-苯并咪唑-3-鎓-氯化物处理而例如以噁唑啉的形式活化后使用,但在同时使用如后面所提及的两种酶的情况下不必活化。供体分子中含有的sg型聚糖具有具备peg接头或具有其部分结构的接头分子的聚糖,其与非还原末端处的唾液酸中含有的羧酸键合。可以采用各种酶作为这种用于转糖基化反应中的酶,只要该酶具有将复杂聚糖转移至n297聚糖的活性。endosd233q是优选的,其为endos的工程改造形式,其通过用gln替代位置233的asp而表现出抑制的水解反应性。使用endosd233q的转糖基化反应描述于wo2013/120066等中。或者,可以利用工程改造的酶诸如endosd233q的另一突变体,即endosd233q/q303l、endosd233q/e350a、endosd233q/e350q、endosd233q/e350d、endosd233q/e350n或endosd233q/d405a。使用endosd233q的这种工程改造形式的转糖基化反应描述于wo2017/010559中。含fc分子的聚糖重建(糖水解和转糖基化反应)后的含fc分子的纯化操作旨在将含fc分子与反应中使用的低分子量化合物和酶分离。这种纯化通常采用凝胶过滤色谱、离子交换色谱、亲和色谱等。添加使用羟基磷灰石柱进一步纯化被证实为改进转糖基化反应效率。具体地,本发明提供了用于产生缀合物的方法,其进一步包括在含fc分子的糖水解后从反应溶液纯化中间体的步骤中使用羟基磷灰石柱的纯化步骤。根据报道的聚糖重塑的情况(j.am.chem.soc.2012,134,12308-12318;和angew.chem.int.ed.2016,55,2361-2367),用水解酶处理的含fc分子的反应溶液仅使用蛋白a柱(亲和色谱柱)纯化。然而,发现该纯化方法不能完全除去水解酶(endos)并且由于残余酶的影响而影响后续转糖基化反应。作为研究本文的纯化方法的结果,使用蛋白a柱和羟基磷灰石柱(cht柱,bio-radlaboratories,inc.)以该顺序纯化用水解酶处理的含fc分子的反应溶液,以由此改进后续转糖基化反应的反应效率,而不受残余酶影响。本发明还提供了通过同时使用两种类型的酶将具有未活化还原末端作为聚糖供体的sgp、(sg-)asn等直接转糖基化至含fc分子的n297聚糖的方法。通常的转糖基化反应需要活化聚糖供体的还原末端。这种活性供体的制备是耗时且昂贵的。本发明的方法可以在转糖基化反应中直接采用天然可获得或商业可得的糖肽等,且因此能够实现有效的聚糖重建。对于使用的两种类型的endo酶,重要的是适当地组合用于作为底物的广泛范围的复杂聚糖的酶a(endom-样酶)和用于作为底物的含fc分子的n297聚糖的酶b(endos-样酶)。酶a的实例可以包括endom、endoom和endocc,以及表现出降低的水解活性的endom突变体、endoom突变体和endocc突变体。酶a优选为endomn175q、endoccn180h或endoomn194q。酶b的实例可以包括endos和endos2(endos49),以及表现出降低的水解活性的endos突变体和endos2(endos49)突变体。酶b优选为endosd233q、endosd233q/q303l、endosd233q/e350a、endosd233q/e350q、endosd233q/e350d、endosd233q/e350n、endosd233q/d405a等。聚糖供体的结构没有特别限制,只要聚糖供体具有被所采用的酶a识别的聚糖结构。可以使用各种分子,诸如天然获得的分子和在化学反应或酶促反应中可合成的分子。可以使用除了r=h以外的任何取代基作为异头物位点处的取代基r。在n-连接的聚糖结构(参见下面给出的式;异头物的取代基r是在最上部结构式的还原末端处的r)的情况下,其实例可以包括酰胺结构和叠氮化物。在o-连接的聚糖结构(参见下面给出的式;异头物的取代基r是在最上部的结构式的还原末端处的r)的情况下,其实例可以包括乙二醇结构、乙醇酸结构和苄基、烯丙基和对硝基苯基,其用作异头物羟基的保护基团。聚糖供体的非还原末端处的结构没有特别限制,只要该结构被酶a识别。可以使用各种结构,诸如天然聚糖结构、天然聚糖结构的非还原末端聚糖缺失突变体、其中任意羟基被磷酸修饰的结构以及与接头结构化学键合的结构。聚糖供体优选为sgp、(sg-)asn、([n3-peg(3)]2-sg)-asn-peg(3)-n3等。用于本发明的方法中的受体分子没有特别限制,只要受体分子是具有n297聚糖的含fc分子。可以适当地选择和使用各种类型,诸如mab、clch和fc片段。反应温度可以根据所用酶的最佳温度适当地选择,且通常为15至50℃,优选25至40℃。在采用两种类型的酶的本发明的方法中,如果所述酶之一在另一种酶的最佳温度下失活,则转移反应不能适当地进行。因此,优选的是在诸如最佳反应温度的条件下选择类似的酶的组合。<peg接头>在本发明中,peg接头意指这样的化学结构,其包含聚乙二醇结构,并介导hanp肽与本发明的缀合物中的含fc分子的连接。peg接头结构通常由不含任何支链结构的线性结构组成,并且在一端结合hanp肽的n末端氨基或c末端羧基,并在另一端结合含fc分子的n297聚糖中的唾液酸的2-位的羧酸。本发明的缀合物中含有的peg接头以线性形式包含约10个或更多个乙二醇单元。该长度对于维持所述缀合物表现出的hanp的活性是重要的。接头结构中含有的乙二醇单元(-ch2-ch2-o-)的数量的上限为约50或更少,优选约40或更少,更优选约35或更少,甚至更优选约30或更少。接头结构中含有的乙二醇单元的数量的下限优选为约15或更多,更优选约20或更多,甚至更优选约25或更多。这种peg接头可以衍生自单一分子,或者可以由多个部分结构(其中多个分子彼此键合)组成。这种充当接头结构的来源的分子被称为“接头分子”。当本发明的peg接头衍生自一个接头分子时,优选的是选择接头分子中含有的官能团,使得peg接头结合hanp肽的n末端,其用于结合含fc分子的sg型n297聚糖中的唾液酸的2-位的羧酸,且用于确保缀合物的均质结构。当本发明的peg接头衍生自多个接头分子时,至少含有结合n297聚糖中唾液酸的2-位的羧酸的官能团的化合物和具有能够结合hanp肽的n末端或c末端的官能团的化合物用作接头分子。这两种类型的接头分子当它们还具有允许它们的直接结合的官能团时可以彼此直接键合,或者可以借助额外化合物连接。考虑到缀合物生产效率或方便性,适当地选择使用的每个接头分子作为具有适当官能团或结构的化合物。因此,只有一种类型的接头分子可以包含peg,或者两个或更多个接头分子可以包含乙二醇单元,只要最终构建的peg接头结构总共包含预定数量的乙二醇单元结构。在由“peg(n)”代表的接头结构中,n代表其中含有的连续乙二醇单元的数目。具有另一种化学结构的插入物的接头结构由peg(n)-peg(m)代表。例如,包含24个单元的聚乙二醇的接头结构被称为peg(24),并且通过包含12个单元的聚乙二醇的接头分子和包含6个单元的聚乙二醇的接头分子之间的结合形成的接头结构被称为peg(12)-peg(6)。例如,通过相同接头分子之间的结合形成的peg(12)-peg(12)部分也可以简称为peg(12)2。这种具有所需长度的peg接头可以例如通过重复脱保护和羧酸活化使用fmoc-peg试剂诸如3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(9h-芴-9-基甲氧基羰基氨基)乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]丙酸(fmoc-peg(12)-cooh)(其在一侧具有fmoc保护的氨基且在另一侧具有羧酸)形成为具有所需peg长度的接头。这种fmoc-peg试剂可作为fmoc-peg(3)-cooh、fmoc-peg(6)-cooh、fmoc-peg(12)-cooh、fmoc-peg(24)-cooh等从novabiochem/merckkgaa商业可得。有机合成化学领域中已知的各种方法可以应用于此类接头分子之间的结合,没有特别限制。通常,该结合可以参考适用于抗体-药物缀合物合成的缀合方法进行(bioconjugatechem.2015,26,2198-2215)。用于通过叠氮化物基团与乙炔基的huisgen反应形成环的方法的实例可以包括形成1,2,3-三唑环的环加成反应[cu(i)-催化的叠氮化物-炔烃1498711133922_0(cuaac)]和由叠氮化物基团和二苄基环辛烯(dbco)形成1,2,3-三唑环的环加成反应[张力促进的叠氮化物-炔烃环加成(spaac)]。在该上下文中,也可以类似地利用其它化合物诸如具有扭曲的环辛炔结构的化合物,双环[6.1.0]壬-4-烯(bcn)(angew.chem.int.ed.2010,49,9422-9425),和在中间环结构上含有杂原子的环炔(angew.chem.int.ed.2015,54,1190-1194)。成环反应的其它实例可以包括1,2,4,5-四嗪环与扭曲的烯烃的diels-alder环加成反应(curr.opin.chem.biol.,2014,21,89-95)。基于与醛基的pictet-spengler反应的环缩合的实例可以包括肼基-pictet-spengler连接(bioconjugatechem.24,846-851)和基于羟氨(oxyamin)的pictet-spengler连接(proc.natl.acad.sci.u.s.a.110,46-51)。其额外实例可以包括氨基和羧基之间的酰胺键,sh基团与马来酰亚胺基团的马来酰亚胺缩合,sh基团与甲基磺酰基苯基噁二唑基团的缩合(angew.chem.,int.ed.52,12592-6),sh基团和碘乙酰基之间的键,通过sh基团与2-吡啶基二硫基的缩合的二硫化物键,醛基与酰肼的腙缩合,和醛基与氨基氧基的肟缩合。可以适当地选择本发明中采用的接头分子作为具有符合任何这些结合模式的官能团的分子,并用于形成接头结构。取决于结合模式,这种结合反应可以形成立体异构体、光学异构体、几何异构体等。这些异构体可以通过已知方法拆分或可以用作混合物。由于最终的缀合物是大分子,因此认为这些部分结构的异构体之间的结构差异很少影响缀合物。这种peg接头的具体实例可以包括诸如以下l(peg)a1至l(peg)h1和l(peg)a2至l(peg)h2的结构。其中“hanp”代表与所述hanp肽的n末端结合,且“n297gly”代表与在n297聚糖的非还原末端处的唾液酸的2-位的羰基结合。这些接头结构具有通过引入键合至含fc分子的n297聚糖中的叠氮化物基团与键合至包含hanp肽的接头分子的dbco基团的点击反应形成的1,2,3-三唑环。在该结构中,形成几何异构体,其中键合至叠氮化物基团的接头结构键合至三唑环的1-或3-位。由于每个含fc分子与两个或四个叠氮化物基团发生反应,所以几何异构结构共存于缀合物的一个分子中。<缀合物及其产生方法>本发明的缀合物可以通过使用有机合成化学领域中已知的反应适当地缀合中间体诸如上面提及的hanp肽、含fc分子和接头分子来产生。其产生顺序没有特别限制,并且可以根据中间体和目标化合物的结构通过常用方法采用各种方法。将每种中间体携带的官能团例如通过根据产生步骤的常见方法适当地活化,失活,用保护基团保护,并脱保护。将每个反应步骤中的中间体或最终产物适当地分离和纯化,并进行下一反应或用作本体药物(bulkpharmaceutical)或试剂。具有上面列出的任何peg接头的缀合物可以例如如下产生。与n297聚糖键合的接头分子是在n297聚糖的非还原末端处具有官能团(例如,叠氮化物基团)的分子,并且通过上面提及的含fc分子的聚糖重建步骤合成。在一侧具有氨基且在另一侧具有另一官能团(例如,叠氮化物基团)的接头分子与sg(10)反应以使接头分子键合至聚糖的非还原末端处的唾液酸的2-位的羧酸。随后,活化该聚糖分子的还原末端处的glcnac以制备聚糖供体分子,然后其在内切糖苷酶存在的情况下与(fucα1,6)glcnac-含fc分子反应以便将官能团引入含fc分子中n297聚糖的非还原末端。键合至hanp肽的n末端的接头分子是在一侧具有羧基且在另一侧具有另一官能团(dbco,保护基团引入的氨基等)的分子。hanp肽可以与接头分子反应,以便将所需官能团引入hanp肽的n末端。因此获得的官能团引入的含fc分子和hanp肽可以直接或经由另一个接头分子彼此连接,以获得本发明的缀合物。本发明的缀合物的实例可以具体包括上述l(peg)-a至l(peg)-h,其中所述hanp肽是hanp(1-28)或(sg-)asn-hanp(1-28),所述含fc分子是由重链和轻链的组合组成的抗体(mab-a),所述重链由来自seqidno:3的氨基酸位置20至474的氨基酸序列组成,且所述轻链由来自seqidno:5的氨基酸位置21至234的氨基酸序列组成;或由重链和轻链的组合组成的clch(clch-a),所述重链由来自seqidno:7的氨基酸位置20至349的氨基酸序列组成,且所述轻链由来自seqidno:9的氨基酸位置21至125的氨基酸序列组成;且sg型n297聚糖是n297-(fuc)sg或n297-(fuc)msg1,且可以更具体地包括实施例3中合成的化合物3-1至化合物3-14。化合物3-1至化合物3-6是优选的。<功能和活性>本发明的缀合物与未修饰的hanp(1-28)相比表现出血液中延长的持续时间和优异的物理特性,并且当皮下施用时,其具有逐渐迁移至血液中的能力,其对于hanp制剂是优选的。天然hanp(1-28)从血液迅速消失,且因此需要在临床实践中通过静脉内输注等连续施用。相反,甚至通过通常的皮下施用,本发明的缀合物也可以持续长时段发挥药理作用。此外,本发明的缀合物甚至在高浓度下也具有低聚集的特性。本发明的缀合物的此类特征允许采用通过常规天然hanp或现有hanp制剂无法实现的施用方法、施用途径和配制技术,并且还使得缀合物能够用于治疗急性心血管疾病以及慢性心血管疾病(高血压、慢性心脏病等)。此外,本发明的缀合物也可用作生物学研究工具。不清楚天然hanp如何或是否迁移至组织中,例如,当持续长时段存在于血液中时。相反,可以通过施用本发明的缀合物来检查这种定位或hanp在生物体上的血液中的长期滞留的影响。可以根据实施例4-2的方法通过将缀合物施用于动物且然后检测外周血中的cgmp浓度和/或在外周血液样品中含有的缀合物来测试本发明的缀合物的血液中的持续时间。本发明的缀合物甚至在体内施用后约24小时也维持升高外周血中的cgmp浓度的效果,更优选地甚至在施用后约48小时也维持该效果,甚至更优选地甚至在施用后约72小时也维持该效果,并且进一步优选地甚至在施用后约96、120、144或168小时也维持该效果。此外,本发明的缀合物在皮下施用后逐渐表现出其活性,并且活性通常在24至48小时后达到峰值。预期血液中的这种动力学能够降低或避免表现低血压的风险。至于在施用缀合物后检测外周血中的缀合物,该缀合物优选地甚至在约24小时后检测到,更优选地甚至在约48小时后检测到,甚至更优选地甚至在约72小时后检测到,且进一步优选地甚至在约96、120、144或168小时后检测到。甚至在具有高浓度的水溶液中,本发明的缀合物也表现出很少聚集的优异物理特性。由于盐在溶液中的影响等,hanp肽易于凝胶化。因此,需要谨慎处理其制剂。本发明的缀合物减少与凝集有关的问题,并且可以以使得增加活性成分的浓度或可以采用各种添加剂的方式应用于各种形式的制剂。具体地,已经证实包含糖基化的hanp肽的缀合物表现出非常有利的非凝集性。本发明的缀合物在血液中的持续时间可以通过如下来测量:将缀合物施用于生物体,以特定时间间隔取血,并检测血液样品中含有的缀合物。各种方法,例如,通过lc-ms和使用特异性识别hanp的环结构的抗体的elisa的检测,可以用作用于检测缀合物的方法。在以产生cgmp升高活性的剂量施用本发明的缀合物的情况下,血液样品的cgmp水平可以使用商业可得的测量试剂盒测量,并与开始施用前测定的血液中的cgmp水平进行比较,以测量血液中缀合物的生物活性的持续时间。或者,所述缀合物可以用放射性同位素标记,并通过sds-page等分离血液样品并检测放射性信号来检测。在本发明中,“延长血液中的持续时间”意味着与天然hanp相比,延长施用后血液中可检测到测试物质的时间。皮下施用于猴的天然hanp在血液中的浓度等于或低于在以200nmol/kg施用后30分钟的测量限值(数据未显示),而甚至在以100nmol/kg施用后7天也从血液检测到所述缀合物。本发明的缀合物还具有对于nep对hanp肽的降解的抗性。这可能部分由于持续时间延长。可以通过已知方法测量这种对nep降解的抗性。本发明的缀合物的cgmp升高活性通过如下测量:用经调节至最高达足够量的浓度梯度的测试物质刺激表达gc-a受体的细胞,然后裂解细胞,测量细胞裂解物中的cgmp浓度,并鉴定最大cgmp浓度(emax)。对于本发明的缀合物描述的短语“维持cgmp升高活性”意味着所述缀合物表现出的最大cgmp浓度与天然hanp的最大cgmp浓度相比为约30%或更高。所述缀合物表现出的最大cgmp浓度优选为约50%或更高,更优选地约70%或更高。本发明的缀合物与天然hanp相比可以以高浓度配制,并且表现出血液中延长的持续时间。因此,基于指数诸如所谓的ec50值来定义本发明的缀合物的活性是不适当的。如果升高浓度的缀合物的最大活性可以等于或大于天然hanp的给定活性,则所述缀合物当在临床实践中连续施用和/或以高浓度施用时可以产生足够的药效。本发明提供了包含本发明的缀合物作为活性成分的药物。<药物>可用作根据本发明的药物的活性成分的物质可以是上面提及的缀合物的药学上可接受的盐。具体地,在本发明中,上面提及的物质的酸(无机酸,例如,盐酸、硫酸或磷酸,或有机酸,例如,甲酸、乙酸、丁酸、三氟乙酸(tfa)、琥珀酸或柠檬酸)-加成盐可用作活性成分。或者,在本发明中,上面提及的物质的金属(例如,钠、钾、锂或钙)盐或基于其有机碱的盐形式可用作活性成分。这种本发明的缀合物的盐可以是基于hanp肽部分的盐,或者可以是在聚糖的结构中形成的盐。本发明的缀合物的盐优选为在hanp肽部分处形成的药学上可接受的盐。根据本发明的药物组合物可以含有与活性成分相关的物质的游离形式或其药学上可接受的盐。可用作根据本发明的药物的活性成分的物质或其药学上可接受的盐优选与已知的药学上可接受的载体、赋形剂、稀释剂等混合,并通过通常用于药物的施用方法施用于个体,所述施用方法即口服施用方法或肠胃外施用方法,诸如经粘膜施用、静脉内施用、肌肉内施用或皮下施用。可用作根据本发明的药物的活性成分的物质的剂量根据疾病的类型、个体(患者)的年龄、体重和病况的严重程度和施用途径等而不同。通常,日剂量的上限为例如约100mg/kg或更低,优选地约50mg/kg或更低,更优选地1mg/kg或更低。日剂量的下限为例如约0.1μg/kg或更高,优选地0.5μg/kg或更高,更优选地1μg/kg或更高。根据本发明的药物的给药频率根据使用的活性成分、施用途径和待治疗的特定疾病而不同。在口服施用例如肽类物质的情况下,优选地开该物质的处方,使得每天的剂量数为4或更少。在肠胃外施用(例如,静脉内施用)的情况下,所述药物可以使用普通注射器注射,或者可以通过使用输注泵、导管等连续施用。或者,通过途径诸如皮下注射或肌肉内注射的施用也是优选的。在这种情况下,可以采用通常使用的各种施用装置。当在溶液中制备用于本发明的药物的活性成分时,可以将本发明的缀合物或其药学上可接受的盐溶解于水性溶剂中,并且如果必要,用稳定剂、ph调节剂、表面活性剂等补充,以制备溶液。在制备冷冻干燥的制剂的情况下,可以将因此制备的溶液冷冻干燥并在使用中溶解于生理盐水、注射用水、葡萄糖溶液等中。将本发明的药物施用于具有可通过活化gc-a和所得的cgmp水平升高治疗的疾病的患者,且由此有效地治疗该疾病。在该上下文中,疾病或其症状的“治疗”意味着预期待通过活化gc-a而正常化的病理病况的进展被延迟、缓解、减少和/或抑制,由此使病况更接近于正常状态。预期本发明的药物对于通过在疾病的早期阶段开始其施用或通过施用于处于该疾病的高风险的个体而预防疾病的恶化或发作是有效的。尽管过去已发展该疾病的患者处于复发或慢性的风险,但可以预期本发明的药物通过连续施用于这种患者来降低复发或慢性的风险。这些效果也包括在治疗的范围内。这种疾病的实例包括高血压、急性心力衰竭(包括急性心力衰竭发作后的医疗病况的管控)、慢性心力衰竭、缺血性心脏病、急性肾炎(包括急性肾炎发作后的医疗病况的管控)、慢性肾炎、急性肾衰竭(包括急性肾衰竭发作后的医疗病况的管控)、慢性肾衰竭、缺血性心脏病(心肌梗塞等)、恶性肿瘤的转移、肝纤维化、肝硬化、由透析引起的组织粘连和纤维化。实施例在下文中,将参照实施例具体描述本发明。实施例中描述的本发明的实施方案仅出于说明目的给出,并且本发明不旨在受这些实施例的限制。实施例1是hanp肽和接头分子或其衍生物(其为产生本发明的缀合物的中间体)的产生实施例。实施例2是抗体分子的制备实施例。实施例3是缀合物的合成实施例。实施例4是用于证实本发明的缀合物的特征或效果的测试实施例。本说明书中描述的每种中间体的质量通过以下方法证实:使用的设备是qexactive(由thermofisherscientificinc.制造)、ultimate3000(由thermofisherscientificinc.制造)和coretecsuplcc18(由watersinc.制造)(1.6μm,2.1x50mm)。在流动相a中使用乙腈,并且在流动相b中使用补充有0.1%甲酸的水溶液。流动相a在4分钟内以2%至95%变化的梯度使用。分析在40℃下以0.4ml/min的流速进行。使用微体积分光光度计xpose(由trineannv制造)测定本文所述的蛋白浓度。通过以下方法证实聚糖重建的抗体分子和缀合物的质量:将聚糖重建的抗体分子或缀合物片段化成重链和轻链。然后,使用分析柱分离它们各自的峰,然后进行质谱分析。使用的设备是qexactive(由thermofisherscientificinc.制造)、ultimate3000(由thermofisherscientificinc.制造)和mabpacrp(由thermofisherscientificinc.制造)(4.0μm,2.1x50mm)。在流动相a中使用乙腈,并且在流动相b中使用补充有0.1%甲酸/0.02%三氟乙酸的水溶液。流动相a在4分钟内以20%至50%变化的梯度使用。分析在80℃下以0.6ml/min的流速进行。[实施例1]各种中间体的合成以下给出的每个结构式中的简单术语“hanp”代表修饰肽中的hanp肽是hanp(1-28)(seqidno:1)。该肽在其n末端ser处与接头分子键合或糖基化。<实施例1-1>dbco-peg(12)2-hanp(1-28)(下式的化合物1-1)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。(1-1a)fmoc-peg(12)-hanp(1-28)的合成向fmoc-peg试剂fmoc-peg(12)-cooh;3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(9h-芴-9-基甲氧基羰基氨基)乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]丙酸(由novabiochem/merckkgaa制造,304mg,0.36mmol)于n,n-二甲基甲酰胺(4.6ml)中的溶液中添加n,n,n',n'-四甲基-o-(n-琥珀酰亚胺基)脲鎓四氟硼酸盐(由tokyochemicalindustryco.,ltd.制造,109mg,0.36mmol)于n,n-二甲基甲酰胺(1.0ml)中的溶液和二异丙基乙胺(131μl,0.75mmol),并将混合物在室温下搅拌1小时。将hanp(1-28)乙酸盐(1000mg)溶解于n,n-二甲基甲酰胺(11ml)和蒸馏水(3ml)中。向溶液中添加二异丙基乙胺(315μl,1.81mmol)。将预先制备的含有活性酯的n,n-二甲基甲酰胺溶液(5.6m)添加至所得溶液中,并将混合物在室温下搅拌1小时。反应完成后,将三氟乙酸(186μl,2.41mmol)添加至反应溶液中,并将混合物转移至20ml闪烁小瓶(由biotagejapanltd.制造)中。使用高速浓缩设备v-10(biotagejapanltd.)除去溶剂。将适量的乙腈添加至小瓶中,并用高速浓缩设备v-10(biotagejapanltd.)除去溶剂,以获得固体物质。将适量的二乙醚添加至固体物质中,并倾析混合物。然后,向其中添加适量的乙腈/二乙醚(1/2),并再次倾析混合物。将获得的固体物质在减压下干燥,以获得含有标题化合物的粗产物。获得的粗产物不经进一步纯化即直接用于下一反应中。(1-1b)h2n-peg(12)-hanp(1-28)的合成将步骤(1-1a)中合成的粗产物(全部量)溶解于n,n-二甲基甲酰胺(16ml)和蒸馏水(3.2ml)的混合溶液中。向溶液中添加哌啶(518μl,5.23mmol),并将混合物在室温下搅拌1小时。反应完成后,将乙酸(451μl,7.88mmol)添加至反应溶液中,并将混合物转移至20ml闪烁小瓶(biotagejapanltd.)中。使用高速浓缩设备v-10(biotagejapanltd.)除去溶剂。将适量的乙腈添加至小瓶中,并用高速浓缩设备v-10(biotagejapanltd.)除去溶剂,以获得固体物质。将适量的二乙醚添加至固体物质中,并倾析混合物。然后,向其中添加适量的乙腈/二乙醚(1/2),并再次倾析混合物。通过添加适量的0.1%三氟乙酸水溶液和乙酸来溶解获得的固体物质,并通过反相hplc分离并以几部分纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥。获得作为无色固体的标题化合物(826mg)。(1-1c)fmoc-peg(12)2-hanp的合成向fmoc-peg试剂fmoc-peg(12)-cooh;3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(9h-芴-9-基甲氧基羰基氨基)乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙氧基]丙酸(由novabiochem/merckkgaa制造,251mg,0.30mmol)于n,n-二甲基甲酰胺(3.8ml)中的溶液中添加n,n,n',n'-四甲基-o-(n-琥珀酰亚胺基)脲鎓四氟硼酸盐(由tokyochemicalindustryco.,ltd.制造,90mg,0.30mmol)于n,n-二甲基甲酰胺(0.8ml)中的溶液和二异丙基乙胺(87μl,0.50mmol),并将混合物在室温下搅拌1小时。通过添加n,n-二甲基甲酰胺(10ml)和蒸馏水(3.5ml)来溶解步骤(1-1b)中合成的化合物(826mg)。向溶液中添加二异丙基乙胺(160μl,0.92mmol)。将预先制备的含有活性酯的n,n-二甲基甲酰胺溶液(4.6m)添加至所得溶液中,并将混合物在室温下搅拌1小时。反应完成后,将三氟乙酸(77μl,1.00mmol)添加至反应溶液中,并将混合物转移至20ml闪烁小瓶(由biotagejapanltd.制造)中。使用高速浓缩设备v-10(biotagejapanltd.)除去溶剂。将适量的乙腈添加至小瓶中,并用高速浓缩设备v-10(biotagejapanltd.)除去溶剂,以获得固体物质。将二乙醚添加至固体物质中,并倾析混合物。然后,向其中添加适量的乙腈/二乙醚(1/2),并再次倾析混合物。将获得的固体物质在减压下干燥,以获得含有标题化合物的粗产物。获得的粗产物不经进一步纯化即直接用于下一反应中。(1-1d)h2n-peg(12)2-hanp(1-28)的合成通过添加n,n-二甲基甲酰胺(14ml)和蒸馏水(1.4ml)来溶解步骤(1-1c)中合成的粗产物(全部量)。向溶液中添加哌啶(395μl,3.99mmol),并将混合物在室温下搅拌30分钟。将蒸馏水(1.0ml)添加至反应溶液中,并将混合物进一步搅拌30分钟。反应完成后,将乙酸(343μl,5.99mmol)添加至反应溶液中,并将混合物转移至20ml闪烁小瓶(由biotagejapanltd.制造)中。使用高速浓缩设备v-10(biotagejapanltd.)除去溶剂。将乙腈添加至小瓶中,并用高速浓缩设备v-10(biotagejapanltd.)除去溶剂,以获得固体物质。将适量的二乙醚添加至固体物质中,并倾析混合物。然后,向其中添加适量的乙腈/二乙醚(1/2),并再次倾析混合物。通过添加适量的0.2%三氟乙酸水溶液和乙酸来溶解获得的固体物质,并通过反相hplc分离并以几部分纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥。获得作为无色固体的标题化合物(694mg)。(1-1e)dbco-peg(12)2-hanp(1-28)的合成通过添加n,n-二甲基甲酰胺(5.5ml)和蒸馏水(1.5ml)来溶解步骤(1-1d)中合成的化合物(520mg)。向溶液中添加二异丙基乙胺(77μl,0.44mmol)。将dbco-nhs酯(由clickchemistrytoolsllc制造,53mg,132mmol)于n,n-二甲基甲酰胺(0.2m)中的溶液添加至所得溶液中,并将混合物在室温下搅拌1小时。反应完成后,将反应溶液转移至20ml闪烁小瓶(由biotagejapanltd.制造)中。使用高速浓缩设备v-10(biotagejapanltd.)除去溶剂。将二乙醚添加至固体物质中,并倾析混合物。然后,向其中添加乙腈/二乙醚(1/2),并再次倾析混合物。通过添加适量的0.1%三氟乙酸水溶液和乙酸来溶解获得的固体物质,并通过反相hplc分离并以几部分纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥,以获得作为无色固体的标题化合物(化合物1-1)(283mg)。esi-ms:c200h322n48o67s3的计算值:[m+4h]4+1142.8(平均值),实测值1142.6;[m+5h]5+914.4(平均值),实测值914.3;[m+6h]6+762.2(平均值),实测值762.0<实施例1-2>dbco-peg(12)2-(sg-)asn-hanp(1-28)(下式的化合物1-2)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。(1-2a)游离形式的fmoc-(sg-)asn的制备将fmoc-(sg-)asn(1s2s-11nc-asn-fmoc,由glytech,inc.制造,2g)溶解于适量的0.1%三氟乙酸水溶液中。通过反相hplc分离并以几部分纯化溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥。获得无色固体(1.8g)。(1-2b)hanp(1-28)tfa盐(三氟乙酸盐)的制备将乙酸卡培立肽(hanp(1-28)乙酸盐)(2.7g)溶解于蒸馏水(200ml)中。向该溶液中添加三氟乙酸(4ml,52.3mmol),并将混合物放置10分钟,且然后冷冻干燥。冷冻干燥完成后,将残余物溶解于蒸馏水(200ml)中。将溶液再次冷冻干燥。获得无色固体(2.7g)。(1-2c)(sg-)asn-hanp(1-28)的合成在冰冷却下向步骤(1-2a)中制备的游离形式的fmoc-(sg-)asn(436mg)于n,n-二甲基甲酰胺(7.2ml)中的溶液中添加hatu(65mg,0.17mmol)于n,n-二甲基甲酰胺(0.8ml)中的溶液和二异丙基乙胺(118μl,0.68mmol),并将混合物搅拌2.5分钟,并立即用于下一反应中(溶液1-2c)。通过添加n,n-二甲基甲酰胺溶液(4.8ml)和蒸馏水(1.2ml)来溶解步骤(1-2b)中制备的hanp(1-28)tfa盐(400mg)。向溶液中添加二异丙基乙胺(118μl,0.68mmol)。将预先制备的含有活性酯的n,n-二甲基甲酰胺溶液1-2c(8.0m)添加至所得溶液中,并将混合物在室温下搅拌30分钟。反应完成后,向其中添加乙酸(82μl,1.36mmol)。将约5ml/管的反应溶液转移至预先补充有乙腈(35ml)的三个离心管(50ml)中。使用小型离心机(hitachikokisalesco.,ltd.,cf16rx)沉淀固体物质,并除去上清液。将三个离心管中的固体物质合并至一个离心管中,用乙腈(30ml)洗涤,然后用二乙醚(30ml)洗涤两次,且然后在减压下干燥,以获得粗产物。将获得的粗产物(全部量)溶解于n,n-二甲基甲酰胺溶液(8.4ml)和蒸馏水(1.4ml)中。向溶液中添加哌啶(224μl,2.26mmol),并将混合物在室温下搅拌30分钟。反应完成后,向其中添加乙酸(194μl,3.39mmol)。将约5ml/管的反应溶液转移至预先补充有乙腈(35ml)的两个离心管(50ml)中。使用小型离心机(hitachikokisalesco.,ltd.,cf16rx)沉淀固体物质,并除去上清液。将两个离心管中的固体物质合并至一个离心管中,用乙腈(30ml)洗涤,用二乙醚(30ml)洗涤两次,且然后在减压下干燥,以获得粗产物。对3批进行上述操作。合并3批中的粗产物。通过添加适量的0.1%三氟乙酸水溶液和乙酸来溶解获得的固体物质,并通过反相hplc分离并以几部分纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥。获得作为无色固体的标题化合物(sg-)asn-hanp(1-28)(1.49g)。esi-ms:c215h345n53o102s3的计算值:[m+4h]4+1351.1(平均值),实测值1351.1;[m+5h]5+1081.1(平均值),实测值1080.9(1-2d)fmoc-peg(12)2-(sg-)asn-hanp的合成向根据wo2014115797a1(实施例2-28a)中描述的方法合成的fmoc-nh-peg(12)2-cooh(122mg,0.085mmol)于n,n-二甲基甲酰胺(1.0ml)中的溶液中添加n,n,n',n'-四甲基-o-(n-琥珀酰亚胺基)脲鎓四氟硼酸盐(tokyochemicalindustryco.,ltd.,23mg,0.077mmol)于n,n-二甲基甲酰胺(1.0ml)中的溶液和二异丙基乙胺(54μl,0.31mmol),并将混合物在室温下搅拌1小时(溶液1-2d)。将(1-2c)中合成的化合物(300mg)溶解于n,n-二甲基甲酰胺(3ml)和蒸馏水(1.4ml)中。向溶液中添加二异丙基乙胺(54μl,0.31mmol)。将预先制备的含有活性酯的n,n-二甲基甲酰胺溶液1-2d(2.0m)添加至所得溶液中,并将混合物在室温下搅拌22小时。在反应完成后,将三氟乙酸(47μl,0.61mmol)添加至反应溶液中,并将约3.2ml/管的反应溶液转移至预先补充有二乙醚/乙腈(30ml/10ml)的两个离心管(50ml)中。使用小型离心机(hitachikokisalesco.,ltd.,cf16rx)沉淀固体物质,并除去上清液。将两个离心管中的固体物质合并至一个离心管中,用适量的二乙醚/乙腈(1/1)洗涤,且然后在减压下干燥,以获得粗产物。获得的粗产物不经进一步纯化即直接使用。对4批进行上述操作,以获得总共1.47g含有标题化合物的粗产物。(1-2e)h2n-peg(12)2-(sg-)asn-hanp(1-28)的合成将步骤(1-2d)中合成的粗产物(0.734g)溶解于n,n-二甲基甲酰胺(7.2ml)和蒸馏水(1.2ml)中。向溶液中添加哌啶(200μl,2.05mmol),并将混合物在室温下搅拌45分钟。在反应完成后,将乙酸(176μl,3.07mmol)添加至反应溶液中,并将约2.1ml/管的反应溶液转移至预先补充有二乙醚/乙腈(30ml/10ml)的四个离心管(50ml)中。使用小型离心机(hitachikokisalesco.,ltd.,cf16rx)沉淀固体物质,并除去上清液。将四个离心管中的固体物质合并至一个离心管中,用适量的二乙醚/乙腈(1/1)和适量的二乙醚洗涤,且然后在减压下干燥,以获得粗产物。通过添加适量的0.2%三氟乙酸水溶液和乙酸来溶解获得的固体物质,并通过反相hplc分离并以几部分纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥。获得作为无色固体的标题化合物(416mg)。(1-2f)dbco-peg(12)2-(sg-)asn-hanp的合成将步骤(1-2e)中合成的化合物(832mg)溶解于n,n-二甲基甲酰胺(12ml)和蒸馏水(2.4ml)中。向溶液中添加二异丙基乙胺(123μl,0.71mmol)。将dbco-nhs酯(由clickchemistrytoolsllc制造,57mg,142mmol)于n,n-二甲基甲酰胺(0.4m)中的溶液添加至所得溶液中,并将混合物在室温下搅拌1小时。在反应完成后,将三氟乙酸(109μl,1.42mmol)添加至反应溶液中,并将约2.5ml/管的反应溶液转移至预先补充有二乙醚/乙腈(30ml/5ml)的六个离心管(50ml)中。使用小型离心机(hitachikokisalesco.,ltd.,cf16rx)沉淀固体物质,并除去上清液。将六个离心管中的固体物质合并至一个离心管中,用适量的二乙醚/乙腈(1/1)洗涤,且然后在减压下干燥,以获得粗产物。通过添加适量的0.2%三氟乙酸水溶液和乙酸来溶解获得的固体物质,并通过反相hplc分离并纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥。获得作为无色固体的标题化合物(620mg)。esi-ms:c288h464n56o130s3的计算值:[m+5h]5+1378.4(平均值),实测值1378.2;[m+6h]6+1148.9(平均值),实测值1148.7;[m+7h]7+984.9(平均值),实测值984.9peg接头的peg长度或缩合的peg的数量可通过选择使用的fmoc-peg试剂、缩合和fmoc脱保护的顺序以及重复的数量来控制。因此,可以根据需要通过将这些因素的选择整合至实施例1-1或1-2的方法中来合成dbco-l(peg)-hanp。在下文中,将给出通过根据与实施例1-1中描述的方法类似的方法改变fmoc-peg试剂、缩合和fmoc脱保护的顺序以及重复的数量来合成的化合物。<实施例1-3>dbco-peg(12)-hanp(1-28)(下式的化合物1-3)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。通过合成在实施例1-1的步骤(1-1e)中使用用步骤(1-1b)中合成的h2n-peg(12)-hanp(1-28)替代的起始材料,获得标题化合物1-3。esi-ms:c173h269n47o54s3的计算值:[m+3h]3+1323.5(平均值),实测值1323.2;[m+5h]5+794.5(平均值),实测值794.4。<实施例1-4>dbco-peg(24)-hanp(1-28)(下式的化合物1-4)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。通过与实施例1-1类似的方法获得标题化合物1-4,除了在步骤(1-1a)中使用fmoc-peg(24)-cooh作为fmoc-peg试剂且使用步骤(1-1b)中获得的h2n-peg(24)-hanp(1-28)作为步骤(1-1e)的起始材料。esi-ms:c197h317n47o66s3的计算值:[m+4h]4+1125.0(平均值),实测值1124.8,[m+5h]5+900.2(平均值),实测值900.0;[m+6h]6+750.4(平均值),实测值750.2。<实施例1-5>dbco-peg(12)-peg(6)-hanp(1-28)(下式的化合物1-5)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。通过合成在实施例1-1的(1-1a)中使用用fmoc-peg(6)-cooh替代的fmoc-peg试剂,获得标题化合物1-5。esi-ms:c288h464n56o130s3的计算值:[m+5h]5+1378.4(平均值),实测值1378.2;[m+6h]6+1148.9(平均值),实测值1148.7;[m+7h]7+984.9(平均值),实测值984.9。<实施例1-6>dbco-peg(6)-peg(12)-hanp(1-28)(下式的化合物1-6)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。通过合成在实施例1-1的(1-1c)中使用用fmoc-peg(6)-cooh替代的fmoc-peg试剂,获得标题化合物1-6。esi-ms:c188h298n48o61s3的计算值:[m+3h]3+1435.3(平均值),实测值1435.0;[m+4h]4+1076.7(平均值),实测值1076.5;[m+5h]5+861.6(平均值),实测值861.4。<实施例1-7>dbco-peg(6)2-hanp(1-28)(下式的化合物1-7)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。通过合成在实施例1-1的步骤(1-1a)和(1-1c)中使用用fmoc-peg(6)-cooh替代的fmoc-peg试剂,获得标题化合物1-7。esi-ms:c176h274n48o55s3的计算值:[m+3h]3+1347.2(平均值),实测值1347.0;[m+4h]4+1010.6(平均值),实测值1010.5;[m+5h]5+808.7(平均值),实测值808.6。<实施例1-8>dbco-peg(6)3-hanp(1-28)(下式的化合物1-8)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。通过合成通过在实施例1-1的步骤(1-1a)和(1-1c)中用fmoc-peg(6)-cooh替代fmoc-peg试剂以及步骤(1-1d)和(1-1e)之间进一步重复步骤(1-1c)和(1-1d),获得标题化合物1-8。esi-ms:c191h303n49o62s3的计算值:[m+3h]3+1459.0(平均值),实测值1458.7;[m+4h]4+1094.5(平均值),实测值1094.3;[m+5h]5+875.8(平均值),实测值875.6。<实施例1-9>dbco-peg(6)4-hanp(1-28)(下式的化合物1-9)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。通过合成通过在实施例1-1的步骤(1-1a)和(1-1c)中用fmoc-peg(6)-cooh替代fmoc-peg试剂以及步骤(1-1d)和(1-1e)之间进一步重复步骤(1-1c)和(1-1d)两次,获得标题化合物1-9。esi-ms:c206h332n50o69s3的计算值:[m+4h]4+1178.3(平均值),实测值1178.3;[m+5h]5+942.9(平均值),实测值942.7;[m+6h]6+785.9(平均值),实测值785.7。<实施例1-10>[n3-peg(3)]2-sg(10)-ox(下式的化合物)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体的示意图中的相应结构。(1-10a)[n3-peg(3)]2-sg(10)的合成向5ml取样管(inaoptikaco.,ltd.)中添加11-叠氮基-3,6,9-三氧杂十一烷-1-胺(sigma-aldrichco.llc,96μl,0.485mmol)和二唾液酸八糖(tokyochemicalindustryco.,ltd.,50mg,0.24mmol)水溶液(0.5ml),并将混合物搅拌1小时,且然后冷冻干燥。向因此冷冻干燥的5ml取样管中添加hatu(92mg,0.24mmol)于n,n-二甲基甲酰胺(0.6ml)中的溶液和二异丙基乙胺(42μl,0.24mmol),并将混合物在37℃下搅拌4小时。在反应完成后,将反应溶液转移至预先补充有二乙醚(20ml)的离心管(50ml)中。使用小型离心机(hitachikokisalesco.,ltd.,cf16rx)沉淀固体物质,并除去上清液。将二乙醚(20ml)添加至残余物中,并倾析混合物。随后,向其中添加乙腈(20ml),并倾析混合物,且然后在减压下干燥,以获得粗产物。将获得的固体物质溶解于适量的0.2%三氟乙酸水溶液中,并通过反相hplc分离并纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥,以获得作为无色固体的标题化合物(42mg)。esi-ms:c92h157n13o61的计算值:[m+2h]2+1211.7(平均值),实测值1211.5;[m-2h]2-1209.6(平均值),实测值1209.5。(1-10b)[n3-peg(3)]2-sg(10)-ox的合成向5ml取样管(由inaoptikaco.,ltd.制造)中添加步骤(1-10a)中合成的化合物(40mg)和2-氯-1,3-二甲基-1h-苯并咪唑-3-鎓-氯化物(cdmbi)(由fushimipharmaceuticalco.,ltd.制造,17.9mg,0.083mmol)水溶液(200μl)。在冰冷却后,将磷酸三钾(52.6mg,0.25mmol)水溶液(200μl)添加至反应溶液中,并将混合物在冰冷却下搅拌2小时。将获得的反应溶液使用amiconultra(ultracel30k,由merckmillipore/merckkgaa制造)超滤以除去固体物质。将流通溶液通过凝胶过滤色谱法纯化。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是hiprep26/10desalting(由gehealthcarejapancorp.制造)。在流动相中使用0.03%nh3水溶液。流速设定为10ml/min,且级分容量设定为10ml。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并。向其中添加1n氢氧化钠水溶液(33μl,0.033mmol),并将混合物冷冻干燥。获得作为无色固体的标题化合物(34mg)。nmr(在d2o中)(图4的图表)。<实施例1-11>[n3-peg(3)]-msg1(9)-ox(下式的化合物)的合成其中结构式右侧的示意图显示图1至3和实施例3的反应方案中显示的缀合物或中间体,或反应方案中的示意图中的相应结构。(1-11a)(msg1-)asn的制备通过反相hplc在下面给出的条件下分离和纯化商业可得的产品单唾液酸-asn游离形式(1s2g/1g2s-10nc-asn,由glytech,inc.制造)(被称为"(msg-)asn")(500mg),以分离作为第一主峰洗脱的(msg1-)asn(保留时间:约15至19分钟)和作为第二主峰洗脱的(msg2-)asn(保留时间:约21至26分钟)。使用的洗脱液是0.1%甲酸水溶液。使用的设备是els-pda触发制备系统(由jascocorp.制造)。使用的柱是inertsilods-3(10um,30φx250mm,由glsciencesinc.制造)。流速被设定为30ml/min。将属于在洗脱期间通过uv(210nm)检测到的第1峰的级分合并并冷冻干燥,以获得作为无色固体的标题化合物(238mg)。可以收集属于在上述洗脱操作中通过uv检测到的第2峰的级分以获得(msg2-)asn。(1-11b)msg1(9)的合成将步骤(1-11a)中获得的化合物(229mg)溶解于200mm磷酸盐缓冲溶液(ph6.25)(1145μl)中。向该溶液中添加endom(由tokyochemicalindustryco.,ltd.制造,1u/ml))水溶液(100μl),并将混合物在35℃下孵育6天。在反应完成后,将反应溶液使用vivaspin15r(hydrosart膜,30k,6,000g)进行超滤,并通过反相hplc分离和纯化获得的流通溶液。使用的洗脱液是0.1%三氟乙酸水溶液。使用的设备是els-pda触发制备系统(由jascocorp.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(210nm)检测到的目标化合物的级分合并并冷冻干燥,以获得作为无色固体的标题化合物(117mg)。(1-11c)[n3-peg(3)]-msg1(9)的合成根据与步骤(1-10a)中相同的方法,使用步骤(1-11b)中合成的化合物(169mg),获得标题化合物(94.2mg)。esi-ms:c73h124n8o51的计算值:[m+h]+1929.9(平均值),实测值1929.7。(1-11d)[n3-peg(3)]-msg1(9)-ox的合成根据与步骤(1-10b)中相同的方法,使用步骤(1-11c)中合成的化合物(100mg),获得标题化合物(89mg)。nmr(在d2o中)(图5的图表)。通过使用作为步骤(1-11a)中的第2峰获得的(msg2-)asn作为步骤(1-11b)的起始材料,且随后进行与该步骤中相同的操作,合成[n3-peg(3)]-msg2(9)-ox(化合物1-12,其中接头键合至化合物1-11中的β-man的1-6支链中的唾液酸)。通过在实施例3-5和3-6中用化合物1-12替代化合物1-11,合成包含连接至不同支链的两个hanp肽部分的缀合物。<实施例1-12>[n3-peg(3)]2-sg(10)-2的合成也通过下面给出的方法合成(1-10a)中合成的[n3-peg(3)]2-sg(10)。可以通过步骤(1-10b)使用通过下面给出的方法获得的化合物合成化合物1-10。(1-12a)([n3-peg(3)]2-sg)-asn-peg(3)-n3(下式的化合物)的合成向步骤(1-2a)中制备的游离形式的fmoc-(sg-)asn(1000mg)于n,n-二甲基甲酰胺(10ml)中的溶液中,向其中添加hatu(891mg,2.34mmol)于n,n-二甲基甲酰胺(3ml)中的溶液和11-叠氮基-3,6,9-三氧杂十一烷-1-胺(tokyochemicalindustryco.,ltd.,511mg,2.34mmol)和二异丙基乙胺(816μl,4.69mmol)于n,n-二甲基甲酰胺(3ml)中的溶液,并将混合物在37℃下搅拌3小时。进一步添加hatu(148mg,0.39mmol)于n,n-二甲基甲酰胺(500μl)中的溶液,并将混合物在37℃下搅拌1小时。然后,向其中添加哌啶(386μl,3.91mmol),并将混合物在37℃下搅拌1小时。在反应完成后,向其中添加乙酸(469μl)。将反应溶液一半转移至两个预先补充有二乙醚(100ml)的巨型锥形管(175ml)中。使用小型离心机(hitachikokisalesco.,ltd.,cf16rx)沉淀固体物质,并除去上清液。将胶状固体物质转移至离心管(50ml)中。向其中添加二乙醚(30ml)和乙腈(10ml),并倾析混合物。将该操作重复两次。类似地,向其中添加适量的乙腈或适量的二乙醚,并倾析混合物,且然后在减压下干燥,以获得粗产物。将获得的固体物质溶解于适量的0.2%三氟乙酸水溶液中,并通过反相hplc分离并纯化该溶液。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥,以获得作为无色固体的标题化合物(637mg)。esi-ms:c112h192n20o70的计算值:[m+3h]3+980.6(平均值),实测值980.4。(1-12b)[n3-peg(3)]2-sg(10)(步骤(1-10a)的化合物)的合成在2ml管中,将步骤(1-12a)中合成的([n3-peg(3)]2-sg)-asn-peg(3)-n3(78.6mg)溶解于100mm磷酸盐缓冲溶液,ph6.0(nacalaitesque,inc.,465μl)中。向该溶液中添加1u/mlendom(tokyochemicalindustryco.,ltd.,70μl),并将混合物在28℃下振荡5小时,且然后在室温下静置4天。在反应完成后,向其中添加适量的0.2%三氟乙酸水溶液,并通过反相hplc分离和纯化混合物。使用0.1%三氟乙酸水溶液和三氟乙酸于乙腈中的0.1%溶液作为洗脱液。使用的设备是purif-rp2(由shokoscientificco.,ltd.制造)。使用的柱是inertsilods-3(由glsciencesinc.制造)。将含有在洗脱期间通过uv(220nm)检测到的目标化合物的级分合并并冷冻干燥,以获得作为无色固体的标题化合物(40mg)。esi-ms:c92h157n13o61的计算值:[m+2h]2+1211.7(平均值),实测值1211.5。[实施例2]载体蛋白的制备<实施例2-1>用于载体的全长抗体(mab-a)的制备选择识别人体内不存在的抗原的抗lps抗体a(下文称为"mab-a")(wo2015/046505中的h#1g5-h1/l1)作为用于载体的全长抗体。以与wo2015/046505的实施例2、5和7等中描述的方法相同的方式制备抗体。最终的保存样品是19.4mg/ml的mab-a溶液(hbsor(25mm组氨酸/5%山梨糖醇,ph6.0))。<实施例2-2>clch-a的制备如下制备缺乏igg1可变区的部分抗体分子clch-a作为载体分子。(2-2a)ch-a表达载体的构建合成dna片段(seqidno:6),其包含编码ch型重链(ch-a)的氨基酸的dna序列,所述ch型重链(ch-a)含有与人重链分泌信号连接的人igg1恒定区。通过pcr扩增合成的dna片段,并与wo2015/046505中描述的pcma-lk的dna片段连接,所述dna片段通过使用in-fusionhdpcr克隆试剂盒(clontechlaboratories,inc.)用xbai和pmei消化,以从其中除去编码κ链分泌信号和人κ链恒定区的dna序列,从而构建ch-a表达载体。获得的表达载体被命名为"pcma/ch"。ch-a的氨基酸序列显示于seqidno:7的氨基酸位置20至349中(氨基酸位置1至19对应于信号序列)。(2-2b)cl-a表达载体的构建合成dna片段(seqidno:8),其包含编码cl型轻链(cl-a)的氨基酸的dna序列,所述cl型轻链(cl-a)含有与人轻链分泌信号连接的人κ链恒定区。以与步骤(2-2a)中相同的方式构建cl表达载体。获得的表达载体被命名为"pcma/cl"。cl-a的氨基酸序列显示于seqidno:9的氨基酸位置21至125中(氨基酸位置1至20对应于信号序列)。(2-2c)clch-a的产生以与wo2015/046505的实施例2中描述的方法相同的方式,使用构建的pcma/ch和pcma/cl的组合以及freestyle293f细胞(由invitrogencorp.制造),获得含有ch-a和cl-a的组合的部分抗体分子的培养上清液。获得的部分抗体分子被命名为“clch-a”。(2-2d)clch-a的纯化通过重组蛋白a亲和色谱和阳离子交换色谱的两个步骤从步骤(2-2c)中获得的培养上清液纯化clch-a。在纯化步骤中和纯化后的缓冲液替代以及浓缩步骤在4至6℃下进行。首先,将培养上清液施加至用pbs平衡的填充有mabselectsure(由gehealthcarebiosciencescorp.制造)的柱。在将全部培养溶液置于柱中后,用pbs以柱体积的两倍或更多倍的量洗涤该柱。随后,用2m精氨酸盐酸盐溶液(ph4.0)进行洗脱。分级分离洗脱液,并基于吸光度收集含有clch-a的级分。通过透析(thermofisherscientificinc.,slide-a-lyzerdialysiscassette),将回收的液体用50mmmes/20mmnacl,ph6.0缓冲液替代。将回收的溶液施加至用50mmmes/20mmnacl,ph6.0平衡的hitrapsphp(由gehealthcarebiosciencescorp.制造)。用50mmmes/20mmnacl,ph6.0洗涤柱。然后,用0至200mm氯化钠进行线性浓度梯度洗脱(柱体积的10倍),并以与上述相同的方式从洗脱液收集含有clch-a的级分。通过透析(thermofisherscientificinc.,slide-a-lyzerdialysiscassette),将回收的液体用hbsor(25mm组氨酸/5%山梨糖醇,ph6.0)缓冲液替代。最后,使用离心uf过滤装置vivaspin20(分子量截止值:uf10k,sartoriusag,在4℃下)浓缩回收的溶液。将浓度调节至20mg/ml或更高以制备纯化的clch-a样品。<实施例2-3>clch-b的制备作为具有clch-a的重链中引入的lala突变的部分抗体分子的clch-b如下制备为用于载体的分子。(2-3a)ch-b表达载体的构建使用下述的引物组(chlala-f和chlala-r)和kod-plus-诱变试剂盒(toyoboco.,ltd.)将用ala-ala取代seqidno:9的氨基酸位置136和137处的leu-leu(对应于基于eu索引的leu234-leu235)的突变引入作为模板的pcma/ch,以构建ch-b表达载体。构建的表达载体被命名为"pcma/ch-b"。编码ch-b的dna的核苷酸序列显示于seqidno:10的核苷酸位置58至1047中(核苷酸位置1至57对应于信号序列)。其氨基酸序列显示于seqidno:11的氨基酸位置20至349中(氨基酸位置1至19对应于信号序列)。(2-3b)clch-b的产生和纯化组合使用步骤(2-2b)中制备的pcma/ch-b和pcma/cl以与步骤(2-2c)中相同的方式获得含有ch-b与cl-a的组合的部分抗体分子的培养上清液(所得部分抗体分子被命名为“clch-b”)。从获得的培养上清液,以与步骤2-2d中相同的方式纯化部分抗体分子,并用作纯化的clch-b样品。<实施例2-4>fc-b(野生型fc)的制备如下制备作为由igg1fc片段组成的部分抗体分子的fc-b作为用于载体的分子。(2-4a)fc-b表达载体的构建合成dna片段(seqidno:14),其包含编码fc-b的氨基酸序列的dna序列,所述fc-b含有与人轻链分泌信号连接的人fc区。以与步骤(2-2a)中相同的方式构建fc-b表达载体。获得的表达载体被命名为"pcma/fc-b"。fc-b的氨基酸序列显示于seqidno:15的氨基酸位置21至243中(氨基酸位置1至20对应于信号序列)。(2-4b)fc-b的产生使用pcma/fc-b,以与实施例(2-2c)中相同的方式获得含有作为部分抗体分子的fc-b的培养上清液。(2-4c)fc-b的纯化通过重组蛋白a亲和色谱(在4至6℃下)和陶瓷羟基磷灰石(在室温下)的两个步骤从步骤(2-4b)中获得的培养上清液纯化fc片段。通过重组蛋白a亲和色谱纯化后和使用陶瓷羟基磷灰石纯化后的缓冲液替代步骤在4至6℃下进行。首先,将培养上清液施加至用pbs平衡的mabselectsure(由gehealthcarebiosciencescorp.制造,hitrap柱)。在将全部培养上清液置于柱中后,用pbs以柱体积的两倍或更多倍的量洗涤该柱。随后,用2m精氨酸盐酸盐溶液(ph4.0)进行洗脱,并收集含有fc片段的级分。通过透析(thermofisherscientificinc.,slide-a-lyzerdialysiscassette)用pbs替代级分中的缓冲液,且然后用5mm磷酸钠/50mmmes,ph7.0的缓冲液稀释5倍。将fc片段溶液施加至用5mmnapi/50mmmes/30mmnacl,ph7.0的缓冲液平衡的陶瓷羟基磷灰石柱(bio-radlaboratories,inc.,bio-scalechtl型羟基磷灰石柱)。用0至2m氯化钠进行线性浓度梯度洗脱,并收集含有fc-b的级分。通过透析(thermofisherscientificinc.,slide-a-lyzerdialysiscassette),将级分中的缓冲液用hbsor(25mm组氨酸/5%山梨糖醇,ph6.0)替代。最后,使用离心uf过滤装置vivaspin20(分子量截止值:uf10k,sartoriusag,在4℃下)浓缩回收的溶液。将浓度调节至10mg/ml以制备纯化的fc-b样品。<实施例2-5>fc-a(lala形式)的制备如下制备作为具有延伸的n末端的fc-b的lala形式的fc-a作为用于载体的分子。(2-5a)fc-a表达载体的构建使用下述的引物组(fclala-f和fc05-r)和kod-plus-诱变试剂盒(toyoboco.,ltd.)将用ala-ala取代seqidno:15的氨基酸位置30和31处的leu-leu(对应于基于eu索引的leu234-leu235)并将4个氨基酸(dkth)添加至n末端的突变引入作为模板的pcma/fc-b,以构建fc-a表达载体。构建的表达载体被命名为"pcma/fc-a"。编码fc-a的dna的核苷酸序列显示于seqidno:16的核苷酸位置61至741中(核苷酸位置1至60对应于信号序列)。其氨基酸序列显示于seqidno:17的氨基酸位置21至247中(氨基酸位置1至20对应于信号序列)。(2-5b)fc-a的产生和纯化使用pcma/fc-a,以与步骤(2-2c)中相同的方式获得含有作为部分抗体分子的fc-a的培养上清液。从获得的培养上清液,以与实施例2-4c中相同的方式纯化部分抗体分子,并用作纯化的fc-a样品。[实施例3]缀合物的制备命名本实施例中产生的每种缀合物以指示其结构特征。尽管该名称通常采用上面在说明书中描述的命名法,但参考附图或结构式来标识完整的结构。在peg接头中,符号"//"代表通过叠氮化物基团与dbco反应形成的1,2,3-三唑环,且符号"-"代表酰胺键。<实施例3-1>mab-a-[peg(3)//peg(12)2-hanp(1-28)]4(化合物3-1)的合成(3-1a)(fucα1,6)glcnac-mab-a的制备使用amiconultra(ultracel30k,由merckmillipore/merckkgaa制造)将19.4mg/ml实施例2-1中制备的mab-a溶液(5%山梨糖醇/25mm组氨酸溶液(ph6.0))(26.0ml)用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代。将2.00mg/ml野生型endos溶液(pbs)(1.26ml)添加至24.5mg/ml获得的mab1溶液(50mm磷酸盐缓冲溶液(ph6.0))(20.0ml)中,并将混合物在37℃下孵育3小时。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,根据以下方法进行通过亲和色谱的纯化和使用羟基磷灰石柱的纯化。(1)通过亲和色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:hitraprproteinaff(5ml)(由gehealthcarejapancorp.制造)流速:5ml/min(进样期间1.25ml/min)以五部分纯化获得的反应溶液。为了与柱结合,将反应溶液添加至柱的上部,并以1.25ml/min以2cv将结合缓冲液(20mm磷酸盐缓冲溶液(ph7.0))注入柱中,并以5ml/min以5cv向其中进一步注入。对于中间洗涤,将洗涤溶液(20mm磷酸盐缓冲溶液(ph7.0)和0.5m氯化钠溶液)以15cv注入柱中。为了洗脱,将洗脱缓冲液(immunopureigg洗脱缓冲液,由pierce/thermofisherscientificinc.制造)以6cv注入柱中。洗脱液立即用1mtris缓冲溶液(ph9.0)中和。使用微体积分光光度计xpose(由trineannv制造)和experion电泳站(由bio-radlaboratories,inc.制造)证实在洗脱期间通过uv(280nm)检测到的级分。使用amiconultra(ultracel30k,由merckmillipore/merckkgaa制造)浓缩含有目标化合物的级分,并用缓冲溶液(5mm磷酸盐缓冲溶液和50mm2-吗啉代乙磺酸(mes)溶液(ph6.8))缓冲液替代。(2)通过羟基磷灰石色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:bio-scaleminichti型盒(5ml)(由bio-radlaboratories,inc.制造)流速:5ml/min(进样期间1.25ml/min)连接两个柱,并将前一步骤(1)中获得的溶液以两部分纯化。将溶液添加至柱的上部,并将溶液a(5mm磷酸盐缓冲溶液和50mm2-吗啉代乙磺酸(mes)溶液(ph6.8))以1.25ml/min以2cv注入柱中,并以5ml/min以3cv向其中进一步注入。然后,使用溶液a和溶液b(5mm磷酸盐缓冲溶液,50mm2-吗啉代乙磺酸(mes)溶液(ph6.8),和2m氯化钠溶液)进行洗脱。洗脱条件涉及溶液a:溶液b=100:0至0:100(15cv)。此外,将洗涤溶液(500mm磷酸盐缓冲溶液(ph6.5))以5cv注入柱中。使用微体积分光光度计xpose(由trineannv制造)和experion电泳站(由bio-radlaboratories,inc.制造)证实在洗脱期间通过uv(280nm)检测到的级分。使用amiconultra(ultracel30k,由merckmillipore/merckkgaa制造)浓缩含有目标化合物的级分,并用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代,以获得23.7mg/ml的(fucα1,6)glcnac-mab-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(19.9ml)。esi-ms:(fucα1,6)glcnac-mab-a(-lys,pyrglu)的重链的计算值,m=50166.6,实测值(m/z),50165.3(去卷积数据)。(fucα1,6)glcnac-mab-a的轻链的计算值,m=23292.9,实测值(m/z),23292.0(去卷积数据)。(3-1b)mab-a-[peg(3)-n3]4的制备其中上面给出的式代表具有在sg型n297聚糖的非还原末端处的唾液酸中引入的叠氮化物基团的接头结构,并且在实施例3中,具有在n297聚糖中引入的叠氮化物基团的所有中间接头结构是与上面给出的式中相同的结构。向23.7mg/ml步骤(3-1a)中获得的(fucα1,6)glcnac-mab-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(8.90ml)中添加50mm磷酸盐缓冲溶液(ph6.0)(1.65ml)、实施例1-10中合成的[n3-peg(3)]2-sg(10)-ox(33.8mg)于50mm磷酸盐缓冲溶液(ph6.0)(0.676ml)中的溶液和2.10mg/ml的endosd233q/q303l溶液(pbs)(1.98ml),并将混合物在30℃下孵育3.5小时。对2批进行上述操作。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,将通过亲和色谱的纯化中使用的结合缓冲液从20mm磷酸盐缓冲溶液(ph7.0)改变为20mm磷酸盐缓冲溶液(ph6.0),并根据与步骤(3-1a)中相同的方法进行通过亲和色谱的纯化和通过羟基磷灰石色谱的纯化。然后,使用amiconultra(ultracel30k,由merckmillipore/merckkgaa制造)浓缩含有目标化合物的级分,且随后用20mm磷酸盐缓冲溶液(ph6.0)缓冲液替代,以获得20.5mg/ml的mab-a-[peg(3)-n3]4溶液(20mm磷酸盐缓冲溶液(ph6.0))(19.0ml)。esi-ms:mab-a-[peg(3)-n3]4(-lys,pyrglu)的重链的计算值,m=52569.9,实测值(m/z),52569.4(去卷积数据)。mab-a-[peg(3)-n3]4的轻链的计算值,m=23292.9,实测值(m/z),23292.1(去卷积数据)。(3-1c)mab-a-[peg(3)//peg(12)2-hanp(1-28)]4(下式的目标化合物:化合物3-1)的制备其中上面给出的式代表化合物3-1中的n297聚糖中的唾液酸、peg接头和hanp肽的结构;通过式中的点击反应形成的三唑环具有几何异构性,且化合物3-1维持具有该式的左右结构的接头的混合物;且由于化合物3-1中所有n297聚糖部分的非还原末端处的唾液酸残基用上面给出的式的接头部分修饰,所以每个缀合物分子连接四个hanp(1-28)分子。向20.5mg/ml的(3-1b)中获得的mab-a-[peg(3)-n3]4溶液(20mm磷酸盐缓冲溶液(ph6.0))(2.44ml)中添加20mm磷酸盐缓冲溶液(ph6.0)(1.56ml)、二甲基亚砜(0.736ml)和作为dbco化合物的实施例1-1中合成的dbco-peg(12)2-hanp(1-28)(12.9mg)于二甲基亚砜(0.264ml)中的溶液,并将混合物在30℃下孵育16小时(点击反应)。用nap25(由gehealthcarejapancorp.制造)和20mm磷酸盐缓冲溶液(ph6.0)部分纯化反应溶液。通过在下面给出的条件下的疏水相互作用色谱、随后通过下面给出的亲和色谱的纯化来证实反应的进展程度。(1)疏水相互作用色谱的分析条件分析设备:hitachid-7000(由hitachi,ltd.制造)柱:tskgelbutyl-npr(4.6x100mm)(由tosohcorp.制造)流动相:溶液a:20mm磷酸盐缓冲溶液(ph7.0)和2m硫酸铵溶液溶液b:20mm磷酸盐缓冲溶液(ph7.0)梯度:a:b=75:25至0:100(0至25min)-0:100(25至30min)温度:25℃波长:214nm流速:1ml/min。(2)通过亲和色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:hitraprproteinaff(5ml)(由gehealthcarejapancorp.制造)流速:5ml/min(进样期间1.25ml/min)。为了与柱结合,将获得的反应溶液添加至柱的上部,并以1.25ml/min以2cv将结合缓冲液(20mm磷酸盐缓冲溶液(ph6.0))注入柱中,并以5ml/min以5cv向其中进一步注入。对于中间洗涤,将洗涤溶液(20mm磷酸盐缓冲溶液(ph7.0)和0.5m氯化钠溶液)以10cv注入柱中。为了洗脱,将洗脱缓冲液(immunopureigg洗脱缓冲液,由pierce/thermofisherscientificinc.制造)以6cv注入柱中。洗脱液立即用1mtris缓冲溶液(ph9.0)中和。使用微体积分光光度计xpose(由trineannv制造)和疏水相互作用色谱证实在洗脱期间通过uv(280nm)检测到的级分。使用amiconultra(ultracel30k,由merckmillipore/merckkgaa制造)浓缩含有目标化合物的级分,用5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)缓冲液替代,并通过过滤器(millex-gv,0.22μm,pvdf,已经灭菌,由merckmillipore/merckkgaa制造)过滤,以获得19.4mg/ml的mab-a-[peg(3)//peg(12)2-hanp]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(2.14ml)。esi-ms:mab-a-[peg(3)//peg(12)2-hanp]4(-lys,pyr-glu)的重链的计算值,m=61708.2,实测值(m/z),61706.4(去卷积数据)。mab-a-[peg(3)//peg(12)2-hanp]4的轻链的计算值,m=23292.9,实测值(m/z),23291.7(去卷积数据)。<实施例3-2>mab-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(下式的目标化合物:化合物3-2)的合成其中上面给出的式代表化合物3-2中的n297聚糖中的唾液酸、peg接头和hanp肽的结构;通过式中的点击反应形成的三唑环具有几何异构性,且化合物3-2维持具有该式的左右结构的接头的混合物;且由于化合物3-2中所有n297聚糖部分的非还原末端处的唾液酸残基用上面给出的式的接头部分修饰,所以每个缀合物分子连接四个hanp(1-28)分子。将20.5mg/ml的步骤(3-1b)中获得的mab-a-[peg(3)-n3]4溶液(20mm磷酸盐缓冲溶液(ph6.0))(5.51ml)、20mm磷酸盐缓冲溶液(ph6.0)(2.49ml)、二甲基亚砜(1.40ml)和步骤(1-2f)中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28)(43.1mg)于二甲基亚砜(0.596ml)中的溶液在30℃下孵育16小时。对2批进行上述操作。以与步骤(3-1c)中相同的方式进行后续程序,以获得21.6mg/ml的mab-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(10.2ml)。esi-ms:mab-a-[peg(3)//peg(12)2-(sg-)asn-hanp]4(-lys,pyrglu)的重链的计算值,m=66348.4,实测值(m/z),66347.5(去卷积数据)。mab-a-[peg(3)//peg(12)2-(sg-)asn-hanp]4的轻链的计算值,m=23292.9,实测值(m/z),23291.9(去卷积数据)。<实施例3-3>clch-a-[peg(3)//peg(12)2-hanp(1-28)]4(化合物3-3)的合成(3-3a)(fucα1,6)glcnac-clch-a的制备使用amiconultra(ultracel10k,由merckmillipore/merckkgaa制造)将21.6mg/ml实施例2-2中制备的clch-a溶液(5%山梨糖醇/25mm组氨酸溶液(ph6.0))(21.0ml)用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代。将2.00mg/ml野生型endos溶液(pbs)(2.27ml)添加至22.0mg/ml获得的clch-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(20.0ml)中,并将混合物在37℃下孵育6小时。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,根据与步骤(3-1a)中相同的方法进行通过亲和色谱的纯化和使用羟基磷灰石柱的纯化。然后,使用amiconultra(ultracel10k,由merckmillipore/merckkgaa制造)浓缩含有目标化合物的级分,并随后用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代,以获得21.1mg/ml的(fucα1,6)glcnac-clch-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(20.4ml)。esi-ms:(fucα1,6)glcnac-clch-a(-lys)的重链的计算值,m=36386.4,实测值(m/z),36386.1(去卷积数据)。(fucα1,6)glcnac-clch-a的轻链的计算值,m=11507.6,实测值(m/z),11506.8(去卷积数据)。(3-3b)clch-a-[peg(3)-n3]4的制备向21.1mg/ml步骤(3-3a)中获得的(fucα1,6)glcnac-clch-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(4.65ml)中添加50mm磷酸盐缓冲溶液(ph6.0)(2.66ml)、实施例1-10中合成的[n3-peg(3)]2-sg(10)-ox(23.5mg)于50mm磷酸盐缓冲溶液(ph6.0)(0.470ml)中的溶液和2.10mg/ml的endosd233q/q303l溶液(pbs)(1.40ml),并将混合物在30℃下孵育3小时。进一步,向其中添加步骤(1-10b)中合成的[n3-peg(3)]2-sg(10)-ox(11.8mg)于50mm磷酸盐缓冲溶液(ph6.0)(0.236ml)中的溶液,并将混合物在30℃下孵育1小时。对3批进行上述操作。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,将使用亲和柱的纯化中使用的结合缓冲液从20mm磷酸盐缓冲溶液(ph7.0)改变为20mm磷酸盐缓冲溶液(ph6.0),并根据与步骤(3-1a)中相同的方法进行通过亲和色谱的纯化和使用羟基磷灰石柱的纯化。然后,使用amiconultra(ultracel10k,由merckmillipore/merckkgaa制造)浓缩含有目标化合物的级分,且随后用5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)缓冲液替代,以获得19.3mg/ml的clch-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(13.8ml)。esi-ms:clch-a-[peg(3)-n3]4(-lys)的重链的计算值,m=38789.6,实测值(m/z),38789.4(去卷积数据)。clch-a-[peg(3)-n3]4的轻链的计算值,m=11507.6,实测值(m/z),11506.7(去卷积数据)。(3-3c)clch-a-[peg(3)//peg(12)2-hanp(1-28)]4(化合物3-3)的制备其中上面给出的式代表化合物3-3中的n297聚糖中的唾液酸、peg接头和hanp肽的结构;通过式中的点击反应形成的三唑环具有几何异构性,且化合物3-3维持具有该式的左右结构的接头的混合物;且由于化合物3-3中所有n297聚糖部分的非还原末端处的唾液酸残基用上面给出的式的接头部分修饰,所以每个缀合物分子连接四个hanp(1-28)分子。向19.3mg/ml的步骤(3-3b)中获得的clch-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(5.18ml)中添加5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)(2.82ml)、二甲基亚砜(1.18ml)和实施例1-1中合成的dbco-peg(12)2-hanp(1-28)(40.1mg)于二甲基亚砜(0.816ml)中的溶液,并将混合物在30℃下孵育16小时。对2批进行上述操作。用nap25(由gehealthcarejapancorp.制造)和20mm磷酸盐缓冲溶液(ph6.0)部分纯化反应溶液。通过在下面给出的条件下的疏水相互作用色谱、随后通过下面给出的亲和色谱的纯化来证实反应的进展程度。(1)疏水相互作用色谱的分析条件分析设备:hitachid-7000(由hitachi,ltd.制造)柱:tskgelbutyl-npr(4.6x100mm)(由tosohcorp.制造)流动相:溶液a:20mm磷酸盐缓冲溶液(ph7.0)和2m硫酸铵溶液溶液b:20mm磷酸盐缓冲溶液(ph7.0)梯度:a:b=75:25至0:100(0至25min)-0:100(25至30min)温度:25℃波长:214nm流速:1ml/min。(2)通过亲和色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:hitraprproteinaff(5ml)(由gehealthcarejapancorp.制造)流速:5ml/min(进样期间1.25ml/min)。将获得的2批反应溶液以三部分纯化。为了与柱结合,将反应溶液添加至柱的上部,并以1.25ml/min以4cv将结合缓冲液(20mm磷酸盐缓冲溶液(ph6.0))注入柱中,并以5ml/min以5cv向其中进一步注入。对于中间洗涤,将洗涤溶液(20mm磷酸盐缓冲溶液(ph7.0)和0.5m氯化钠溶液)以10cv注入柱中。为了洗脱,将洗脱缓冲液(immunopureigg洗脱缓冲液,由pierce/thermofisherscientificinc.制造)以6cv注入柱中。洗脱液立即用1mtris缓冲溶液(ph9.0)中和。使用微体积分光光度计xpose(由trineannv制造)和疏水相互作用色谱证实在洗脱期间通过uv(280nm)检测到的级分。使用amiconultra(ultracel10k,由merckmillipore/merckkgaa制造)浓缩含有目标化合物的级分,用5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)缓冲液替代,并通过过滤器(millex-gv,0.22μm,pvdf,已经灭菌,由merckmillipore/merckkgaa制造)过滤,以获得21.3mg/ml的clch-a-[peg(3)//peg(12)2-hanp(1-28)]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(9.60ml)。esi-ms:clch-a-[peg(3)//peg(12)2-hanp]4(-lys)的重链的计算值,m=47928.0,实测值(m/z),47928.4(去卷积数据)。clch-a-[peg(3)//peg(12)2-hanp]4的轻链的计算值,m=11507.6,实测值(m/z),11506.7(去卷积数据)。<实施例3-4>clch-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(下式的目标化合物:化合物3-4)的合成其中上面给出的式代表化合物3-4中的n297聚糖中的唾液酸、peg接头和hanp肽的结构;通过式中的点击反应形成的三唑环具有几何异构性,且化合物3-4维持具有该式的左右结构的接头的混合物;且由于化合物3-4中所有n297聚糖部分的非还原末端处的唾液酸残基用上面给出的式的接头部分修饰,所以每个缀合物分子连接四个hanp(1-28)分子。将19.3mg/ml的步骤(3-3b)中获得的clch-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(2.59ml)、5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)(1.41ml)、二甲基亚砜(0.602ml)和实施例1-2中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28)(28.7mg)于二甲基亚砜(0.398ml)中的溶液在30℃下孵育16小时。以与步骤(3-3c)中相同的方式进行后续程序,以获得20.0mg/ml的clch-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(2.46ml)。esi-ms:clch-a-[peg(3)//peg(12)2-(sg-)asn-hanp]4(-lys)的重链的计算值,m=52568.2,实测值(m/z),52568.4(去卷积数据)。clch-a-[peg(3)//peg(12)2-(sg-)asn-hanp]4的轻链的计算值,m=11507.6,实测值(m/z),11506.7(去卷积数据)。<实施例3-5>mab-a-[peg(3)//peg(12)2-(sg-)asn-hanp]2(化合物3-5)的合成(3-5a)mab-a-[peg(3)-n3]2的制备使用实施例1-11中合成的[n3-peg(3)]-msg1(9)-ox作为聚糖供体,将步骤(3-1a)中合成的化合物在30℃下孵育3小时。进行与步骤(3-1b)中相同的操作,以获得14.1mg/ml的mab-a-[peg(3)-n3]2溶液(20mm磷酸盐缓冲溶液(ph6.0))(5.90ml)。esi-ms:mab-a-[peg(3)-n3]2(-lys,pyrglu)的重链的计算值,m=52078.4,实测值(m/z),52077.0(去卷积数据)。mab-a-[peg(3)-n3]2的轻链的计算值,m=23292.9,实测值(m/z),23291.7(去卷积数据)。(3-5b)mab-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]2(下式的目标化合物:化合物3-5)的制备其中上面给出的结构式代表化合物3-5中的n297聚糖中的唾液酸、peg接头和hanp肽的结构;通过式中的点击反应形成的三唑环具有几何异构性,且化合物3-5维持具有该式的左右结构的接头的混合物;且由于化合物3-5中所有n297聚糖部分的非还原末端处的唾液酸残基用上面给出的式的接头部分修饰,所以每个缀合物分子连接两个hanp(1-28)分子。使用4当量的实施例1-2中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28),对步骤(3-5a)中合成的化合物进行与实施例3-2中相同的操作,以获得17.9mg/ml的mab-a-[peg(3)//peg(12)2-hanp(1-28)]2溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(2.55ml)。esi-ms:mab-a-[peg(3)//peg(12)2-hanp]2(-lys,pyrglu)的重链的计算值,m=58967.7,实测值(m/z),58968.1(去卷积数据)。mab-a-[peg(3)//peg(12)2-hanp]2的轻链的计算值,m=23292.9,实测值(m/z),23291.8(去卷积数据)。<实施例3-6>clch-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]2(化合物3-6)的合成(3-6a)clch-a-[peg(3)-n3]2的制备使用实施例1-11中合成的[n3-peg(3)]-msg1(9)-ox作为聚糖供体,将步骤(3-3a)中合成的化合物在30℃下孵育3小时。进行与步骤(3-3b)中相同的操作,以获得14.6mg/ml的clch-a-[peg(3)-n3]2溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(3.98ml)。esi-ms:n3-msg-clch-a(-lys)的重链的计算值,m=38298.1,实测值(m/z),38297.7(去卷积数据)。n3-msg-clch-a的轻链的计算值,m=11507.6,实测值(m/z),11506.8(去卷积数据)。(3-6b)clch-a-[peg(3)//peg(12)2-(sg-)asn-anp(1-28)]2的制备其中上面给出的式代表化合物3-6中的n297聚糖中的唾液酸、peg接头和hanp肽的结构;通过式中的点击反应形成的三唑环具有几何异构性,且化合物3-6维持具有该式的左右结构的接头的混合物;且由于化合物3-6中所有n297聚糖部分的非还原末端处的唾液酸残基用上面给出的式的接头部分修饰,所以每个缀合物分子连接两个hanp(1-28)分子。使用4当量的实施例1-2中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28),对步骤(3-6a)中合成的化合物进行与实施例3-4中相同的操作,以获得19.2mg/ml的clch-a-[peg(3)//peg(12)2-(sg-)asn-anp(1-28)]2溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(2.62ml)。esi-ms:clch-a-[peg(3)//peg(12)2-(sg-)asn-anp]2(-lys)的重链的计算值,m=45187.4,实测值(m/z),45188.0(去卷积数据)。clch-a-[peg(3)//peg(12)2-(sg-)asn-anp]2的轻链的计算值,m=11507.6,实测值(m/z),11506.8(去卷积数据)。<实施例3-7>具有不同接头结构的缀合物(化合物3-7至3-14)的合成通过使用实施例3-1中描述的方法从单克隆抗体(mab-b)合成mab-b-[peg(3)-n3]4。随后,通过使用实施例1中合成的不同化合物1-1、1-3、1-4、1-5、1-6、1-7、1-8和1-9各自作为用于点击反应中的dbco化合物合成peg接头部分的结构不同的mab-b-[l(peg)-hanp(1-28)]4分子。表1显示所用dbco化合物和目标缀合物的结构之间的对应关系(对应于每种化合物的l(peg)为化合物3-7:l(peg)b,化合物3-8:l(peg)e,化合物3-9:l(peg)a,化合物3-10:l(peg)d,化合物3-11:l(peg)c,化合物3-12:l(peg)f,化合物3-13:l(peg)g,或化合物3-14:l(peg)h);在所有化合物中,如在化合物3-1至3-6中,通过式中的点击反应形成的三唑环具有几何异构性,且所述化合物维持具有几何异构结构的接头的混合物;且每个缀合物分子连接四个hanp(1-28)分子)。[表1]表1.mab-b中使用的peg接头的类型化合物编号peg接头的类型dbco化合物3-7mab-b-[peg(3)//peg(12)-peg(12)-hanp(1-28)]4化合物1-13-8mab-b-[peg(3)//peg(12)-hanp(1-28)]4化合物1-33-9mab-b-[peg(3)//peg(24)-hanp(1-28)]4化合物1-43-10mab-b-[peg(3)//peg(12)-peg(6)-hanp(1-28)]4化合物1-53-11mab-b-[peg(3)//peg(6)-peg(12)-hanp(1-28)]4化合物1-63-12mab-b-[peg(3)//peg(6)-peg(6)-hanp(1-28)]4化合物1-73-13mab-b-[peg(3)//peg(6)-peg(6)-peg(6)-hanp(1-28)]4化合物1-83-14mab-b-[peg(3)//peg(6)-peg(6)-peg(6)-peg(6)-hanp(1-28)]4化合物1-9<实施例3-8>clch-b-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(其中上述化合物3-2的结构中的clch-a用clch-b替代的目标化合物:化合物3-15)的合成本实施例的每个步骤中的反应方案和物质结构对应于其中clch-a用clch-b替代的实施例3-2的相应方案中的那些。(3-8a)(fucα1,6)glcnac-clch-b的制备根据步骤(3-3a)中描述的方法通过用20.6mg/ml的实施例2-3中制备的clch-b溶液(5%山梨糖醇/25mm组氨酸溶液(ph6.0))替代步骤(3-3a)中描述的clch-a溶液来获得19.8mg/ml的(fucα1,6)glcnac-clch-b溶液(50mm磷酸盐缓冲溶液(ph6.0))(16ml)。esi-ms:(fucα1,6)glcnac-clch-b(-lys)的重链的计算值,m=36303.1;实测值36302.6(去卷积数据)。(fucα1,6)glcnac-clch-b的轻链的计算值,m=11507.8;实测值11506.7(去卷积数据)。(3-8b)clch-b-[peg(3)-n3]4的制备根据步骤(3-3b)中描述的方法通过用19.8mg/ml的步骤(3-8a)中获得的(fucα1,6)glcnac-clch-b溶液(50mm磷酸盐缓冲溶液(ph6.0))替代步骤(3-3b)中描述的(fucα1,6)glcnac-clch-a溶液来获得19.3mg/ml的clch-b-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(15.7ml)。esi-ms:clch-b-[peg(3)-n3]4(-lys)的重链的计算值,m=38706.3;实测值38704.7(去卷积数据)。clch-b-[peg(3)-n3]4的轻链的计算值,m=11507.8;实测值11506.8(去卷积数据)。(3-8c)clch-b-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(化合物3-15)的制备将19.3mg/ml的步骤(3-8b)中获得的clch-b-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(3.90ml)、5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)(4.1ml)、二甲基亚砜(1.4ml)和实施例1-2中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28)(43.4mg)于二甲基亚砜(0.6ml)中的溶液混合并在30℃下孵育16小时。对4批进行上述操作。以与(3-3c)中相同的方式进行后续程序,以获得27.1mg/ml的clch-b-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(11.5ml)。esi-ms:clch-b-[peg(3)//peg(12)2-(sg-)asn-hanp]4(-lys)的重链的计算值,m=52484.9;实测值52484.4(去卷积数据)。clch-b-[peg(3)//peg(12)2-(sg-)asn-hanp]4的轻链的计算值,m=11507.8;实测值11506.7(去卷积数据)。<实施例3-9>fc-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(下式的目标化合物:化合物3-16)的合成(3-9a)(fucα1,6)glcnac-fc-a的制备其中上面给出的式代表衍生自制备的载体蛋白中所含的聚糖结构缺失突变体的混合物;且除了n=1以外,可以包括n=0。使用vivaspin20(10,000mwco,由sartoriusag制造)将10.4mg/ml的实施例2-5中制备的fc-a溶液(5%山梨糖醇/25mm组氨酸溶液(ph6.0))(80.0ml)用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代并调节至约50ml。将获得的fc-a溶液(50mm磷酸盐缓冲溶液(ph6.0))分至两个violamo离心管(vio-50b)中。向其中添加7.70mg/ml野生型endos溶液(pbs)(0.8ml),并将混合物在37℃下孵育2小时。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,根据以下方法进行通过亲和色谱的纯化和使用羟基磷灰石柱的纯化。(1)通过亲和色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:mediascoutvalichrom25mmidx100mmh;v=50.0ml柱填料:kanekakancapa流速:40ml/min(样品添加期间10ml/min)将获得的反应溶液(×2管)通过0.45μmpvdf过滤器过滤,调节至约40ml(x2),并以两部分纯化。为了与柱结合,将反应溶液添加至柱的上部,并以10ml/min以4.4cv将结合缓冲液(20mm磷酸盐缓冲溶液(ph7.0))注入柱中,并以40ml/min以5cv向其中进一步注入。对于中间洗涤,将洗涤溶液(20mm磷酸盐缓冲溶液(ph7.0)和0.5m氯化钠溶液)以10cv注入柱中。为了洗脱,将洗脱缓冲液(immunopureigg洗脱缓冲液,由pierce/thermofisherscientificinc.制造)以6cv注入柱中。洗脱液立即用1mtris缓冲溶液(ph9.0)中和。根据需要使用微体积分光光度计xpose(由trineannv制造)和experion电泳站(由bio-radlaboratories,inc.制造)证实在洗脱期间通过uv(280nm)检测到的级分。使用vivaspin20(10,000mwco,由sartoriusag制造)浓缩含有目标化合物的级分,并用缓冲溶液(5mm磷酸盐缓冲溶液和50mm2-吗啉代乙磺酸(mes)溶液(ph7.0))缓冲液替代。(2)通过羟基磷灰石色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:mediascoutvalichrom25mmidx100mmh;v=50.0ml柱填料:bio-radchti型40um流速:20ml/min(样品添加期间10ml/min)将前一步骤(1)中获得的溶液通过0.45μmpvdf过滤器过滤并分成两部分(各自约40ml)。通过以下步骤将这些部分以两部分纯化:将溶液添加至柱的上部,并将溶液a(5mm磷酸盐缓冲溶液和50mm2-吗啉代乙磺酸(mes)溶液(ph7.0))以10ml/min以4.2cv注入柱中,并以20ml/min以2cv向其中进一步注入。然后,使用溶液a和溶液b(5mm磷酸盐缓冲溶液,50mm2-吗啉代乙磺酸(mes)溶液(ph7.0),和2m氯化钠溶液)进行洗脱。洗脱条件涉及溶液a:溶液b=100:0至0:100(15cv)。此外,将洗涤溶液(500mm磷酸盐缓冲溶液(ph6.5))以5cv注入柱中。根据需要使用微体积分光光度计xpose(由trineannv制造)和experion电泳站(由bio-radlaboratories,inc.制造)证实在洗脱期间通过uv(280nm)检测到的级分。使用vivaspin20(10,000mwco,由sartoriusag制造)浓缩含有目标化合物的级分,并用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代。将所得溶液分至4个容器(各自为24ml)中并用作以下溶液a至d。溶液a:7.24mg/ml的(fucα1,6)glcnac-fc-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(24ml)(173.8mg)溶液b:7.42mg/ml的(fucα1,6)glcnac-fc-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(24ml)(178.1mg)溶液c:7.18mg/ml的(fucα1,6)glcnac-fc-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(24ml)(172.3mg)溶液d:7.19mg/ml的(fucα1,6)glcnac-fc-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(24ml)(172.6mg)esi-ms:(fucα1,6)glcnac-fc-a(-lys)的链的计算值,m=25685.0,实测值25681.9(去卷积数据)。(3-9b)fc-a-[peg(3)-n3]4的制备其中上面给出的式代表衍生自制备的载体蛋白中所含的聚糖结构缺失突变体的混合物;且除了n=1以外,可以包括n=0。将步骤(3-8a)中获得的四种(fucα1,6)glcnac-fc-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(24ml)各自分成两部分以制备总共八个50ml离心管。向一种(fucα1,6)glcnac-fc-a溶液(50mm磷酸盐缓冲溶液(ph6.0))(12ml)中添加50mm磷酸盐缓冲溶液(ph6.0)(1.65ml)、实施例1-10中合成的[n3-peg(3)]2-sg(10)-ox(56.0mg)于50mm磷酸盐缓冲溶液(ph6.0)(1.0ml)中的溶液和4.3mg/ml的endosd233q/q303l溶液(pbs)(1.16ml),并将混合物在30℃下孵育4小时。对8批进行上述操作。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,将通过亲和色谱的纯化中使用的结合缓冲液从20mm磷酸盐缓冲溶液(ph7.0)改变为20mm磷酸盐缓冲溶液(ph6.0),并根据与步骤(3-9a)中相同的方法进行通过亲和色谱的纯化和通过羟基磷灰石色谱的纯化。将含有目标化合物的所有级分合并,且然后使用配备有超滤膜(pelliconxl50cassetteultracel10kda(使用两个膜))的超滤设备(cogentμscaletffsystem)浓缩至约100ml。使用vivaspin20(10,000mwco,由sartoriusag制造)将浓缩物用5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)缓冲液替代。将获得的溶液分至两个容器中,并用作以下溶液a和b。溶液a:12.45mg/ml的fc-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(19.7ml)(245.3mg)溶液b:14.95mg/ml的fc-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(19.5ml)(291.5mg)esi-ms:fc-a-[peg(3)-n3]4(-lys)的链的计算值,m=28088.3;实测值28086.4(去卷积数据)。(3-9c)fc-a-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(化合物3-16)的制备其中上面给出的式代表衍生自制备的载体蛋白中所含的聚糖结构缺失突变体的混合物;且除了n=1以外,可以包括n=0。其中上面给出的式代表化合物3-16中的n297聚糖中的唾液酸、peg接头和hanp肽的结构;通过式中的点击反应形成的三唑环具有几何异构性,且化合物3-16维持具有该式的左右结构的接头的混合物;且由于化合物3-16中所有n297聚糖部分的非还原末端处的唾液酸残基用上面给出的式的接头部分修饰,所以每个缀合物分子连接四个用于正常形式fc-a的hanp(1-28)分子和两个用于聚糖缺失突变体fc-a的hanp(1-28)分子。将12.45mg的作为步骤(3-9b)中制备溶液a的fc-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(4.0ml)、5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)(6.0ml)、二甲基亚砜(1.9ml)和实施例1-2中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28)(45.1mg)于二甲基亚砜(0.62ml)中的溶液混合并在30℃下孵育16小时。对4批溶液a进行上述操作。此外,将14.95mg的作为步骤(3-9b)中制备的溶液b的fc-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(3.3ml)、5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)(6.7ml)、二甲基亚砜(1.9ml)和实施例1-2中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28)(45.1mg)于二甲基亚砜(0.62ml)中的溶液混合并在30℃下孵育16小时。对4批溶液b进行上述操作。用nap25(由gehealthcarejapancorp.制造)和20mm磷酸盐缓冲溶液(ph6.0)部分纯化因此获得的总共8批的反应溶液。通过在下面给出的条件下的疏水相互作用色谱、随后通过下面给出的亲和色谱的纯化来证实反应的进展程度。(1)疏水相互作用色谱的分析条件分析设备:hitachid-7000(由hitachi,ltd.制造)柱:tskgelbutyl-npr(4.6x100mm)(由tosohcorp.制造)流动相:溶液a:20mm磷酸盐缓冲溶液(ph7.0)和2m硫酸铵溶液溶液b:20mm磷酸盐缓冲溶液(ph7.0)梯度:a:b=75:25至0:100(0至25min)-0:100(25至30min)温度:25℃波长:214nm流速:1ml/min。(2)通过亲和色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:mediascoutvalichrom25mmidx100mmh;v=50.0ml柱填料:kanekakancapa流速:40ml/min(样品添加期间10ml/min)。将获得的溶液通过0.45μmpvdf过滤器过滤,分成两部分(各自约80ml),并通过以下操作以两部分纯化:为了与柱结合,将反应溶液添加至柱的上部,并以10ml/min以5cv将结合缓冲液(20mm磷酸盐缓冲溶液(ph6.0))注入柱中,并以40ml/min以5cv向其中进一步注入。对于中间洗涤,将洗涤溶液(20mm磷酸盐缓冲溶液(ph7.0)和0.5m氯化钠溶液)以10cv注入柱中。为了洗脱,将洗脱缓冲液(immunopureigg洗脱缓冲液,由pierce/thermofisherscientificinc.制造)以6cv注入柱中。洗脱液立即用1mtris缓冲溶液(ph9.0)中和。根据需要使用微体积分光光度计xpose(由trineannv制造)和疏水相互作用色谱证实在洗脱期间通过uv(280nm)检测到的级分。将来自两部分的含有目标化合物的所有级分合并,然后使用配备有超滤膜(pelliconxl50cassetteultracel10kda(使用两个膜))的超滤设备(cogentμscaletffsystem)浓缩至约40ml,且然后用5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)缓冲液替代。最后,将溶液通过过滤器(millex-gv,0.22μm,pvdf,已经灭菌,由merckmillipore/merckkgaa制造)过滤,以获得7.81mg/ml的fc-a-[peg(3)//peg(12)2-hanp(1-28)]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(56ml)。fc-a-[peg(3)//peg(12)2-hanp(1-28)]4(-lys)的链的计算值,m=41866.8;实测值41863.8(去卷积数据)。下面将给出通过没有片段化的质谱法分析缀合物的结果。fc-a-[peg(3)//peg(12)2-hanp(1-28)]4(-lys)的计算值,m=83713.5;实测值83712.6(去卷积数据)。fc-a-[peg(3)//peg(12)2-hanp(1-28)]2(-lys)的计算值,m=67186.4;实测值67184.8(去卷积数据)。<实施例3-10>fc-b-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(具有其中化合物3-16的结构中的fc-a用fc-b替代的结构的化合物:化合物3-17)的合成本实施例的每个步骤中的反应方案和物质结构对应于其中fc-a用fc-b替代的实施例3-9的相应方案中的那些。(3-10a)(fucα1,6)glcnac-fc-b的制备使用vivaspin20(10,000mwco,由sartoriusag制造)将13.0mg/ml的实施例2-4中制备的fc-b溶液(5%山梨糖醇/25mm组氨酸溶液(ph6.0))(20.0ml)用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代。向20.0mg/ml的获得的fc-b溶液(50mm磷酸盐缓冲溶液(ph6.0))(13.0ml)中添加50mm磷酸盐缓冲溶液(ph6.0)(7.0ml),然后添加2.00mg/ml的野生型endos溶液(pbs)(1.92ml),并将混合物在37℃下孵育2小时。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,根据以下方法进行通过亲和色谱的纯化和使用羟基磷灰石柱的纯化。(1)通过亲和色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:hitraprproteinaff(5ml)(由gehealthcarejapancorp.制造)流速:5ml/min(进样期间1.25ml/min)将获得的反应溶液分成五部分,并通过以下方法以五部分进行纯化:为了与柱结合,将反应溶液添加至柱的上部,并以1.25ml/min以2cv将结合缓冲液(20mm磷酸盐缓冲溶液(ph7.0))注入柱中,并以5ml/min以5cv向其中进一步注入。对于中间洗涤,将洗涤溶液(20mm磷酸盐缓冲溶液(ph7.0)和0.5m氯化钠溶液)以10cv注入柱中。为了洗脱,将洗脱缓冲液(immunopureigg洗脱缓冲液,由pierce/thermofisherscientificinc.制造)以6cv注入柱中。洗脱液立即用1mtris缓冲溶液(ph9.0)中和。根据需要使用微体积分光光度计xpose(由trineannv制造)和experion电泳站(由bio-radlaboratories,inc.制造)证实在洗脱期间通过uv(280nm)检测到的级分。将来自五部分的含有目标化合物的所有级分合并,然后使用vivaspin20(10,000mwco,由sartoriusag制造)浓缩,用缓冲溶液(5mm磷酸盐缓冲溶液和50mm2-吗啉代乙磺酸(mes)溶液(ph6.5))缓冲液替代,并调节至12ml。(2)通过羟基磷灰石色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:bio-scaleminichti型盒(5ml)(由bio-radlaboratories,inc.制造)流速:5ml/min(进样期间1.25ml/min)。连接两个柱,并将前一步骤(1)中获得的溶液分成三部分,并通过以下方法以三部分纯化:将溶液添加至柱的上部,并将溶液a(5mm磷酸盐缓冲溶液和50mm2-吗啉代乙磺酸(mes)溶液(ph6.5))以1.25ml/min以2cv注入柱中,并以5ml/min以3cv向其中进一步注入。然后,使用溶液a和溶液b(5mm磷酸盐缓冲溶液,50mm2-吗啉代乙磺酸(mes)溶液(ph6.5),和2m氯化钠溶液)进行洗脱。洗脱条件涉及溶液a:溶液b=100:0至0:100(15cv)。此外,将洗涤溶液(500mm磷酸盐缓冲溶液(ph6.5))以5cv注入柱中。根据需要使用微体积分光光度计xpose(由trineannv制造)和experion电泳站(由bio-radlaboratories,inc.制造)证实在洗脱期间通过uv(280nm)检测到的级分。将来自三部分的含有目标化合物的所有级分合并,然后使用vivaspin20(10,000mwco,由sartoriusag制造)浓缩,并用50mm磷酸盐缓冲溶液(ph6.0)缓冲液替代,以获得7.17mg/ml的(fucα1,6)glcnac-fc-b溶液(50mm磷酸盐缓冲溶液(ph6.0))(35.4ml)。esi-ms:(fucα1,6)glcnac-fc-b(-lys)的链的计算值,m=25287.7;实测值25286.8(去卷积数据)。(3-10b)fc-b-[peg(3)-n3]4的制备向7.17mg/ml的步骤(3-10a)中获得的(fucα1,6)glcnac-fc-b溶液(50mm磷酸盐缓冲溶液(ph6.0))(6.0ml)中添加实施例1-10中合成的[n3-peg(3)]2-sg(10)-ox(28.6mg)溶液(50mm磷酸盐缓冲溶液(ph6.0))(0.50ml)和2.10mg/ml的endosd233q/q303l溶液(pbs)(1.2ml),并将混合物在30℃下孵育3.5小时。向该反应溶液中进一步添加步骤1-10中合成的[n3-peg(3)]2-sg(10)-ox(6.1mg)溶液(50mm磷酸盐缓冲溶液(ph6.0))(0.10ml),并将混合物在30℃下孵育0.5小时。对5批进行上述操作。使用experion电泳站(由bio-radlaboratories,inc.制造)证实反应的进展程度。反应完成后,将通过亲和色谱的纯化中使用的结合缓冲液从20mm磷酸盐缓冲溶液(ph7.0)改变为20mm磷酸盐缓冲溶液(ph6.0),并根据与步骤(3-10a)中相同的方法进行通过亲和色谱的纯化和通过羟基磷灰石色谱的纯化。将分离操作中获得的部分合并,并使用vivaspin20(10,000mwco,由sartoriusag制造)浓缩含有目标化合物的所有级分,且然后使用nap25(由gehealthcarejapancorp.制造)用5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)缓冲液替代,以获得13.06mg/ml的fc-b-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(13.7ml)。esi-ms:fc-b-[peg(3)-n3]4(-lys)的链的计算值,m=27691.0;实测值27690.4(去卷积数据)。(3-10c)fc-b-[peg(3)//peg(12)2-(sg-)asn-hanp(1-28)]4(化合物3-17)的制备将13.06mg/ml的步骤(3-10b)中制备的fc-b-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(3.1ml)、5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)(4.9ml)、二甲基亚砜(1.5ml)和实施例1-2中合成的dbco-peg(12)2-(sg-)asn-hanp(1-28)(36.6mg)于二甲基亚砜(0.51ml)中的溶液混合并在30℃下孵育16小时。对4批进行上述操作。用nap25(由gehealthcarejapancorp.制造)和20mm磷酸盐缓冲溶液(ph6.0)部分纯化反应溶液。通过在下面给出的条件下的疏水相互作用色谱、随后通过下面给出的亲和色谱的纯化来证实反应的进展程度。(1)疏水相互作用色谱的分析条件分析设备:hitachid-7000(由hitachi,ltd.制造)柱:tskgelbutyl-npr(4.6x100mm)(由tosohcorp.制造)流动相:溶液a:20mm磷酸盐缓冲溶液(ph7.0)和2m硫酸铵溶液溶液b:20mm磷酸盐缓冲溶液(ph7.0)梯度:a:b=75:25至0:100(0至25min)-0:100(25至30min)温度:25℃波长:214nm流速:1ml/min。(2)通过亲和色谱的纯化纯化设备:aktapure150(由gehealthcarejapancorp.制造)柱:hitraprproteinaff,5ml流速:5ml/min(样品添加期间1.25ml/min)。将获得的溶液分成六部分,并通过以下方法以六部分进行纯化:为了与柱结合,将反应溶液添加至柱的上部,并以1.25ml/min以4cv将结合缓冲液(20mm磷酸盐缓冲溶液(ph6.0))注入柱中,并以5ml/min以5cv向其中进一步注入。对于中间洗涤,将洗涤溶液(20mm磷酸盐缓冲溶液(ph7.0)和0.5m氯化钠溶液)以10cv注入柱中。为了洗脱,将洗脱缓冲液(immunopureigg洗脱缓冲液,由pierce/thermofisherscientificinc.制造)以6cv注入柱中。洗脱液立即用1mtris缓冲溶液(ph9.0)中和。根据需要使用微体积分光光度计xpose(由trineannv制造)和疏水相互作用色谱证实在洗脱期间通过uv(280nm)检测到的级分。将来自六部分的含有目标化合物的所有级分合并,使用vivaspin20(10,000mwco,由sartoriusag制造)浓缩,且然后用5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5)缓冲液替代。最后,将溶液通过过滤器(millex-gv,0.22μm,pvdf,已经灭菌,由merckmillipore/merckkgaa制造)过滤,以获得13.03mg/ml的fc-a-[peg(3)//peg(12)2-hanp(1-28)]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(13.6ml)。fc-b-[peg(3)//peg(12)2-hanp(1-28)]4(-lys)的链的计算值,m=41469.4;实测值41467.9(去卷积数据)。下面将给出通过没有片段化的质谱法分析缀合物的结果。fc-b-[peg(3)//peg(12)2-hanp(1-28)]4(-lys)的计算值,m=82918.8;实测值82917.0(去卷积数据)。fc-b-[peg(3)//peg(12)2-hanp(1-28)]2(-lys)的计算值,m=66391.7;实测值66390.8(去卷积数据)。[实施例4]各种缀合物的评估<实施例4-1>对缀合物的cgmp升高活性的测试通过以下方法测量实施例3中制备的化合物3-1至3-6的cgmp升高活性。将cho/人gc-a细胞(其为引起组成型表达人gc-a的cho细胞)以2×105个细胞/ml悬浮于α-mem、10%fbs和1%青霉素-链霉素中,以20μl/孔(4×103个细胞/孔)接种于384-孔板(由corninginc.,3826制造)上,并在co2培养箱中培养过夜。第二天,从该板去除培养基,且然后向其中以10至15μl/孔添加1.6mmibmx/krb缓冲液。将板在板振荡器上搅拌,且然后在室温下孵育10分钟。随后,向其中以5μl/孔添加溶解于0.1%bsa/pbs中的测试物质(制备稀释系列,使得每种缀合物或天然hanp(1-28)的最终浓度为0.0001、0.001、0.01、0.1、1、10和100nm)。将板在板振荡器上搅拌,且然后在co2培养箱中孵育15分钟。然后,使用cgmp试剂盒(由cisbiobioassays制造)根据所附方案测量每个孔中的cgmp水平。当仅补充有溶剂的孔的测量值被定义为0且补充有1nm天然hanp的孔的测量值被定义为1时,校正每种浓度下测试物质的活性值(t/c)。基于该校正,计算每种测试物质的比活性(测试物质的ec50值/天然hanp的ec50值)和emax(测试物质在浓度范围内的最大活性值)(表2)。从表2的结果可见,所有缀合物都表现出cgmp升高活性。此外,所述缀合物具有几乎等于天然hanp的emax。证实糖基化的hanp-mab-a缀合物(化合物3-2)具有比hanp-mab-a缀合物(化合物3-1)的比活性更弱的比活性。此外,证实糖基化的hanp-clch-a缀合物(化合物3-4)具有比hanp-clch-a缀合物(化合物3-3)的比活性更弱的比活性。这表明向hanp肽引入聚糖倾向于降低体外活性。与具有四个hanp肽部分的糖基化hanp-mab-a缀合物(化合物3-2)相比,具有两个hanp肽部分的糖基化hanp-mab-a缀合物(化合物3-5)的比活性减弱4.5倍。此外,与具有四个hanp肽部分的糖基化hanp-clch-a缀合物(化合物3-4)相比,具有两个hanp肽部分的糖基化hanp-clch-a缀合物(化合物3-6)的比活性减弱3.2倍。这表明具有更大量hanp肽部分的缀合物倾向于表现出更有利的活性。利用clch-b(clch的llca形式)作为载体分子的化合物3-15、利用fc-a(fc片段的lala形式)作为载体分子的化合物3-16和利用fc-b(野生型fc片段)作为载体分子的化合物3-17全部表现出与天然hanp相同水平的体外活性。[表2]<测试实施例4-2>对大鼠血液中缀合物的持续时间的测试通过以下方法检查实施例3中制备的每种缀合物在大鼠血液中的持续时间(可持续升高血液中的cgmp的效果和测试物质在血液中可检测的时间)。(1)血浆样品的制备异氟烷:日本药典异氟烷用于血液收集的针头和注射器:用于结核菌素的terumosyringe25gx1sr用于血液收集的管:capiject微量收集管edta-2na500μl用于样品储存的管:matrix4170样品追踪管0.75ml对每只8-至9-周龄雄性slc:sd大鼠进行异氟烷吸入麻醉(escain吸入麻醉剂的吸入保持在1至2%的浓度)。将根据需要用pbs稀释的测试物质(每种缀合物:化合物3-1至3-6)的溶液以100nmol/kg(1ml/kg)的剂量快速皮下施用于大鼠。在施用前起为每种化合物选择的时间点和施用后0.25、0.5、1、2、4、8、24、48、72、96、120和168小时,随着时间从颈静脉将血液取样(200μl/取样)。将血液样品立即置于冰上。使用离心机(sigma4k15,转子:nr12130-h)将收集的血液样品在4℃下以5000rpm离心5分钟。将分离的血浆样品分成两种类型(用于pk测量的样品和用于cgmp测量的样品)并保存在-80℃,直至测量。(2)血浆cgmp浓度的测量根据试剂盒所附的方案,使用amershamcgmp酶免疫测定biotrak(tm)(eia)系统(双重范围)测量100倍稀释的血浆样品的血浆cgmp浓度。获得的血浆cgmp浓度的变化显示于图6中。(3)血浆样品中的测试物质的检测将10μl段落(1)中制备的每种大鼠血浆样品用rexxiphn缓冲液(gyrosab制造)稀释10倍,并用与上述相同的缓冲液进一步适当稀释10倍(最终:100倍稀释),或者,根据需要,将所得稀释液用1%血浆/rexxiphn缓冲液进一步稀释10倍(最终:1000倍稀释),以制备用于测量的血浆样品。使用的一抗是用于测量样品中的所有含人fc分子(fc/fc)的山羊抗人igg生物素缀合物(southernbiotech),和用于测量样品中具有与之键合的hanp(1-28)的缀合物(anp/fc)的小鼠抗anpigg(genetexinc.)。将每种一抗用0.1%pbs-t调节至700nm。对于两种一抗,用rexxipf缓冲液(gyrosab)将dylight-标记的山羊抗人igg(southernbiotech)作为用于检测的二抗调节至10nm。将每种溶液上样于自动elisa设备gyrolabxp工作站中并注入bioaffy200cd(gyrosab)。通过夹心elisa测量测试物质(fc/fc和anp/fc)的含量以计算血浆浓度。获得的血浆浓度的变化显示于图7中。从图6和7可见,与天然hanp相比,这些各种缀合物被证实是具有以下全部的gc-a活化剂:逐渐迁移至血液中,在血液中长期保留,以及长药理作用持续时间。从图6证实了以下内容。化合物3-1(其为hanp-mab-a缀合物)表现出最高的emax。而且,化合物3-1表现出比作为hanp-clch-a缀合物的化合物3-3的药理活性持续时间更长的药理活性持续时间。化合物3-2(其为糖基化的hanp-mab-a缀合物)和化合物3-4(其为糖基化的hanp-clch-a缀合物)表现出比化合物3-1更慢的达到emax的时间和衰减速率。这可能意味着糖基化的hanp-mab-a缀合物表现出更长的药理活性持续时间。化合物3-5和3-6(其为具有两个hanp肽部分的缀合物)表现出比化合物3-2和3-4(其为具有四个hanp肽部分的缀合物)更弱的emax。这可能意味着具有更大量hanp肽部分的糖基化hanp-mab-a缀合物表现出更强的药理活性。从图7证实了以下内容。证实了血液中hanp缀合物的浓度,甚至在施用后168小时。与化合物3-3(其为hanp-clch-a缀合物)相比,化合物3-1(其为hanp-mab-a缀合物)具有fc/fc和anp/fc之间的小差异,在施用后168小时的高血浆浓度,和缓慢的衰减速率。这表明,化合物3-1在血液中的保留时间长于化合物3-3在血液中的保留时间。与化合物3-1(其为hanp-mab-a缀合物)相比,化合物3-2(其为糖基化的hanp-mab-a缀合物)具有fc/fc和anp/fc之间的小差异,在施用后168小时的高血浆浓度,和缓慢的衰减速率。这表明,化合物3-2在血液中的保留时间长于化合物3-1在血液中的保留时间。与化合物3-3(其为hanp-clch-a缀合物)相比,化合物3-4(其为糖基化的hanp-clch-a缀合物)具有fc/fc和anp/fc之间的小差异,在施用后168小时的高血浆浓度,和缓慢的衰减速率。这表明,化合物3-4在血液中的保留时间长于化合物3-3在血液中的保留时间。<实施例4-3>peg接头对缀合物的cgmp升高活性的影响进行本实验,其目的是使用实施例3-7中合成的缀合物研究peg接头部分对cgmp升高活性的影响。以与实施例4-1中描述的方法相同的方式计算天然hanp和测试物质的比活性和emax。结果显示于表3中。关于peg接头的长度或类型对体外活性的影响,证实了以下内容。所有化合物都在与hanp中相同的程度上保持emax。相对于hanp的比活性范围为0.41至2.84倍。因此,所有化合物在给定或更高水平具有cgmp升高活性。具有peg接头中含有的更大量的乙二醇单元的化合物倾向于表现出更有利的活性。另一方面,据表明,当接头结构整体具有几乎相等的长度时,具有其中键合较少量的具有长peg的接头分子的接头结构的化合物倾向于表现出比具有其中键合较大量的具有短peg(例如,peg(6))的接头分子的接头结构的化合物更有利的活性。[表3]<测试实施例4-4>peg接头对大鼠血液中缀合物的持续时间的影响进行本实验,其目的是使用实施例3-7中合成的缀合物研究peg接头部分对动物血液中的持续时间的影响。以与实施例4-2中描述的方法相同的方式检查在将测试物质皮下施用于大鼠后血浆cgmp浓度的时间依赖性变化。结果显示于图8中。关于peg接头的长度或类型对体外活性的影响,证实了以下内容。具有peg(12)-peg(12)的化合物3-7具有最高的emax,而具有peg(24)的化合物3-9具有最弱的活性和短持续时间,这与体外结果相反。<测试实施例4-5>缀合物的物理特性评估已知天然hanp(1-28)高度凝集。例如,当将天然hanp(1-28)于pbs中的7mg/ml溶液在30℃下孵育时,在24小时内证实凝胶样沉积物,并证实整个溶液在48小时内变成凝胶。如果基于所含hanp的摩尔数以几乎相同的浓度进行测试,则可以间接比较hanp缀合物的凝集。进行以下测试,其目的是证实本发明的缀合物的凝集。(1)加速劣化测试将每种样品使用vivapore5(由vivascienceag制造)浓缩,针对25mmacona/5%山梨糖醇,ph5.5(absor溶液)或pbs透析,且然后调节至70mg/ml。将溶液通过spin-x0.22μm离心过滤器(由costargroup,inc.制造)过滤,并以80μl/管分配至0.5ml无菌细管(由sumitomobakeliteco.,ltd.制造),然后将其密封,且然后在40℃下静置2周。使用由agilent1260lc,dawnheleos8和eclipse3+构建的sec-mals系统(由wyatttechnologycorp.制造)测量劣化之前和之后的样品,并使用astra软件(由wyatttechnologycorp.制造)进行分析。在以下条件下进行测量:柱:nanofilmsec-2507.8'300mm(由sepaxtechnologies,inc.制造),缓冲液:0.2mkpi/0.2mkcl,ph7.0,流速:0.5ml/min,柱温:30℃,和检测波长:280nm。结果显示于表4中。天然hanp于pbs中的全部溶液在48小时内变成凝胶,而证实所述缀合物甚至在2周后还具有溶液流动性并且表现出改善的溶液稳定性。加速劣化处理使化合物3-3的高分子量形式在absor溶液中从8.2%增加至14.2%,在pbs溶液中从9.2%增加至45.7%。对于具有在化合物3-3的hanp位点附近引入的聚糖的化合物3-4,加速劣化处理使高分子量形式在absor溶液中从1.8%增加至4.5%,在pbs溶液中从2.8%增加至12.8%。因此,发现将聚糖引入hanp位点附近显著减少凝集。[表4]<测试实施例4-6>对猴血液中缀合物的持续时间的测试通过根据以下方法检查在血液中可检测到测试物质的时间,检查实施例3中制备的其载体分子不同的缀合物在猴血液中的持续时间,以研究载体分子对缀合物在血液中的持续时间的影响。(1)血浆样品的制备用于血液收集的针头和注射器:用于结核菌素的terumosyringe25gx1sr用于血液收集的管:capiject微量收集管edta-2na或venoject真空血液收集容器edta-2na用于样品储存的管:matrix4170样品追踪管0.75ml对每只3-至7-岁的雄性食蟹猴进行试验物质的施用和在没有麻醉的情况下收集血液。将根据需要用pbs稀释的测试物质(每种缀合物:化合物3-2、3-4、3-15、3-16和3-17)的溶液以10nmol/kg(1ml/kg)的剂量快速皮下施用于猴。在施用前的每个时间点和施用后0.25、0.5、1、2、4、8、24、48、96、168、336、528(或504)和696(或672)小时,随着时间从静脉将血液取样(约500μl/取样)。将血液样品立即置于冰上。将收集的血液样品离心,并将因此获得的血浆样品保存在-80℃下,直至测量。(2)血浆样品中的测试物质的检测向10μl段落(1)中制备的每种猴血浆样品中添加90μlrexxiphn缓冲液(gyrosab制造),且然后混合(10倍稀释的样品,mrd:10),或将所得稀释液用rexxiphn缓冲液进一步稀释10倍(100倍稀释的样品,mrd:100),并用1%血浆/rexxiphn缓冲液进一步稀释10倍(1000倍稀释的样品,mrd:100)以制备血浆样品用于测量。使用的一抗是用于测量样品中的所有含人fc分子(fc/fc)的山羊抗人igg生物素缀合物(southernbiotech),和用于测量样品中具有与之键合的hanp(1-28)的缀合物(anp/fc)的小鼠抗anpigg(genetexinc.)。将每种一抗用0.1%pbs-t调节至700nm。对于两种一抗,用rexxipf缓冲液(gyrosab)将dylight-标记的山羊抗人igg(southernbiotech)作为用于检测的二抗调节至10nm。将每种溶液上样于自动elisa设备gyrolabxp工作站中并注入bioaffy200cd(gyrosab)。通过夹心elisa测量测试物质(fc/fc和anp/fc)的含量以计算血浆浓度。获得的血浆浓度的变化显示于图9中。从图9证实在该测试中使用的hanp缀合物甚至在施用后672小时(28天)在血液中具有缀合物浓度,而没有降解。与具有mab-a作为载体分子的化合物3-2相比,分别具有clch-a和clch-b作为载体分子的化合物3-4和3-15从血液快速衰减。另一方面,分别具有fc-a和fc-b作为载体分子的化合物3-16和3-17比化合物3-2更逐渐地从血液衰减。因此,发现糖基化的hanp-fc缀合物在猴的血液中具有最长的持续时间。[实施例5]使用两种类型的endo酶的转糖基化反应酶a(endom样酶)和酶b(endos样酶)可以适当地组合为使用的两种类型的endo酶。酶a的实例可以包括endom、endoom和endocc,以及表现出降低的水解活性的endom突变体、endoom突变体和endocc突变体。酶b的实例可以包括endos和endos2(endos49),以及表现出降低的水解活性的endos突变体和endos2(endos49)突变体。在聚糖供体部分的结构中,除了r=h以外的任何取代基可以用作异头物位点的取代基r,只要所得分子可在化学反应或酶促反应中合成。<实施例5-1>使用sgp作为聚糖供体测量转糖基化率商业可得的曲妥珠单抗用作mab-c。通过使用实施例3-1中描述的方法,将曲妥珠单抗(440mg/小瓶,由genentechinc.制造)制备成51.3mg/ml的(fucα1,6)glcnac-曲妥珠单抗溶液(50mmtris缓冲溶液(ph7.4))(1.65ml)。向51.3mg/ml的(fucα1,6)glcnac-曲妥珠单抗溶液(50mmtris缓冲溶液(ph7.4))(19.5μl)中添加50mmtris缓冲溶液(ph7.4)(0.5μl)、sgp(由tokyochemicalindustryco.,ltd.制造)(5.82mg)溶液(50mmtris缓冲溶液(ph7.4))(29.1μl)、1u/ml的endomn175q溶液(由tokyochemicalindustryco.,ltd.制造)(5.0μl)和2.00mg/ml的endosd233q溶液(pbs)(10.0μl),并将混合物在28℃下孵育48小时。在反应开始后2小时、4小时、8小时、24小时和48小时的每个时间点,收集该反应溶液的一部分,并使用experion电泳站(由bio-radlaboratories,inc.制造)测量反应的进展程度。为了测量,根据附属于设备的手册制备测量样品。在此过程中,由于将收集的反应溶液暴露于含有二硫苏糖醇的溶液并将反应溶液在95℃下加热5分钟的操作,立即停止转糖基化反应。将获得的测量样品转移至experionpro260芯片,并根据附属于experion电泳站的手册进行测量。从获得的色谱图,未反应的产物和转糖基化的形式被证实为分开的峰。根据来自未反应产物和转糖基化形式之间的峰面积比的以下表达计算转糖基化率。转糖基化率(%)=[衍生自sg-曲妥珠单抗的h链的峰面积]/{[衍生自(fucα1,6)glcnac-曲妥珠单抗的h链的峰面积+[衍生自sg-曲妥珠单抗的h链的峰面积]}×100类似地,使用各种endo酶的不同组合进行反应,并计算每个反应的每个时间点的转糖基化率(表5)。<实施例5-2>使用sg-asn作为聚糖供体测量转糖基化率向51.3mg/ml的(fucα1,6)glcnac-曲妥珠单抗溶液(50mmtris缓冲溶液(ph7.4))(19.5μl)中添加50mmtris缓冲溶液(ph7.4)(0.5μl)、sg-asn铵盐(4.74mg)溶液(50mmtris缓冲溶液(ph7.4))(23.7μl)、1u/ml的endomn175q溶液(5.0μl)和2.00mg/ml的endosd233q溶液(pbs)(10.0μl),并将混合物在28℃下孵育48小时。以与实施例5-1中相同的方式进行后续程序,以计算反应的每个时间点的转糖基化率(表5)。<实施例5-3>使用endocc作为酶a测量转糖基化率向51.3mg/ml的(fucα1,6)glcnac-曲妥珠单抗溶液(50mmtris缓冲溶液(ph7.4))(19.5μl)中添加50mmtris缓冲溶液(ph7.4)(0.5μl)、sgp(由tokyochemicalindustryco.,ltd.制造)(5.82mg)溶液(50mmtris缓冲溶液(ph7.4))(29.1μl)、1.16u/ml的endocc溶液或2.21u/ml的endoccn180h溶液(fushimipharmaceuticalco.,ltd.)(15.0μl)和2.00mg/ml的endosd233q溶液(pbs)(10.0μl),并将混合物在28℃或37℃下孵育48小时。以与实施例5-1中相同的方式进行后续程序,以计算反应的每个时间点的转糖基化率(表5)。表5.[表5]表5.反应条件和转糖基化率的时间依赖性变化当噁唑啉形式不用作聚糖供体时,通过以适当比率混合两种类型的endo酶突变体,证实转糖基化反应有效地进行。当酶a具有高水解活性时,即使使用表现出降低的水解活性的突变体作为酶b,转糖基化率也较低。表现出降低的水解活性的酶b在反应时间上不同,直到转糖基化率超过90%,这取决于其程度。据报道,其最佳反应温度为50℃的endocc倾向于在较高反应温度下产生较高的转移率。通过选择适当化学修饰的聚糖供体,可以将化学修饰的聚糖结构引入含fc分子中。例如,如果使用(n3-peg(3)-sg-)asn-peg(3)-n3代替聚糖供体sgp或(sg-)asn,相应的聚糖可以转移至含fc分子。而且,mab、clch或fc可以适当地用作含fc分子。<实施例5-4>使用([n3-peg(3)]2-sg-)asn-peg(3)-n3制备mab-a-[peg(3)-n3]4通过以下方法使用([n3-peg(3)]2-sg-)asn-peg(3)-n3作为聚糖供体制备mab-a-[peg(3)-n3]4。向51.27mg/ml的(fucα1,6)glcnac-mab-a溶液(50mmtris缓冲溶液(ph7.4))(200μl)中添加步骤(1-12a)中制备的([n3-peg(3)]2-sg-)asn-peg(3)-n3(50.0mg)溶液(50mmtris缓冲溶液(ph7.4))(240μl)、1u/ml的endomn175q溶液(由tokyochemicalindustryco.,ltd.制造)(50μl)和2.10mg/ml的endosd233q/q303l溶液(pbs)(100μl),并将混合物在28℃下孵育20小时。然后,根据实施例3-1中描述的方法选择适合于反应规模的纯化方法(纯化设备和柱)和超滤方法(超滤膜),以获得14.78mg/ml的mab-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(500μl)。esi-ms:mab-a-[peg(3)-n3]4(-lys,pyrglu)的重链的计算值,m=52569.9;实测值52570.0(去卷积数据)。mab-a-[peg(3)-n3]4的轻链的计算值,m=23292.9;实测值23292.0(去卷积数据)。<实施例5-5>使用([n3-peg(3)]2-sg-)asn-peg(3)-n3制备clch-b-[peg(3)-n3]4通过以下方法使用([n3-peg(3)]2-sg-)asn-peg(3)-n3作为聚糖供体制备clch-b-[peg(3)-n3]4。向39.2mg/ml的(fucα1,6)glcnac-clch-b溶液(50mmtris缓冲溶液(ph7.4))(150μl)中添加50mmtris缓冲溶液(ph7.4)(50μl)、步骤(1-12a)中制备的([n3-peg(3)]2-sg-)asn-peg(3)-n3(53.0mg)溶液(50mmtris缓冲溶液(ph7.4))(240μl)、1u/ml的endomn175q溶液(由tokyochemicalindustryco.,ltd.制造)(50μl)和2.10mg/ml的endosd233q/q303l溶液(pbs)(100μl),并将混合物在28℃下孵育20小时。然后,根据实施例3-8中描述的方法选择适合于反应规模的纯化方法(纯化设备和柱)和超滤方法(超滤膜),以获得8.92mg/ml的clch-b-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(500μl)。esi-ms:clch-b-[peg(3)-n3]4(-lys)的重链的计算值,m=38706.3;实测值38706.1(去卷积数据)。clch-b-[peg(3)-n3]4的轻链的计算值,m=11507.8;实测值11506.7(去卷积数据)。<实施例5-6>使用([n3-peg(3)]2-sg-)asn-peg(3)-n3制备fc-a-[peg(3)-n3]4通过以下方法使用([n3-peg(3)]2-sg-)asn-peg(3)-n3作为聚糖供体制备fc-a-[peg(3)-n3]4。向24.8mg/ml的(fucα1,6)glcnac-fc-a溶液(50mmtris缓冲溶液(ph7.4))(140μl)中添加50mmtris缓冲溶液(ph7.4)(60μl)、步骤(1-12a)中制备的([n3-peg(3)]2-sg-)asn-peg(3)-n3(60.0mg)于50mmtris缓冲溶液(ph7.4)(240μl)中的溶液、1u/ml的endomn175q溶液(由tokyochemicalindustryco.,ltd.制造)(50μl)和2.10mg/ml的endosd233q/q303l溶液(pbs)(100μl),并将混合物在28℃下孵育20小时。然后,根据实施例3-9中描述的方法选择适合于反应规模的纯化方法(纯化设备和柱)和超滤方法(超滤膜),以获得4.06mg/ml的fc-a-[peg(3)-n3]4溶液(5%山梨糖醇/10mm乙酸盐缓冲溶液(ph5.5))(500μl)。fc-b-[peg(3)-n3]4(-lys)的链的计算值,m=28088.3;实测值28085.6(去卷积数据)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1