阿齐沙坦中间体、阿齐沙坦及它们的制造方法与流程
本发明涉及阿齐沙坦中间体、阿齐沙坦及它们的制造方法。
更具体地,涉及作为阿齐沙坦中间体的、2-乙氧基-1-[[2'-(肟基胺基)联苯-4-基]甲基]-1h-苯并咪唑-7-羧酸烷基酯和2-乙氧基-1-[[2'-(2,5-二氢-5-氧代-1,2,4-噁二唑-3-基)联苯-4-基]甲基]苯并咪唑-7-羧酸烷基酯;作为阿齐沙坦的、2-乙氧基-1-[[2'-(2,5-二氢-5-氧代-1,2,4-噁二唑-3-基)联苯-4-基]甲基]苯并咪唑-7-羧酸及它们的制造方法。
背景技术:
由下述式(5)
【化1】
表示的阿齐沙坦(别名:1-[[2'-(4,5-二氢-5-氧代-1,2,4-噁二唑-3-基)[1,1'-联苯-4-基]甲基]-2-乙氧基-1h-苯并咪唑-7-羧酸)是作为治疗药非常有用的化合物,所述治疗药作为血管紧张素ii受体拮抗药显示出优异的效果(专利文献1)。
该阿齐沙坦采用以下这样的制造方法合成。
【化2】
即,首先,使羟基胺和/或羟基胺酸盐与由上述式(1)表示的1-[(2'-氰基联苯-4-基)甲基]-2-乙氧基苯并咪唑-7-羧酸烷基酯(以下也有时简称为“腈化合物”)反应,制造由上述式(2)表示的2-乙氧基-1-[[2'-(肟基胺基)联苯-4-基]甲基]-1h-苯并咪唑-7-羧酸烷基酯(以下也有时简称“偕胺肟化合物”)。
接下来,将偕胺肟化合物直接用于环化反应,或者制成将该偕胺肟化合物的羟基用酯保护基保护的由上述式(3)表示的2-乙氧基-1-[[2'-(烷氧基-羰氧基脒基)联苯-4-基]甲基]-1h-苯并咪唑-7-羧酸烷基酯(以下也有时简称为“含有酯保护基的化合物”)后,进行环化反应,制造由上述式(4)表示的2-乙氧基-1-[[2'-(2,5-二氢-5-氧代-1,2,4-噁二唑-3-基)联苯-4-基]甲基]苯并咪唑-7-羧酸烷基酯(以下也有时简称为“阿齐沙坦烷基酯”)。
然后,最后通过将该阿齐沙坦烷基酯水解,从而制造由上述式(5)表示的阿齐沙坦(例如参照专利文献1~3、非专利文献1)。
对于阿齐沙坦这样的原药,希望减少杂质。但是,由于如上所述经过多个工序制造,因此在各工序中有时生成与目标物不同的杂质。该杂质由于结构与目标物相似,因此如果其量增多,则精制等变得非常困难。因此,在制造原药的情况下,即使是中间体的制造,也希望减少杂质。
现有技术文献
专利文献
专利文献1:日本专利26459962号公报
专利文献2:日本特表2014-506898号公报
专利文献3:日本特表2014-505097号公报
专利文献4:日本特表2014-530805号公报
非专利文献
非专利文献1:journalofmedicinalchemistry,(美国),1996年,第39卷,第5228-5235页
技术实现要素:
发明要解决的课题
(1)本发明的第一课题
根据本发明人等的研究,获知在制造由上述式(2)表示的偕胺肟化合物时,在现有技术中在以下方面有改善的余地。
例如,在专利文献1中,使用羟基胺盐酸盐,在作为碱的甲醇钠的存在下、在二甲基亚砜的反应溶剂中实施反应。根据该方法,主生成物为由下述式(7)
【化3】
表示的腈化合物的脱乙基体(以下也有时简称为“腈脱乙基体”),上述偕胺肟化合物的生成比例小。
另外,在非专利文献1中记载的方法中,使用羟基胺盐酸盐,在作为碱的三乙胺这样的有机碱的存在下、在二甲基亚砜的反应溶剂中实施反应。根据本发明人等的研究,在该方法中,上述偕胺肟化合物的生成比例增加。但是,获知同时副产与偕胺肟体同等量的由下述式(8)
【化4】
表示的酰胺体(以下也有时简称为“酰胺体”)。进而,获知如果延长反应时间,则由下述式(9)
【化5】
表示的偕胺肟化合物的脱乙基体(以下也有时简称为“偕胺肟脱乙基体”)和由下述式(10)
【化6】
表示的酰胺体的脱乙基体(以下也有时简称为“酰胺脱乙基体”)倾向于增加。
在专利文献2中记载的方法中,使用羟基胺的水溶液,在二甲基亚砜这样的非质子性极性溶剂等反应溶剂中实施反应。根据本发明人等的研究,在该方法中,使上述酰胺体减少,能够增加目标的偕胺肟化合物。但是,在该方法中,上述偕胺肟化合物的纯度为70%左右,含有10%左右的上述酰胺体。根据本发明人等的研究,获知即使在使上述偕胺肟化合物成为结晶而从反应体系中取出的情况下,上述偕胺肟化合物的纯度也为约85%左右,含有约5%左右的上述酰胺体。而且,获知如果延长反应时间,则上述偕胺肟脱乙基体和上述酰胺脱乙基体倾向于增加。
应予说明,在本发明中,上述偕胺肟化合物的纯度、其他化合物的纯度和杂质的含有比例为采用实施例中记载的高效液相色谱(hplc)的测定条件测定的各峰的面积%。
另外,除此之外,在专利文献3中记载的方法中,使用羟基胺盐酸盐,在作为碱的碳酸氢钠的存在下、二甲基亚砜的反应溶剂中实施反应。
不仅是该专利文献3中记载的方法,如上所述,在非专利文献1、专利文献1和2中记载的方法中,作为反应溶剂,也主要使用二甲基亚砜。根据本发明人的研究获知:作为反应溶剂,如果二甲基亚砜成为主成分,则上述偕胺肟化合物的结晶难以析出。因此,对于使用二甲基亚砜作为反应溶剂的现有的方法而言,一般采用在反应后的溶液中加水来使偕胺肟化合物结晶化的方法。在该方法中,只能得到低纯度的偕胺肟化合物的结晶。而且,为了取得高纯度的偕胺肟化合物的结晶,必须另外进行重结晶等精制操作,存在着操作变得烦杂的问题。
因此,本发明的第一目的在于提供能够采用简便的操作以高收率得到高纯度的上述偕胺肟化合物的、上述偕胺肟化合物的制造方法。进而在于提供使用采用该方法制造的上述偕胺肟化合物来制造高纯度的阿齐沙坦的方法。
(2)本发明的第二课题
根据本发明人等的研究,获知在现有技术中在制造阿齐沙坦烷基酯时在以下的方面有改善的余地。
例如,在非专利文献1中,使用二甲苯作为反应溶剂,在该二甲苯中、回流温度(反应溶液的回流温度;约130℃)下对r2为2-乙基己基的含有酯保护基的化合物进行环化反应,制造阿齐沙坦甲酯。采用该方法,能够用比较短的反应时间得到阿齐沙坦甲酯(收率:52%)。但是,根据本发明人等的研究,获知在非专利文献1中记载的方法中结构不清楚、但在液相色谱质量分析计(lc-mass)的分析结果中在阿齐沙坦甲酯的分子量上加上10的分子量的杂质增加。
另一方面,在专利文献1中,使用二甲苯作为反应溶剂,在该二甲苯中、回流温度(反应溶液的回流温度;约130℃)下对r2为乙基的含有酯保护基的化合物进行环化反应,制造阿齐沙坦甲酯。但是,获知在该方法中上述杂质也增加。进而,对于上述专利文献1中记载的方法而言,收率低达23%左右,有改善的余地。
另外,在专利文献1中,也示出了在醋酸乙酯中、碱(碳酸钾、二氮杂双环十一碳烯)的存在下进行环化反应的方法。但是,在该反应中,阿齐沙坦甲酯在反应中途析出。因此,以包含该碱的状态的固体得到阿齐沙坦甲酯。其结果在精制工序变得烦杂的方面有改善的余地。
因此,本发明的第二目的在于提供以高收率制造高纯度的阿齐沙坦烷基酯的方法。此外,在于提供能够使作为后工序的精制工序变得容易的阿齐沙坦烷基酯的制造方法。而且,最终在于提供使用采用该方法制造的阿齐沙坦烷基酯来制造高纯度的阿齐沙坦的方法。
(3)本发明的第三课题
另外,根据本发明人等的研究,获知在现有技术中在制造阿齐沙坦烷基酯时在以下的方面有改善的余地。
例如,在专利文献1中记载了以下的方法。其为如下方法:首先,在二甲苯中对r1为甲基、r2为乙基的含有酯保护基的化合物实施环化反应,合成阿齐沙坦甲酯。接下来,在反应液中加入醋酸乙酯,水洗干燥后,将二甲苯馏除,将残渣用硅胶色谱精制,将得到的粗结晶从醋酸乙酯和异丙醚中重结晶。
另外,在非专利文献1中记载了以下的方法。其为如下方法:首先,在二甲苯中对r1为甲基、r2为2-乙基己基的含有酯保护基的化合物实施环化反应,合成阿齐沙坦甲酯。接下来,将二甲苯馏除,使用醋酸乙酯进行重结晶。
但是,根据本发明人等的研究,获知在上述专利文献1和上述非专利文献1中记载的方法中在由下述式(11)
【化7】
(式中,r1为烷基)
表示的阿齐沙坦烷基酯的水解物(以下也有时简称为“脱乙基体”)、或由下述式(12)
【化8】
(式中,r1为烷基)
表示的阿齐沙坦烷基酯的二聚体(以下也有时简称为“二聚体”)、进而结构不清楚但在液相色谱质量分析计(lc-mass)的分析结果中在阿齐沙坦甲酯的分子量上加上10的分子量的杂质不能减少的方面有改善的余地。
即,在用包含醋酸乙酯的溶剂重结晶的情况下,在杂质减少的方面有改善的余地。
另外,以往获知的阿齐沙坦甲酯的熔点为高达190~200℃的温度。因此,认为只要能够进一步制造熔点更低的阿齐沙坦甲酯,就易溶解于溶剂,在制成阿齐沙坦的情况下,不会使不需要的杂质增加。因此,需要开发具有新型的晶型的阿齐沙坦甲酯。
因此,本发明的第三目的在于提供制造高纯度的阿齐沙坦烷基酯的方法。此外,在于提供具有低熔点部分的新型的晶型的阿齐沙坦甲酯。而且,最终在于提供使用采用该方法制造的纯度高的阿齐沙坦烷基酯、和/或、纯度高且为新型的晶型的上述阿齐沙坦甲酯来制造高纯度的阿齐沙坦的方法。
用于解决课题的手段
本发明人为了解决上述(1)~(3)的课题,反复认真研究。
而且,如下所述推定了在制造上述偕胺肟化合物时在现有技术中使用二甲基亚砜这样的非质子性极性溶剂的理由。即,认为如果使用质子性溶剂等,则由上述式(1)表示的腈化合物中的、-or(r:碳原子数1~4的烷基)、-oet(et:乙基)部分发生酯交换反应,反应有可能变得更为复杂。
由此考虑如果即使使用质子性极性溶剂,在-or、-oet中也不发生酯交换反应,是否以高收率得到高纯度的上述偕胺肟化合物,进行了研究。
其结果,发现通过使用包含特定的醇的反应溶剂,从而以高收率得到高纯度的上述偕胺肟化合物,完成了第一本发明。
接下来,针对在进行环化反应时对于含有酯保护基的化合物和阿齐沙坦烷基酯的溶解性高、在环化反应中能够不使含有酯保护基的化合物和阿齐沙坦烷基酯分解地促进反应的条件进行了研究。其结果,发现通过在特定的反应溶剂、即包含碳原子数1~8的醇的反应溶剂中进行环化反应,从而能够解决上述课题,完成了第二本发明。
进而,使用各种溶剂进行了阿齐沙坦烷基酯的重结晶的研究。其结果获知:使用丙酮、或者丙酮与醇的混合溶剂进行了阿齐沙坦烷基酯的结晶化,结果能够有效率地减少阿齐沙坦烷基酯的脱乙基体、或二聚体的杂质。此外发现,将阿齐沙坦甲酯在丙酮、或者丙酮与醇的混合溶剂中结晶化,结果得到现有结晶中不存在的、具有低熔点部分的新型的结晶,完成了第三本发明。
即,第一本发明为通过在包含碳原子数2~7的醇的反应溶剂中使由下述式(1)
【化9】
(式中,r1为碳原子数1~4烷基)
表示的腈化合物与羟基胺和/或羟基胺酸盐反应,从而制造由下述式(2)
【化10】
(式中,r1与上述式(1)中的r1同义)表示的偕胺肟化合物的方法。
第一本发明中,优选在碱的存在下进行上述反应。通过在碱的存在下进行反应,从而能够进一步抑制上述酰胺体和上述酰胺脱乙基体的副产。其中,为了更高度地抑制上述酰胺体和上述酰胺脱乙基体的副产,并且提高操作性,优选上述碱包含有机碱,特别优选地,相对于由上述式(1)表示的腈化合物1摩尔,该有机碱的配合量设为0.01~0.5摩尔。
另外,本发明中,为了能够制造高纯度的上述偕胺肟化合物,并且进一步提高操作性,优选上述醇为碳原子数3~7的直链状或分支状醇。更优选地,使用羟基胺,并且上述反应溶剂包含水。
采用本发明的方法得到的偕胺肟化合物为高纯度,杂质少。因此,采用本发明的方法得到的偕胺肟化合物能够适合在由上述式(4)表示的阿齐沙坦烷基酯和由上述式(5)表示的阿齐沙坦的制造中采用。
第二本发明为通过在包含碳原子数1~8的醇的反应溶剂中使由下述式(3)
【化11】
(式中,r1为烷基,r2为保护羟基的保护基)
表示的含有酯保护基的化合物进行环化反应,从而制造由下述式(4)
【化12】
(式中,r1与上述式(3)中的r1同义)
表示的阿齐沙坦烷基酯的方法。
本发明中,为了进一步提高收率,优选在50℃以上且反应溶液的回流温度以下进行上述环化反应。应予说明,上述回流温度与反应溶剂回流的温度大致相同。不过,之所以称为“反应溶液的回流温度”,是因为由于副产的r2-oh(具有r2基的醇)、溶解的阿齐沙坦烷基酯浓度等的不同,与反应溶剂的回流温度产生一些差异。本发明中,所谓反应溶液,是指在反应溶剂中含有酯基的化合物和/或阿齐沙坦烷基酯溶解、包含副产的r2-oh的反应溶液。应予说明,在根据需要使用碱的情况下,当然在反应溶液中也包含碱。
本发明中,优选上述醇为碳原子数3~8的直链状或分支状醇。通过使用该醇,不仅以高收率得到高纯度的阿齐沙坦烷基酯,而且在反应完成后变得容易在反应溶剂中使该阿齐沙坦烷基酯结晶化。其结果能够使后处理工序变得容易。
进而,在本发明中,为了用短时间提高收率,优选在碱存在下实施上述环化反应。其中,相对于由上述式(3)表示的含有酯保护基的化合物1摩尔,优选上述碱的使用量为0.01~5摩尔。进而,优选上述碱为有机碱。
根据第二本发明得到的阿齐沙坦烷基酯成为新型的结晶结构的阿齐沙坦烷基酯。具体地,能够得到在使用cu-kα线的x射线衍射中至少在2θ=9.9±0.2°、10.9±0.2°具有特征峰的阿齐沙坦甲酯。
另外,第二本发明包含在采用该方法制造阿齐沙坦烷基酯后通过将得到的阿齐沙坦烷基酯水解从而制造由下述式(5)
【化13】
表示的阿齐沙坦的方法。根据本发明,能够制造更高纯度的阿齐沙坦烷基酯,因此能够制成高纯度的阿齐沙坦。
第三本发明为阿齐沙坦烷基酯的制造方法,其特征在于,在丙酮、或者丙酮与醇的混合溶剂中使由下述式(4)
【化14】
(式中,r1为烷基)
表示的阿齐沙坦烷基酯结晶化。
第三本发明在上述式(4)中式中r1为甲基的情况下,即,为2-乙氧基-1-[[2'-(2,5-二氢-5-氧代-1,2,4-噁二唑-3-基)联苯-4-基]甲基]苯并咪唑-7-羧酸甲酯(以下也有时简称为“阿齐沙坦甲酯”)的情况下,尤其发挥效果。而且,能够使该阿齐沙坦甲酯成为具有熔点低的部分的、准结晶状态的阿齐沙坦甲酯。
该阿齐沙坦甲酯根据使用cu-kα线的x射线衍射,至少在2θ=9.2±0.2°、15.8±0.2°、22.1±0.2°具有特征峰。另外,阿齐沙坦甲酯优选成为至少在150~165℃的温度范围和185~195℃的温度范围至少具有熔点的化合物。
进而,第三本发明包含将采用该方法制造的阿齐沙坦烷基酯、和/或阿齐沙坦甲酯水解而制造阿齐沙坦的方法。通过将它们作为原料,从而能够制造纯度更高的阿齐沙坦。
第一~第三本发明的阿齐沙坦的制造方法包含附加使用活性炭将作为杂质含有的阿齐沙坦二聚体除去的工序的制造方法。
另外,第一~第三本发明的阿齐沙坦的制造方法包含附加在使阿齐沙坦溶解于二甲基甲酰胺而得到的溶液中加入酮类或酯类的溶剂、使该阿齐沙坦析出的工序的制造方法。
进而,本发明包含如下制造方法:采用第一本发明的制造方法由上述腈化合物得到上述偕胺肟化合物;
接下来,由上述偕胺肟化合物得到上述含有酯保护基的化合物;
采用第二本发明的制造方法由上述含有酯保护基的化合物得到上述阿齐沙坦烷基酯;
采用第三本发明的制造方法由上述阿齐沙坦烷基酯得到上述阿齐沙坦。
发明的效果
根据第一本发明的方法,能够采用更简便的操作以高收率得到高纯度的偕胺肟化合物。其结果,通过使用本发明中得到的偕胺肟化合物来制造阿齐沙坦烷基酯和阿齐沙坦,也能够使它们成为高纯度的产物。
根据第二本发明的方法,能够采用更简便的操作以高收率得到更高纯度的阿齐沙坦烷基酯。其结果,通过将本发明中得到的阿齐沙坦烷基酯水解来制造阿齐沙坦,从而能够得到高纯度的阿齐沙坦。此外,由于更容易将阿齐沙坦烷基酯作为结晶取出,因此操作性也能够提高。
根据第三本发明的方法,能够得到高纯度的阿齐沙坦烷基酯、特别是高纯度的阿齐沙坦甲酯。其结果,通过将本发明中得到的阿齐沙坦烷基酯、和/或阿齐沙坦甲酯水解来制造阿齐沙坦,从而能够得到高纯度的阿齐沙坦。此外,能够使采用本发明的方法得到的阿齐沙坦甲酯可以制成具有熔点低的部分的准结晶。
这些方法特别是能够提高最终得到的、作为原药使用的阿齐沙坦的纯度,因此其工业上的利用价值高。
附图说明
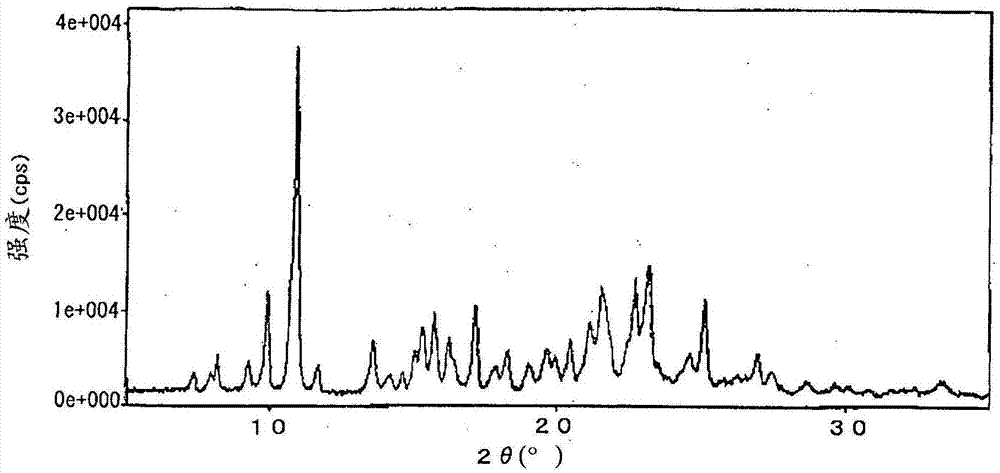
图1为实施例10中制造的本发明的阿齐沙坦甲酯新型结晶的x射线衍射图。
图2为比较例4中制造的现有的阿齐沙坦甲酯结晶的x射线衍射图。
图3为实施例18中制造的本发明的阿齐沙坦甲酯的新型结晶(准结晶)的x射线衍射图。
图4为比较例5中制造的现有的阿齐沙坦甲酯结晶的x射线衍射图。
图5为实施例18中制造的本发明的阿齐沙坦甲酯的新型结晶(准结晶)的dsc图。
图6为比较例5中制造的现有的阿齐沙坦甲酯结晶的dsc图。
图7为实施例33中制造的本发明的阿齐沙坦m型结晶的x射线衍射图。
具体实施方式
1.第一本发明
第一本发明的特征在于,在使由下述式(1)
【化15】
(式中,r1为碳原子数1~4烷基)
表示的腈化合物与羟基胺和/或羟基胺酸盐反应而制造由下述式(2)
【化16】
(式中,r1与上述式(1)中的r1同义)
表示的偕胺肟化合物时,在包含碳原子数2~7的醇的反应溶剂中进行该反应。以下按顺序进行说明。
(原料化合物;腈化合物)
对由上述式(1)表示的腈化合物并无特别限制,能够采用公知的方法制造。具体地,能够采用专利文献1中记载的方法,即通过在3-氨基-2-[[(2'-氰基联苯-4-基)甲基]氨基]苯甲酸烷基酯的原碳酸乙酯的溶液中加入醋酸,在80℃下边搅拌1小时边使其反应而制造(参照专利文献1的实施例1b)。
(原料化合物:羟基胺和/或羟基胺酸盐)
本发明中,使上述腈化合物的腈部分与羟基胺和/或羟基胺酸盐反应,制造上述偕胺肟化合物。
对使用的羟基胺和/或羟基胺酸盐并无特别限制,能够使用市售的产品。应予说明,“和/或”当然是指羟基胺单独、羟基胺酸盐单独、羟基胺与羟基胺酸盐的混合物。以下将它们汇总表示时,也有时记为羟基胺类。
羟基胺酸盐可以列举出羟基胺盐酸盐、羟基胺硫酸盐、羟基胺磷酸盐、羟基胺草酸盐等。
这些羟基胺酸盐也能够使用碱进行中和处理而作为羟基胺使用。
对羟基胺和/或羟基胺酸盐的使用量并无特别限制,相对于上述腈化合物1摩尔,优选设为1~10摩尔,更优选设为2~7摩尔。
在羟基胺和/或羟基胺酸盐中,为了减少上述腈脱乙基体和上述酰胺体,提高得到的偕胺肟化合物的纯度,优选使用羟基胺。在使用羟基胺的情况下,从获得的容易性的方面出发,优选使用羟基胺水溶液,例如羟基胺的浓度为30~50质量%的水溶液。本发明中,使用羟基胺的优点并不明显。不过,本发明人认为由于能够在本反应中的适当的ph条件下进行反应,因此能够提高上述偕胺肟化合物的纯度。再有,将羟基胺作为水溶液用于反应的情况下,反应溶剂至少含有碳原子数2~7的醇和水。本发明的反应溶剂可以是该含水的反应溶剂。
(反应溶剂)
本发明的最大特征在于使用包含碳原子数2~7的醇的反应溶剂这点。通过使用该反应溶剂,从而能够减少上述酰胺体、上述脱乙基体的副产量,能够制造高纯度的上述偕胺肟化合物。
如果例示碳原子数2~7的醇,可列举出乙醇、1-丙醇、异丙醇、1-丁醇、2-甲基-1-丙醇、2-丁醇、2-甲基-2-丙醇、1-戊醇、3-甲基-1-丁醇、1-己醇、2-甲基-1-戊醇、3-甲基-1-戊醇、2-甲基-2-戊醇、2,4-二甲基-3-戊醇、3-乙基-3-戊醇等。其中,从得到的偕胺肟化合物的收率、纯度和所含的杂质的比例以及最终将反应溶剂除去的方面出发,优选碳原子数3~7的直链状或分支状醇。具体地,优选1-丙醇、异丙醇、1-丁醇、2-丁醇,特别优选1-丙醇、1-丁醇。
以上例示的醇能够使用1种,也能够使用2种以上的混合物。在作为混合物使用的情况下,使用的量的基准是以混合物的总量为对象。
本发明中,反应溶剂只要包含碳原子数2~7的醇,则可包含其他溶剂。具体地,能够配合如上所述使用羟基胺水溶液时的水、新配合的水,此外与碳原子数2~7的醇相容的溶剂。如果考虑反应的后处理等,其他溶剂的含量在反应溶剂的总量100质量%中优选成为不到50质量%,更优选成为30质量%以下(当然反应溶剂的其余部分为碳原子数2~7的醇。)。反应溶剂的总量可以为碳原子数2~7的醇,在将羟基胺水溶液在反应中使用的情况下,将反应溶剂的总量设为100质量%时,优选将碳原子数2~7的醇设为70~99质量%,将水设为1~30质量%。
本发明中,对反应溶剂的使用量并无特别限制,只要使用如下的量即可:在反应中为能够将原料化合物充分地混合的状态,并且能够将原料化合物和生成的上述偕胺肟化合物充分地溶解。其中,为了使得容易将得到的偕胺肟化合物作为结晶取出,相对于上述腈化合物1g,优选使用5~50ml的反应溶剂,更优选使用6~30ml。应予说明,该反应溶剂的使用量为23℃下的体积。
(反应方法)
本发明中,在包含碳原子数2~7的醇的反应溶剂中,通过使上述腈化合物与羟基胺类接触,从而能够使其反应。因此,在该反应溶剂中,可将上述腈化合物与羟基胺类进行搅拌混合来使作为原料化合物的两者接触。
(碱)
在本发明的方法中,使作为原料化合物的两者接触(反应)时,也能够在碱的存在下实施。通过使用碱,能够进一步提高上述偕胺肟化合物的纯度,特别是能够抑制上述酰胺体和上述酰胺脱乙基体的副产量。
作为使用的碱,能够使用公知的碱。具体地,能够使用碳酸氢钠、碳酸氢钾、碳酸氢钙、碳酸钠、碳酸钾、碳酸铯、碳酸钙、碳酸锂、氢氧化钠、氢氧化钾、氢氧化钡、氢氧化锂等这样的无机碱,以及甲胺、乙胺、三甲胺、三乙胺、二异丙胺、三丙胺、二异丙基乙基胺、吡啶、哌嗪、吡咯烷、苯胺、n,n-二甲基氨基吡啶、二氮杂双环十一碳烯、n-甲基吗啉等这样的有机碱。这些碱能够单独使用,也能够同时使用多种碱。在同时使用多种碱的情况下,成为基准的配合量是以多种碱的合计量为基准。
在以上这样的碱中,特别是在想要抑制上述酰胺体和上述酰胺脱乙基体的副产量的情况下,优选使用有机碱。通过使用有机碱,该有机碱的除去也变得容易。在该有机碱中,如果考虑工业上的生产,优选使用三乙胺、吡啶、二异丙基乙基胺。
对碱的使用量并无特别限制,可设为通常的催化剂量。其中,在使用有机碱的情况下,相对于由上述式(1)表示的腈化合物1摩尔,优选有机碱的使用量为0.01~0.5摩尔。通过使有机碱的使用量为上述范围,从而能够进一步抑制上述酰胺体和上述酰胺脱乙基体的副产量。进而,为了抑制上述酰胺体和上述酰胺脱乙基体的副产量,相对于由上述式(1)表示的腈化合物1摩尔,优选有机碱的使用量为0.1~0.5摩尔,特别优选为0.2~0.5摩尔。
(反应条件)
本发明中,对其他反应条件并无特别限制,优选采用以下的条件实施。
将原料化合物、反应溶剂和根据需要配合的碱在反应容器中混合时,将各成分导入反应容器中的顺序并无特别限制。具体地,能够采用将原料化合物、反应溶剂和根据需要配合的碱同时导入反应容器中的方法。另外,也能够将一方的原料化合物和反应溶剂预先导入反应容器,接下来,将另一方的原料化合物(根据需要可用反应溶剂稀释)和根据需要配合的碱(可用反应溶剂稀释)导入反应容器中。此时,后面添加的原料化合物可分为数次导入。其中,为了使操作更为简便,在反应容器中首先装入上述腈化合物和反应溶剂。接下来,导入根据需要配合的碱,进而导入羟基胺类。此时,羟基胺类当然可以用反应溶剂稀释。另外,在使用羟基胺的情况下,也能够使用30~50质量%的浓度的水溶液。
本发明中,对反应温度(将全部成分混合后的反应溶液的温度)并无特别限制,优选在50℃以上且该反应溶液的回流温度以下来实施。反应溶液的回流温度因使用的反应溶剂的种类、原料化合物的浓度等而异,因此不能一概地限定。不过,为了抑制上述偕胺肟化合物的分解,反应溶液的温度优选设为50~100℃以下,更优选设为60~95℃。
本发明中,对反应时间(将全部成分混合后的时间)并无特别限制,可边确认上述腈化合物的消耗比例、得到的偕胺肟化合物的生成比例等边决定。不过,如果反应时间过长,有可能发生上述偕胺肟化合物的分解等,因此通常反应时间优选设为1~20小时。此外,对反应时的气氛也无特别限制,能够在空气存在下或惰性气体存在下实施。另外,能够在减压下、加压下或大气压下实施反应。其中,为了提高操作性,优选在空气存在下、大气压下进行反应。
(后处理工序)
按照上述方法,能够制造上述偕胺肟化合物。就反应溶液中生成的偕胺肟化合物而言,优选将反应溶液冷却或者将反应溶剂馏除,使其结晶化而取出。其中,优选地,在使反应溶液的温度成为50℃以上且反应溶液的回流温度以下的情况下,优选冷却到30℃以下,更优选冷却到10~30℃的温度,在使用的反应溶剂中使上述偕胺肟化合物的结晶析出。其中,为了提高得到的偕胺肟化合物的纯度,优选使从反应温度成为30℃以下时的冷却速度为5~50℃/小时。另外,为了提高收率,优选以30℃以下的温度放置1小时以上、优选2小时以上且10小时以下。
对于析出的偕胺肟化合物的结晶能够采用公知的方法进行处理。通常,优选通过过滤将结晶取出,进行清洗、干燥。另外,在要得到纯度更高的羟基脒基化合物的情况下,可用反应溶剂进行重结晶。
根据本发明,能够使得到的偕胺肟化合物成为纯度90.0~98.0%、酰胺体0.1~3.0%、上述腈脱乙基体0.0(未检测出)~0.5%、上述偕胺肟脱乙基体0.1~1.0%、上述酰胺脱乙基体0.05~1.0%。通过进一步调整条件,优选地,也能够使其成为纯度94.0~98.0%、上述酰胺体0.1~2.0%、上述腈脱乙基体0.0(未检测出)~0.1%、上述偕胺肟脱乙基体0.1~1.0%、上述酰胺脱乙基体0.05~0.5%的高纯度的产物。更优选地,也能够使其成为纯度94.0~98.0%、上述酰胺体0.1~1.0%、上述腈脱乙基体0.0(未检测出)~0.1%、上述偕胺肟脱乙基体0.1~1.0%、上述酰胺脱乙基体0.05~0.5%的高纯度的产物。
再有,上述的偕胺肟化合物的纯度、上述酰胺体、上述腈脱乙基体、上述偕胺肟脱乙基体、上述酰胺脱乙基体比例由于也存在还包含其他成分的情形,因此合计未必成为100%。
因此,得到的偕胺肟化合物能够适合作为阿齐沙坦烷基酯和阿齐沙坦的原料使用。
(阿齐沙坦甲酯的制造方法)
本发明中,能够由采用上述方法得到的偕胺肟化合物制造由下述式(4)
【化17】
(式中,r与上述式(1)中的r同义)
表示的阿齐沙坦烷基酯。
制造阿齐沙坦烷基酯的方法能够无限制地采用公知的方法。
例如,如org.process.res.dev2013,17中记载那样,通过在包含1,1'-羰基咪唑、二氮杂双环十一碳烯、二甲基亚砜的反应溶剂中使其反应,从而能够由上述偕胺肟化合物直接实施环化反应,能够制造上述阿齐沙坦烷基酯。
另外,能够采用以下的方法。为非专利文献1、专利文献1中记载的方法。具体地,首先,通过使上述偕胺肟化合物与氯甲酸烷基酯(烷基为羟基的保护基,具体地,为2-乙基己基或乙基)在有机碱(吡啶或三乙胺)存在下、在二甲基甲酰胺、四氢呋喃/二氯甲烷溶剂中反应,从而得到由下述式(3)
【化18】
(式中,r2为作为羟基的保护基的烷基,具体地,为2-乙基己基或乙基)表示的含有酯保护基的化合物。接下来,通过将得到的含有酯保护基的化合物在二甲苯溶剂中置于回流温度下(约130℃),从而进行环化反应,能够制造阿齐沙坦烷基酯。
对得到的阿齐沙坦烷基酯并无特别限制,可采用org.process.res.dev2013,17、非专利文献1和专利文献1中记载的方法来实施精制等。
具体地,可列举出从丙酮、醋酸乙酯、醋酸乙酯/异丙醚、氯仿/醋酸乙酯中重结晶的方法。为了使得到的阿齐沙坦烷基酯的纯度成为更高纯度,优选选择从丙酮中重结晶的方法。
(阿齐沙坦制造方法)
本发明中,通过将采用上述方法得到的阿齐沙坦烷基酯水解,从而能够制造由下述式(5)
【化19】
表示的阿齐沙坦。使用的阿齐沙坦烷基酯可以是新型的结晶,也可以是用包含酮溶剂的溶剂进行重结晶的产物。
对水解的条件并无特别限制,能够采用公知的方法例如专利文献1中记载的方法。具体地,可通过在碱或酸的存在下进行水解,从而使-coor1(烷基酯基)成为-cooh(羧酸)。
第二本发明的特征在于,在进行由下述式(3)
【化20】
(式中,r1为烷基,r2为保护羟基的保护基)
表示的含有酯保护基的化合物的环化反应,制造由下述式(4)
【化21】
(式中,r1与上述式(3)中的r1同义)
表示的阿齐沙坦烷基酯时,在包含碳原子数1~8的醇的反应溶剂中进行该环化反应。
应予说明,上述式中,r1为烷基。如果考虑成为原料的含有酯保护基的化合物、阿齐沙坦烷基酯的稳定性和阿齐沙坦的制造的容易性,r1优选为碳原子数1~4的烷基。具体地,可列举出甲基、乙基、1-丙基、异丙基、1-丁基、异丁基等,特别优选为甲基。
以下按顺序进行说明。
(原料化合物;含有酯保护基的化合物)
对由上述式(3)表示的含有酯保护基的化合物并无特别限制,能够采用公知的方法制造。具体地,能够采用非专利文献1、专利文献1中记载的方法制造。具体地,能够按照以下的反应式制造。
【化22】
由上述式(2)表示的偕胺肟化合物是公知的化合物,其制造方法记载于非专利文献1、专利文献1中。另外,也优选使用根据第一本发明制造的产物。在碱的存在下使由上述式(2)表示的偕胺肟化合物与由xcoor2表示的化合物反应,能够制造由上述式(3)表示的含有酯保护基的化合物。
上述反应式中,与由上述式(2)表示的化合物反应的xcoor2的x为卤素原子,r2与由上述式(3)表示的含有酯保护基的化合物中的r2相同,是保护羟基的保护基。
在xcoor2中,x可列举出氯原子、溴原子、碘原子。其中,如果考虑工业上获得的容易性、反应性等,优选为氯原子。
r2可列举出保护羟基的、一般的保护基。具体地,可列举出可具有取代基的烷基、苄基、可具有取代基的苯基等。其中,如果考虑工业上的获得的容易性、含有酯保护基的化合物中的职能、最终除去等,优选为碳原子数1~8的未取代烷基。该未取代烷基可以为直链状的烷基,也可以为分支状的烷基。
如果具体地例示xcoor2,则可列举出氯甲酸甲酯、氯甲酸乙酯、氯甲酸丙酯、氯甲酸异丙酯、氯甲酸丁酯、氯甲酸异丁酯、氯甲酸戊酯、氯甲酸-2-乙基己酯、氯甲酸己酯、氯甲酸庚酯、氯甲酸氯甲酯、氯甲酸-2-氯乙酯、氯甲酸苄酯、氯甲酸苯酯、氯甲酸-4-氯苯酯等。其中,如果考虑工业上的获得的容易性、反应性和含有酯保护基的化合物中的职能等,优选使用氯甲酸甲酯、氯甲酸乙酯、氯甲酸丙酯等。
对xcoor2的使用量并无特别限制。具体地,相对于由上述式(2)表示的化合物1摩尔,xcoor2的使用量可设为1~5摩尔。
上述反应在碱的存在下进行。如果例示使用的碱,能够列举出碳酸氢钠、碳酸氢钾、碳酸氢钙、碳酸钠、碳酸钾、碳酸铯、碳酸钙、碳酸锂、氢氧化钠、氢氧化钾、氢氧化钡、氢氧化锂等无机碱;
甲胺、乙胺、三甲胺、三乙胺、二异丙胺、三丙胺、二异丙基乙基胺、吡啶、哌嗪、吡咯烷、苯胺、n,n-二甲基氨基吡啶、二氮杂双环十一碳烯、n-甲基吗啉等有机碱。
其中,如果考虑反应的进行性、除去容易性、后工序中的处理等,优选为三乙胺、吡啶、二异丙基乙基胺的有机碱。
就上述的碱而言,能够使用1种,也能够使用多种碱。在使用多种碱的情况下,成为基准的碱的量为多种碱的合计量。
对上述碱的使用量并无特别限制。具体地,相对于由上述式(2)表示的化合物1摩尔,上述碱的使用量可规定为1~5摩尔。再有,如后所述,在将含有酯基的化合物环化时,优选在碱的存在下实施。因此,在将该反应中得到的含有酯基的化合物环化的情况下,也能够在上述碱残存的状态下实施环化反应。
另外,使用的溶剂可从不与xcoor2反应的非质子性溶剂中选择。具体地,能够列举出苯、甲苯、二氯甲烷、氯仿、1,4-二噁烷等。这些反应溶剂可使用1种,也可使用2种以上的混合溶剂。
就反应而言,优选在碱的存在下、溶剂中、以由上述式(2)表示的偕胺肟化合物和xcoor2充分地接触的方式进行搅拌混合。对将这些成分导入反应容器中的顺序并无特别限制。作为优选的方法,优选预先在溶剂中加入由上述式(2)表示的偕胺肟化合物和上述碱,接下来,加入根据需要用溶剂稀释的xcoor2。此时,为了防止急剧的发热,优选滴入xcoor2。
此外,对进行上述反应时的条件并无特别限制。反应温度优选为-10~10℃。另外,就反应时间而言,只要是0.5~15小时则足以。
通过在以上这样的条件下使其反应,从而能够制造上述含有酯保护基的化合物。将上述含有酯保护基的化合物从反应体系中取出的方法并无特别限制。具体地,通过使上述含有酯保护基的化合物在醋酸乙酯、甲苯、氯仿、二氯甲烷这样的在水中难溶的溶剂中溶解,进行水洗、浓缩、干燥等,从而能够将上述含有酯保护基的化合物取出。再有,在溶剂中使用上述在水中难溶的溶剂的情况下,也能够直接对溶液进行水洗。
对在以上这样的条件下得到的含有酯基的化合物并无特别限制,能够制成纯度为90.0~99.5%的化合物。另外,也能够通过调整水洗,从而在取出的该含有酯基的化合物含有碱的状态下实施接下来的环化反应。
(环化反应)
本发明的特征在于,进行上述含有酯保护基的化合物的环化反应来制造由下述式(4)
【化23】
(式中,r1与上述式(3)中r1的同义)
表示的阿齐沙坦烷基酯时,在包含碳原子数1~8的醇的反应溶剂中实施。该环化反应时副产r2-oh。
就该环化反应而言,通过进行加热,从而能够使该反应进行。具体地,通过加热将上述含有酯保护基的化合物溶解于碳原子数1~8的醇而成的反应溶液,从而促进环化反应,能够使上述含有酯保护基的化合物成为阿齐沙坦烷基酯。该环化反应时,优选将上述含有酯保护基的化合物溶解于反应溶剂中,边搅拌混合边加热。应予说明,当然也可边搅拌上述含有酯保护基的化合物和反应溶剂边加热而制成反应溶液,就这样将该反应溶液加热。
(反应溶剂)
该环化反应中使用的反应溶剂为包含碳原子数1~8的醇的溶剂。如果具体地例示碳原子数1~8的醇,可列举出甲醇、乙醇、1-丙醇、异丙醇、1-丁醇、2-甲基-1-丙醇、2-丁醇、2-甲基-2-丙醇、1-戊醇、3-甲基-1-丁醇、1-己醇、2-甲基-1-戊醇、3-甲基-1-戊醇、2-甲基-2-戊醇、2,4-二甲基-3-戊醇、3-乙基-3-戊醇、辛醇等。
其中,作为能够提高环化反应时的温度、提高反应速度并且能够减少杂质的溶剂,优选使用碳原子数3~8的直链状或分支状的醇。具体地,优选使用1-丙醇、异丙醇、1-丁醇、2-丁醇,特别优选1-丙醇、1-丁醇。
这些碳原子数1~8的醇能够使用1种,也能够使用多种的混合溶剂。在使用混合溶剂的情况下,成为基准的该醇的量为混合溶剂的合计量。
再有,反应溶剂也能够以不到50质量%的比例包含碳原子数1~8的醇以外的其他溶剂,如果考虑精制的容易性等,其他溶剂优选为10质量%以下,更优选为0质量%。
本发明中,对反应溶剂中的碳原子数1~8的醇的使用量并无特别限制。如果考虑反应的效率化、杂质的减少和后工序的操作性,相对于上述含有酯保护基的化合物1g,优选反应溶剂中的碳原子数1~8的醇的量为3~30ml。通过满足该范围,在环化反应结束后,进行冷却,容易将阿齐沙坦甲酯作为结晶取出。为了进一步发挥上述效果,相对于上述含有酯保护基的化合物1g,更优选反应溶剂中的碳原子数1~8的醇的量为5~20ml。应予说明,反应溶剂的上述体积为23℃下的体积。
就环化反应的反应温度而言,为了提高反应速度并且减少杂质,优选规定为50℃以上且反应溶液的回流温度以下,更优选规定为60℃以上且反应溶液的回流温度以下,进一步优选规定为70℃以上且反应溶液的回流温度以下。反应溶液的回流温度因使用的反应溶剂、上述含有酯保护基的化合物的浓度、副产的r2-oh的种类而异,因此不能一概地限定。不过,为了进一步抑制杂质的生成,反应温度优选规定为100℃以下。
(碱)
本发明中,能够按照上述条件来促进环化反应。其中,为了进一步缩短反应时间,优选在碱的存在下实施。具体地,只要是上述反应溶液中含有碱的状态即可。
对环化反应中能够使用的碱并无特别限制,能够使用上述的无机碱或有机碱。其中,为了得到的阿齐沙坦烷基酯的精制的容易性、提高操作性,优选使用三乙胺、吡啶、二异丙基乙基胺等有机碱。
这些碱能够使用1种,也能够使用多种碱。在使用多种碱的情况下,成为基准的碱的量为多种碱的合计量。
再有,该碱如上所述,在制造上述含有酯保护基的化合物时使用碱的情况下,也能够使用将该含有酯保护基的化合物取出时残存的碱。
本发明中,即使不使用碱,也能够使环化反应进行。不过,在使用碱的情况下,相对于上述含有酯保护基的化合物1摩尔,使用的碱的量优选规定为0.01~5摩尔。通过在该范围使用碱,从而能够提高反应速度,并且能够提高阿齐沙坦烷基酯的收率和纯度。为了进一步提高该效果,相对于上述含有酯保护基的化合物1摩尔,使用的碱的量更优选规定为0.1~1摩尔。
本发明中,在使用碱的情况下,也能够在反应溶剂中预先加入碱和上述含有酯保护基的化合物,边加热边搅拌混合。另外,也能够在边搅拌混合边加热的反应溶液中从中途追加该碱以促进反应。在从中途追加碱的情况下,使用的碱的总量成为基准的量。
(阿齐沙坦烷基酯的取出方法)
通过在以上这样的条件下进行环化反应,从而能够制造阿齐沙坦烷基酯。对将得到的阿齐沙坦烷基酯从反应体系取出的方法并无特别限制,能够采用非专利文献1、专利文献1中记载的方法。
其中,在本发明中,由于在反应溶剂中使用包含1~8的醇的溶剂,因此优选采用以下的方法。具体地,优选将反应溶液冷却,或者从反应溶液中将一部分反应溶剂馏除,在包含碳原子数1~8的醇的反应溶剂中使阿齐沙坦烷基酯的结晶析出,将该结晶取出。特别优选将反应溶液冷却,使结晶析出。
在反应溶剂中使阿齐沙坦烷基酯的结晶析出的情况下,并无特别限制。具体地,相对于阿齐沙坦烷基酯1g,优选使碳原子数1~8的醇的量为3~30ml。通过碳原子数1~8的醇满足上述范围,从而操作性提高,并且能够提高纯度。为了进一步提高该效果,相对于阿齐沙坦烷基酯1g,优选使碳原子数1~8的醇的量为5~20ml。应予说明,碳原子数1~8的醇的上述量为23℃下的体积。
环化反应优选加热进行。而且,在更优选的实施方式中,使反应溶液的温度(反应温度)成为50℃以上。因此,优选将反应结束后的反应溶液冷却到30℃以下的范围,更优选冷却到-10~30℃的范围,特别优选冷却到-10~10℃的范围。本发明中,由于使用碳原子数1~8的醇,因此在上述冷却温度的范围中,阿齐沙坦烷基酯的结晶容易析出。另外,在使结晶析出时也能够使用种晶。而且,在本发明中,只要进行冷却以阿齐沙坦烷基酯的结晶析出的方式进行调整,则该结晶难以收进副产物和根据需要所配合的碱。
为了进一步提高得到的阿齐沙坦烷基酯的纯度,优选以10~30℃/小时的冷却速度冷却反应结束后的反应溶液,使其成为30℃以下、优选0~30℃、更优选-10~30℃、特别优选-10~20℃的温度。进而,为了提高得到的阿齐沙坦烷基酯的收率,优选在30℃以下、优选0~30℃、更优选-10~30℃、特别优选-10~20℃的温度下放置1小时以上、优选2小时以上且20小时以下。
对于析出的阿齐沙坦烷基酯的结晶,能够采用公知的方法进行处理。通常优选通过过滤将结晶取出,进行清洗、干燥。另外,在要得到更高纯度的阿齐沙坦烷基酯的情况下,优选使用碳原子数1~8的醇进行重结晶。
(新型的阿齐沙坦烷基酯的结晶;新型的结晶的制造方法)
如上所述得到的阿齐沙坦烷基酯即使再次用碳原子数1~8的醇重结晶,也成为包含碳原子数1~8的醇的溶剂合物的结晶。即,认为将碳原子数1~8的醇的一部分收进结晶内部。
在碳原子数1~8的醇中使用1-丙醇的情况下r1为甲基的阿齐沙坦甲酯能够制成在使用cu-kα线的x射线衍射中至少在2θ=9.9±0.2°、10.9±0.2°具有特征峰的结晶。另外,该阿齐沙坦甲酯是除此以外在2θ=13.6±0.2°、17.2±0.2°、23.2±0.2°等具有峰的结晶。具有以上这样的峰的结晶在现有技术中不存在,是新型的晶型。该结晶也能够包含0.5~5质量%的1-丙醇(以下也有时将该结晶简称为“新型的结晶”。)。
在此,本发明中的上述特征峰是指在2θ=9.9±0.2°具有强度成为最大的峰、同时在2θ=10.9±0.2°出现相对于最大峰强度(2θ=9.9±0.2°的峰强度)具有5%以上的强度的峰。相对于最大峰强度具有不到5%的强度的峰视为噪音等,不相当于本发明中的特征峰。
(阿齐沙坦烷基酯的重结晶)
本发明中,也能够将上述阿齐沙坦烷基酯直接水解来合成阿齐沙坦。不过,为了制成纯度更高的阿齐沙坦烷基酯,优选用包含酮溶剂的溶剂对采用上述方法得到的新型的晶型的阿齐沙坦烷基酯进行重结晶。当然,新型的晶型的该阿齐沙坦烷基酯可以为包含0.5~5质量%的1-丙醇的溶剂合物。
如果例示使用的酮溶剂,可以列举出丙酮、甲乙酮、二乙基酮、甲基异丙基酮、甲基丁基酮、甲基异丁基酮等。其中,为了提高纯度,改善操作性,优选使用丙酮。这些酮溶剂能够使用1种,也能够使用多种的混合溶剂。在使用混合溶剂的情况下,成为基准的酮溶剂的量为混合溶剂的合计量。
再有,包含酮溶剂的溶剂也能够以不到50质量%的比例包含酮溶剂以外的其他溶剂,如果考虑精制的容易性等,其他溶剂优选为10质量%以下,更优选为0质量%。
对使用的酮溶剂的量并无特别限制。具体地,相对于上述阿齐沙坦烷基酯的结晶1g,酮溶剂的量优选设为3~30ml,更优选设为5~20ml。
作为重结晶的方法,使上述阿齐沙坦烷基酯的结晶溶解于包含酮溶剂的溶剂中。优选地,加热到溶液的回流温度(约60℃),使上述阿齐沙坦烷基酯的结晶溶解。接下来,优选以10~30℃/小时的冷却速度进行冷却,在0~30℃、更优选-10~30℃、特别优选-10~20℃的温度范围内放置一定时间。
(阿齐沙坦的制造方法)
本发明中,通过与上述第一发明同样地将采用上述方法得到的阿齐沙坦烷基酯水解,从而能够制造由下述式(5)
【化24】
表示的阿齐沙坦。
第三本发明为阿齐沙坦烷基酯的制造方法,其特征在于,在丙酮、或者丙酮与醇的混合溶剂中使由下述式(4)
【化25】
(式中,r1为烷基)
表示的阿齐沙坦甲酯结晶化。以下按顺序进行说明。
(对象阿齐沙坦烷基酯)
首先,对于成为结晶化的对象的阿齐沙坦烷基酯(以下也有时称为对象阿齐沙坦烷基酯)进行说明。应予说明,上述式中,r1为烷基。如果考虑成为原料的含有酯保护基的化合物、阿齐沙坦烷基酯的稳定性和阿齐沙坦的制造的容易性,r1优选为碳原子数1~4的烷基。具体地,可列举出甲基、乙基、丙基、异丙基、丁基、异丁基等,特别优选为甲基。
(对象阿齐沙坦烷基酯的制造)
对本发明中使用的对象阿齐沙坦烷基酯并无特别限制,能够使用采用公知的方法合成的产物。例如,优选采用通过上述第二本发明中说明的制造方法制造的产物。
即,本发明中,对象阿齐沙坦烷基酯优选使用如第二本发明中说明那样在包含碳原子数1~8的醇的反应溶剂中进行环化反应而得到的产物。采用该方法得到的对象阿齐沙坦烷基酯与在二甲苯溶剂中进行环化反应,在包含醋酸乙酯的溶剂中析出,与现有的阿齐沙坦烷基酯相比,能够减少结构不明的杂质。该现有的阿齐沙坦烷基酯倾向于包含分子量比阿齐沙坦烷基酯大的杂质(在阿齐沙坦甲酯的情况下,倾向于包含分子量比阿齐沙坦甲酯大10的杂质。)。因此,在本发明中,优选使用通过在包含碳原子数1~8的醇的反应溶剂中进行环化反应而使杂质减少的对象阿齐沙坦烷基酯。不过,在本发明中,由于如以下将详述那样能够制成高纯度的阿齐沙坦烷基酯,因此也当然可以将包含结构不明、分子量大的杂质的、现有的阿齐沙坦烷基酯作为对象阿齐沙坦烷基酯。
(阿齐沙坦烷基酯的结晶化方法;准结晶的制造方法)
(溶剂:丙酮、或者丙酮与醇的混合溶剂)
本发明中,为了提高上述对象阿齐沙坦烷基酯的纯度,并且使晶型变形而制成熔点低的阿齐沙坦烷基酯,必须在丙酮、或者丙酮与醇的混合溶剂中使对象阿齐沙坦烷基酯结晶化。通过使用丙酮、或者丙酮与醇的混合溶剂,从而与使用醋酸乙酯的现有的方法相比,能够高效率地减少阿齐沙坦烷基酯的脱乙基体或二聚体的杂质。
本发明中使用的溶剂为丙酮、或者丙酮与醇的混合溶剂,将丙酮与醇的合计的容积设为100时,优选丙酮的容积比率成为100~50,醇的容积比率成为0~50。根据本发明人等的研究,获知即使在以容积比率50%以下的范围包含醇的情况下,也能够与不含醇的(只是丙酮的)情形同样地得到新型的晶型。因此,只要是醇的容积比率没有变得比使用的丙酮的容积比率大的范围,就不必严格地管理对象阿齐沙坦烷基酯中的醇残量。即,在使用在包含碳原子数1~8的醇的反应溶剂中进行环化反应而得到的对象阿齐沙坦烷基酯的情况下,不必严格地将该醇除去,因此能够提高操作性。因此,在对象阿齐沙坦烷基酯为包含醇的粗体的情况下,只要测定其醇量来调整使用的丙酮的量即可。
本发明中,在使用醇的情况下,该醇优选为碳原子数1~8的醇。如果具体地例示,可列举出甲醇、乙醇、1-丙醇、异丙醇、1-丁醇、2-甲基-1-丙醇、2-丁醇、2-甲基-2-丙醇、1-戊醇、3-甲基-1-丁醇、1-己醇、2-甲基-1-戊醇、3-甲基-1-戊醇、2-甲基-2-戊醇、2,4-二甲基-3-戊醇、3-乙基-3-戊醇、辛醇等。其中,如果考虑收率和阿齐沙坦烷基酯的脱乙基体或二聚体的杂质的减少效果,优选使用碳原子数3~8的直链状、或分支状的醇。具体地,优选使用1-丙醇、异丙醇、1-丁醇、2-丁醇,特别优选1-丙醇、1-丁醇。这些醇能够使用1种,也能够将多种混合使用。本发明中,在使用醇的情况下,优选丙酮的容积比率成为99~51,醇的容积比率成为1~49,更优选丙酮的容积比率成为90~60,醇的容积比率成为10~40。
(结晶化的方法)
另外,对使用的丙酮、或者丙酮与醇的混合溶剂的量并无特别限制,相对于上述对象阿齐沙坦烷基酯的结晶1g,优选设为3~30ml,更优选设为5~20ml。应予说明,使用的溶剂的上述体积为23℃下的体积。另外,在使用混合溶剂的情况下,成为基准的溶剂的量为丙酮和醇的合计量。
作为重结晶的方法,优选在丙酮、或者丙酮与醇的混合溶剂中使上述对象阿齐沙坦烷基酯溶解,优选加热到得到的溶液的回流温度(约60℃),接下来,以10~30℃/小时的冷却速度冷却,优选在0~30℃、更优选在-10~30℃、特别优选在-10~20℃的温度范围保持一定时间。
(阿齐沙坦烷基酯、新型阿齐沙坦甲酯)
在丙酮、或者丙酮与醇的混合溶剂中结晶化而得到的阿齐沙坦烷基酯中,适合的化合物即r1成为甲基的阿齐沙坦甲酯成为根据使用cu-kα线的x射线衍射至少在2θ=9.2±0.2°、15.8±0.2°、22.1±0.2°具有特征峰的新型的结晶。成为作为其他峰在2θ=18.2±0.2°、25.1±0.2°、27.4±0.2°具有峰的结晶。
另外,在丙酮、或者丙酮与醇的混合溶剂中结晶化而得到的阿齐沙坦甲酯成为存在至少2个熔点的、新型的晶型(以下也有时将该熔点低的结晶简称为“准结晶”)。其中,在丙酮、或者丙酮与醇的混合溶剂中使其结晶化而得到的阿齐沙坦甲酯成为如下的结晶:至少在150~165℃、185~195℃观察到用实施例中所示的差示扫描量热计的条件测定的熔点。应予说明,用该差示扫描量热(dsc)测定确定的熔点为吸热峰的峰顶温度。该新型的准结晶由于熔点比以往已知的阿齐沙坦烷基酯低,因此认为并非完全结晶体。因此,由于纯度高,能够容易地溶解,因此能够适合在接下来的水解反应中使用。
准结晶的阿齐沙坦甲酯的熔解热量如图5中所示的图表那样,不能通过对吸热峰画出完全的切线而求出。推测这是因为,该阿齐沙坦甲酯为不稳定的状态的结晶。因此,该熔解热量不是正确的值,在如图5那样求出了熔解热量的情况下,优选与150~165℃的温度范围的熔点(以下也有时简称为“低熔点”)有关的熔解热量为2~15j/g,与185~195℃的温度范围的熔点(以下也有时简称为“高熔点”)有关的熔解热量为40~70j/g。更优选与低熔点有关的熔解热量为4~10j/g,与高熔点有关的熔解热量为50~60j/g。
通过对这样在包含碳原子数1~8的醇的溶剂中得到的新型的结晶进一步用丙酮、或者丙酮与醇的混合溶剂进行重结晶,不仅能够提高纯度,而且能够制成能够容易地溶解的熔点低的准结晶。
(阿齐沙坦制造方法)
本发明中,通过与上述第一发明同样地将采用上述方法得到的阿齐沙坦烷基酯水解,从而能够制造由下述式(5)
【化26】
表示的阿齐沙坦。
对得到的阿齐沙坦并无特别限制,可采用公知的方法进行精制而制成原药。例如可列举出采用重结晶或再淤浆、柱色谱等方法的方法。
(阿齐沙坦二聚体的除去)
根据第一本发明~第三本发明制造的阿齐沙坦有时包含由下述式(6)
【化27】
表示的阿齐沙坦二聚体作为杂质。在这种情况下,在使包含阿齐沙坦二聚体的粗阿齐沙坦的溶液与活性炭接触后,可以附加将阿齐沙坦的结晶从该溶液中分离的工序。
(粗阿齐沙坦)
所谓粗阿齐沙坦,意指包含阿齐沙坦二聚体作为杂质的阿齐沙坦。粗阿齐沙坦可以是在高效液相色谱(hplc)分析中96.0~99.0%纯度的阿齐沙坦。另外,在成为精制的对象的粗阿齐沙坦中可包含0.01~0.50%的上述阿齐沙坦二聚体。
成为要减少的对象的阿齐沙坦二聚体认为是如下所述副产的。即,作为原料使用的偕胺肟化合物(式(2)的化合物)与认为在将该偕胺肟化合物环化时已生成的阿齐沙坦(式(5)的化合物)首先反应,生成由下述式(12)
【化28】
(式中,r1为烷基)
表示的阿齐沙坦烷基酯二聚体。接下来,认为由该阿齐沙坦烷基酯二聚体得到阿齐沙坦二聚体。
(溶解有粗阿齐沙坦的溶液与活性炭的接触方法)
以下对于使包含阿齐沙坦二聚体的粗阿齐沙坦的溶液与活性炭接触的方法进行说明。
对使用的活性炭并无特别限制,优选采用bet法求出的比表面积为1000~3500m2/g,并且累计细孔容积为0.6~1.5ml/g。通过使用具有该范围的物性的活性炭,从而能够更有效地减少上述阿齐沙坦二聚体。
对使用的活性炭的赋活(活化)方法并无特别限制,采用药品赋活法得到的氯化锌炭、采用水蒸汽赋活法得到的水蒸汽炭均能够适合地使用。另外,对活性炭的种类也无特别限制,粉末炭、破碎炭、粒状炭、颗粒炭、成形炭等,只要满足上述物性就能够使用。其中,如果考虑处理容易性或活性炭自身的除去效率等,优选使用粉末炭、粒状炭。对于活性炭,如果具体地例示,则能够列举出精制白鹭、特性白鹭、粒状白鹭、白鹭a、白鹭p、白鹭c、白鹭m(以上为大阪瓦斯化学制造)、太阁a、太阁ca、太阁k、太阁m(以上为futamurachemicalco.,ltd.制造)等。
与活性炭接触的粗阿齐沙坦的溶液只要是包含作为杂质的阿齐沙坦二聚体的粗阿齐沙坦溶解的溶液,则并无特别限制。因此,在粗阿齐沙坦的溶液中使用的溶剂只要是该粗阿齐沙坦能够溶解的溶剂,则可以是有机溶剂,也可以是水。其中,如上所述,优选使阿齐沙坦烷基酯水解而得到的包含粗阿齐沙坦的溶液(水解反应后得到的包含粗阿齐沙坦的溶液)与活性炭接触。这种情况下,包含粗阿齐沙坦的溶液能够包含碱。另外,以减少阿齐沙坦二聚体为目的,也能够使从该溶液取出的阿齐沙坦再次溶解于碱性水溶液等而成的溶液与活性炭接触。不过,如果考虑作业性,则优选将水解反应后得到的包含粗阿齐沙坦的溶液作为对象。
对使粗阿齐沙坦的溶液与上述活性炭接触的方法并无特别限定。例如,能够采用将粗阿齐沙坦、活性炭和能够溶解粗阿齐沙坦的溶剂同时地混合的方法;准备粗阿齐沙坦溶解的溶液,在该溶液中添加活性炭而混合的方法;或者使该溶液通过填充活性炭的柱的方法等。其中,从操作的容易性出发,优选采用在该溶液中添加活性炭并进行混合的方法。
活性炭的使用量可根据活性炭的种类、杂质量等而适当地确定。在使用采用上述方法得到的粗阿齐沙坦溶解的溶液的情况下,相对于粗阿齐沙坦1g,优选使用0.03~0.2g的活性炭。此时,该溶液与活性炭的混合优选通过搅拌来进行。另外,就搅拌混合时的温度而言,优选在15~35℃下进行,特别优选在20~30℃下进行。另外,对与活性炭的接触时间并无特别限制,通常,如果在该温度下以1~5小时的范围进行则足以。
(活性炭的除去方法)
如上所述,使粗阿齐沙坦的溶液与活性炭接触后,接下来,从该混合液中将活性炭分离,将分离液回收。对将活性炭分离的方法并无特别限制,能够采用公知的方法实施。例如,可采用倾析、过滤、离心过滤等分离方法。此时,为了提高过滤的效率,也能够使用沸石、钠沸石等过滤助剂。
(阿齐沙坦的分离)
本发明中,必须从上述活性炭处理后得到的分离液中将阿齐沙坦的结晶分离。对于从分离液中将阿齐沙坦的结晶分离的方法,也无特别限制,能够采用公知的方法实施。例如,能够无特别限制地采用:通过从分离液中将溶剂直接馏除从而将阿齐沙坦的结晶分离的方法、或将分离液中和而使阿齐沙坦的结晶析出的方法。
采用上述方法析出的阿齐沙坦的结晶能够采用公知的方法进行分离(分取)。具体地,可采用倾析、减压/加压过滤、离心过滤等分离方法。另外,就分离的阿齐沙坦的结晶而言,优选使用与上述溶剂同种的溶剂进行清洗。这样得到的阿齐沙坦的结晶为湿体,通过在30~50℃下干燥3~20小时,从而得到阿齐沙坦的结晶的干燥体。
如上所述,使包含阿齐沙坦二聚体作为杂质的粗阿齐沙坦与活性炭接触后,通过将阿齐沙坦的结晶从该溶液中分离,从而能够得到特别是阿齐沙坦二聚体的含量减少的、高纯度的阿齐沙坦的结晶。进而,作为上述活性炭,通过使用采用bet法求出的比表面积为1000~3500m2/g、累计细孔容积为0.6~1.5ml/g的活性炭,从而能够进一步使上述阿齐沙坦二聚体的含量减小,得到更高纯度的阿齐沙坦的结晶。
(阿齐沙坦m型结晶)
根据上述第一本发明~第三本发明制造的阿齐沙坦的各结晶对于有机溶剂具有非常难溶性。因此,在使用有机溶剂进行精制操作的情况下,需要大量的有机溶剂。为了对其进行改善,也优选使根据上述第一本发明~第三本发明制造的阿齐沙坦成为对于包含甲醇或乙醇等醇类或醋酸乙酯等酯类的各种溶剂的溶解度非常高的结晶形态。
具体地,优选制成具有如下结晶结构的阿齐沙坦:根据使用cu-kα线的x射线衍射,至少在2θ=9.4±0.2°、11.5±0.2°、13.3±0.2°、14.8±0.2°、26.0±0.2°给予特征峰。应予说明,在本说明书中,有时将具有该结晶结构的本发明的阿齐沙坦称为“阿齐沙坦m型结晶”。应予说明,作为x射线衍射角的测定误差的±0.2°包含通过四舍五入而成为±0.2°的范围。将该阿齐沙坦m型结晶的x射线衍射测定结果示于图7中。
其中,本发明中的特征峰是指在2θ=9.4±0.2°具有强度成为最大的峰、同时在2θ=11.5±0.2°、13.3±0.2°、14.8±0.2°、26.0±0.2°出现相对于最大峰强度(2θ=9.4±0.2°的峰强度)具有7%以上的强度的峰。相对于最大峰强度具有不到7%的强度的峰视为噪音等,不相当于本发明中的特征峰。
阿齐沙坦m型结晶与专利文献1~4、非专利文献1中记载的已知的阿齐沙坦结晶相比,对于甲醇或乙醇等醇类;醋酸乙酯等酯类;丙酮等酮类;四氢呋喃等醚类的有机溶剂的溶解性得到了改善。具体地,在室温下,阿齐沙坦m型结晶与已知的阿齐沙坦结晶相比,在等量的甲醇中能够溶解约7~10倍。
另外,阿齐沙坦m型结晶显示出比已知的阿齐沙坦结晶低的熔点。具体地,由差示扫描量热(dsc)测定所确定的熔点为115℃以上且135℃以下。本发明中,由差示扫描量热(dsc)测定所确定的熔点是指通过测定得到的吸热峰的峰顶温度。
(阿齐沙坦m型结晶的制造方法)
阿齐沙坦m型结晶能够通过如下制造:在通过将阿齐沙坦溶解于二甲基甲酰胺而得到的溶液中加入酮类和/或酯类的溶剂,使阿齐沙坦m型结晶析出。
在阿齐沙坦为水合物或溶剂合物的情况下,对水或溶剂的分子数并无特别限制。另外,由于在阿齐沙坦m型结晶的制造时使用二甲基甲酰胺和酮类和/或酯类的溶剂,因此可以是包含该有机溶剂的湿体,对于其他溶剂,可在结晶化时不产生影响的范围内残留。具体地,可以以该阿齐沙坦的50质量%以下的量残留。最优选不含该有机溶剂以外的溶剂。另外,对使用的阿齐沙坦的纯度并无特别限制,能够直接使用根据第一本发明~第三本发明得到的阿齐沙坦。
(阿齐沙坦溶液的制备方法)
阿齐沙坦m型结晶的制造方法首先通过将阿齐沙坦溶解于二甲基甲酰胺而得到阿齐沙坦溶液。此时,对使用的二甲基甲酰胺并无特别限制,能够直接使用市售的产品。二甲基甲酰胺的使用量可根据使用的阿齐沙坦的晶型适当地确定,一般地,相对于阿齐沙坦1g,可规定为0.5ml以上且10ml以下。如果二甲基甲酰胺的使用量增多,则收率降低,因此优选规定为0.5ml以上且5ml以下。应予说明,溶剂的体积规定为25℃下的体积。另外,使阿齐沙坦溶解时的温度可根据使用的阿齐沙坦的晶型或二甲基甲酰胺的量适当地确定,优选在10℃以上且50℃以下的范围内使其溶解。再有,当然在存在没有完全地溶解阿齐沙坦的情况下,也能够将没有溶解的阿齐沙坦过滤分离来处理。进而,对得到阿齐沙坦溶液的方法并无特别限制,可将阿齐沙坦和二甲基甲酰胺混合来调整溶液,对混合的方法或顺序也无特别限制。
(阿齐沙坦m型结晶的结晶化)
阿齐沙坦m型结晶的制造方法,其特征在于,在得到的阿齐沙坦溶液中加入酮类和/或酯类的溶剂,使阿齐沙坦m型结晶析出。通过采用本方法,从而能够以高收率得到在有机溶剂中的溶解度得到了改善的阿齐沙坦m型结晶。在阿齐沙坦溶液中加入的溶剂能够从丙酮、甲乙酮、甲基异丁基酮、二异丁基酮、环己酮等酮类;和/或醋酸甲酯、醋酸乙酯、醋酸丙酯、醋酸异丙酯、醋酸丁酯、醋酸异丁酯等酯类中选择。为了得到更高纯度的阿齐沙坦,优选加入酯类的溶剂,其中最优选使用醋酸乙酯。另外,在本发明中,也能够将这些酮类的溶剂与酯类的溶剂混合而加入。本发明中,通过加入酮类和/或酯类的溶剂而使阿齐沙坦析出,从而能够使对于有机溶剂的溶解性提高的阿齐沙坦m型结晶析出。
加入阿齐沙坦溶液中的酮类和/或酯类的溶剂的使用量可根据选择的溶剂的种类适当地确定。通常,相对于上述阿齐沙坦溶液的调制中使用的二甲基甲酰胺1ml,可规定为1ml以上且50ml以下,如果考虑收率、操作性,优选规定为5ml以上且20ml以下。此时,对加入酮类和/或酯类的溶剂的温度并无特别限制,也能够在确认阿齐沙坦溶解于二甲基甲酰胺后在该温度下立即加入,更优选在30℃以下加入。通过在30℃以下加入,从而能够抑制热分解引起的杂质的增加,得到的阿齐沙坦m型结晶的纯度也成为更高纯度。另外,对加入酮类和/或酯类的溶剂的方法也无特别限制,一次加入全部量的方法、分数次加入的方法的任一种都能够采用。本发明中,加入酮类和/或酯类的溶剂后,通过在一定温度下进行搅拌,从而使阿齐沙坦m型结晶析出。此时保持的温度可规定为-5℃以上且30℃以下,为了以更高收率得到阿齐沙坦,优选保持在0℃以上且10℃以下。另外,保持的时间可根据保持的温度适当地确定,优选通常规定为5小时以上。另外,此时,在阿齐沙坦的结晶难以析出的情况下,也能够添加种晶。
就这样析出的阿齐沙坦m型结晶而言,通过过滤或离心分离等固液分离后,通过采用自然干燥、送风干燥、真空干燥等方法进行干燥,从而能够离析。
采用本方法得到的阿齐沙坦是具有新型的结晶结构的阿齐沙坦m型结晶。就本发明的阿齐沙坦m型结晶而言,对于有机溶剂的溶解度得到改善,与已知的晶型相比,对于醇类、酯类、酮类、醚类的溶剂的溶解度极高。因此,将阿齐沙坦m型结晶作为对象进行精制操作的情况下,能够使用醇类、酯类、酮类、醚类的各溶剂容易地进行重结晶等精制操作。
实施例
以下列举实施例对本发明详细地说明,这些实施例为具体例,本发明并不由这些实施例所限定。
应予说明,实施例和比较例中的纯度评价采用使用以下的高效液相色谱(hplc)的方法进行。
<hplc的测定条件>
装置:高效液相色谱(hplc)。
机种:2695-2489-2998(waters公司制造)
检测器:紫外吸光光度计(测定波长:210nm)
柱:kromasilc18、内径4.6mm、长15cm(粒径5μm)(阿克苏诺贝尔公司制造)
柱温度:30℃恒定
样品温度:25℃恒定
流动相a:乙腈
流动相b:15mm磷酸二氢钾水溶液(ph=2.5用磷酸调整)
流动相的送液:如表1那样改变流动相a、b的混合比,进行浓度梯度控制。
【表1】
流速:1.0ml/分钟
测定时间:0~90分钟
在上述条件下,确认上述偕胺肟脱乙基体在约1.8分钟、上述偕胺肟化合物在约2.8分钟、上述酰胺脱乙基体在约4.0分钟、上述酰胺体在约8.5分钟、上述腈脱乙基体在约11.2分钟、上述腈化合物在约25.0分钟、上述含有酯保护基的化合物a(r1:甲基,r2:乙基)在约16.2分钟、上述含有酯保护基的化合物b(r1:甲基,r2:2-乙基己基)在约52.3分钟、上述阿齐沙坦甲酯在约14.5分钟、上述脱乙基体在约7.0分钟、上述阿齐沙坦在约7.3分钟、上述二聚体在约49.1分钟、分子量比阿齐沙坦甲酯大10的杂质在约5.5分钟有峰。以下的实施例、比较例中,上述偕胺肟化合物、上述含有酯保护基的化合物、上述阿齐沙坦甲酯、上述阿齐沙坦的各纯度全部为相对于上述条件下所测定的全部峰的面积值(不包括来自溶剂的峰)的合计的各化合物的峰面积值的比例。
<阿齐沙坦烷基酯、阿齐沙坦的晶型的测定>
装置:x射线衍射装置(xrd)
机种:smartlab(株式会社リガク制造)
测定方法:asc6bbdtex
x射线输出:40kv-30ma
波长:
<阿齐沙坦烷基酯的熔点的测定>
装置:差示扫描量热计(dsc)
机种:dsc6200(siinanotechnologyinc.制造)
升温条件:10℃/分钟
气体:氩气。
<阿齐沙坦的熔点的测定>
装置:差示扫描量热计(dsc)
机种:dsc6200(siinanotechnologyinc.制造)
升温条件:5℃/分钟
气体:氩气
a.关于第一本发明的实施例和比较例
[实施例1]
在直径2.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取上述腈化合物5g(12.2mmol),加入1-丙醇50ml、市售的50质量%羟基胺水溶液4.0g(60.8mmol),加热到回流温度(约92℃)后,在该温度下进行12小时反应。为上述偕胺肟化合物纯度:82.2%、上述酰胺体:9.1%、上述腈化合物:2.2%、上述偕胺肟脱乙基体:6.0%、上述酰胺脱乙基体:0.3%、上述腈脱乙基体:0.05%。
将反应后的溶液以30℃/小时的速度冷却到20℃,在20℃下搅拌整夜。接下来,对得到的浆液进行减压过滤,分取析出的结晶,在50℃下干燥,得到4.0g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:94.7%、上述酰胺体:3.0%、上述腈化合物:0.2%、上述偕胺肟脱乙基体:0.7%、上述酰胺脱乙基体:0.3%、上述腈脱乙基体:未检测出)(收率:77.1%)。将结果汇总于表2、3中。
[实施例2]
在直径10cm的具备2片搅拌叶片的1l三口烧瓶中量取上述腈化合物70g(170.1mmol),加入1-丙醇700ml、三乙胺5.16g(51.0mmol)、市售的50质量%羟基胺水溶液56.2g(850.5mmol),加热到回流温度(约92℃)后,在该温度下进行13小时反应。为上述偕胺肟化合物的纯度:83.9%、上述酰胺体:2.4%、上述腈化合物:2.4%、上述偕胺肟脱乙基体:7.6%、上述酰胺脱乙基体:0.2%、上述腈脱乙基体:0.01%。
将反应后的溶液以20℃/小时的速度冷却到20℃,在20℃下搅拌13小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在50℃下干燥,得到61.0g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:96.9%、上述酰胺体:0.5%、上述腈化合物:0.1%、上述偕胺肟脱乙基体:0.5%、上述酰胺脱乙基体:0.1%、上述腈脱乙基体:未检测出)(收率:81.0%)。将结果汇总于表2、3中。
[实施例3]
在直径2.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取腈化合物5g(12.2mmol),加入1-丙醇50ml、三乙胺0.62g(6.1mmol)、市售的50质量%羟基胺水溶液4.0g(60.8mmol),加热到回流温度(约92℃)后,在该温度下进行13小时反应。为上述偕胺肟化合物的纯度:84.2%、上述酰胺体:1.8%、上述腈化合物:2.6%、上述偕胺肟脱乙基体:7.4%、上述酰胺脱乙基体:0.2%、上述腈脱乙基体:0.01%。
将反应后的溶液以20℃/小时的速度冷却到20℃,在20℃下搅拌13小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在50℃下干燥,得到4.1g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:97.1%、上述酰胺体:0.3%、上述腈化合物:0.1%、上述偕胺肟脱乙基体:0.5%、上述酰胺脱乙基体:0.1%、上述腈脱乙基体:未检测出)(收率:77.4%)。将结果汇总于表2、3中。
[实施例4]
在实施例3中,除了将反应溶剂从1-丙醇变为1-丁醇,将三乙胺的使用量从0.62g(6.1mmol)变为0.37g(3.66mmol)以外,进行同样的操作。
得到4.4g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:97.0%、上述酰胺体:0.4%、上述腈化合物:0.1%、上述偕胺肟脱乙基体:0.6%、上述酰胺脱乙基体:0.1%、上述腈脱乙基体:未检测出)(收率:81.1%)。将结果汇总于表2、3。
[实施例5]
在实施例3中,除了将使用的碱从三乙胺变为吡啶,将碱的使用量从0.62g(6.1mmol)变为0.37g(3.66mmol)以外,进行同样的操作。
得到4.3g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:96.5%、上述酰胺体:0.4%、上述腈化合物:0.2%、上述偕胺肟脱乙基体:0.6%、上述酰胺脱乙基体:0.1%、上述腈脱乙基体:未检测出)(收率:79.4%)。将结果汇总于表2、3。
[实施例6]
在实施例3中,除了将三乙胺的使用量从0.62g(6.1mmol)变为0.12g(1.22mmol)以外,进行同样的操作。
得到4.3g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:95.3%、上述酰胺体:1.5%、上述腈化合物:0.2%、上述偕胺肟脱乙基体:0.6%、上述酰胺脱乙基体:0.3%、上述腈脱乙基体:未检测出)(收率:80.2%)。将结果汇总于表2、3。
[实施例7]
在实施例1中,除了将1-丙醇的使用量从50ml变为75ml以外,进行同样的操作。
得到3.9g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:94.9%、上述酰胺体:2.8%、上述腈化合物:0.2%、上述偕胺肟脱乙基体:0.7%、上述酰胺脱乙基体:0.3%、上述腈脱乙基体:未检测出)(收率:73.1%)。将结果汇总于表2、3。
[实施例8](阿齐沙坦甲酯的合成)
在直径10cm的具备2片搅拌叶片的1l三口烧瓶中量取实施例2中得到的上述偕胺肟化合物40g,加入二甲基亚砜350ml、1,1'-羰基咪唑17.5g、二氮杂双环十一碳烯15.5g,在室温下边搅拌4小时边进行反应。在另外准备的3l三口烧瓶中量取水1500ml,慢慢地滴入该反应液。在得到的溶液中加入5%盐酸水溶液,将ph调节到约4后,通过减压过滤分取析出的阿齐沙坦甲酯的结晶,在50℃下干燥后,由丙酮400ml进行重结晶,对得到的浆液进行减压过滤,分取析出的结晶,在50℃下干燥,得到32.0g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:99.4%)。
[实施例9](阿齐沙坦的合成)
在直径10cm的具备2片搅拌叶片的1l三口烧瓶中量取实施例8中得到的阿齐沙坦甲酯20g,加入1.25m氢氧化钠水溶液200ml,加热到50℃后,在该温度下进行3小时反应。将反应液冷却到45℃后,在该温度下加入丙酮100ml、醋酸75ml、水70ml,使阿齐沙坦的结晶析出。将反应液以20℃/小时的速度冷却到20℃后,在该温度下搅拌5小时。接下来,对得到的浆液进行减压过滤,分取析出的结晶,在40℃下干燥,得到16.6g的阿齐沙坦的结晶(阿齐沙坦的纯度:99.4%)。
[比较例1](采用非专利文献1中记载的方法的上述偕胺肟化合物的制造)
在直径7.5cm的具备2片搅拌叶片的500ml三口烧瓶中量取上述腈化合物5g(12.2mmol),加入二甲基亚砜50ml、羟基胺盐酸盐4.2g(60.8mmol)、三乙胺6.15g(60.8mmol),加热到90℃后,在该温度下进行16小时反应。为上述偕胺肟化合物纯度:42.6%、上述酰胺体:37.0%、上述腈化合物:4.5%、上述偕胺肟脱乙基体:9.3%、上述酰胺脱乙基体:4.8%、上述腈脱乙基体:1.0%。
将反应后的溶液冷却到20℃,搅拌12小时,但上述偕胺肟化合物的结晶没有析出。因此,在反应液中加入水125ml,使上述偕胺肟化合物的结晶析出。接下来,对得到的浆液进行减压过滤,分取析出的结晶,在50℃下干燥,得到3.1g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:49.9%、上述酰胺体:46.4%、上述腈化合物:0.5%、上述偕胺肟脱乙基体:1.7%、上述酰胺脱乙基体:0.9%、上述腈脱乙基体:0.1%)(收率:57.1%)。将结果汇总于表2、3。
[比较例2](采用专利文献2中记载的方法的偕胺肟化合物的制造)
在直径7.5cm的具备2片搅拌叶片的500ml三口烧瓶中量取上述腈化合物5g(12.2mmol),加入二甲基亚砜50ml、市售的50质量%羟基胺水溶液2.0g(30.4mmol),加热到90℃后,在该温度下进行15小时反应。为上述偕胺肟化合物的纯度:72.0%、上述酰胺体:9.8%、上述腈化合物:0.5%、上述偕胺肟脱乙基体:9.3%、上述酰胺脱乙基体:1.0%、上述腈脱乙基体:0.2%。
将反应后的溶液冷却到20℃,搅拌12小时,但上述偕胺肟化合物的结晶没有析出。因此,在反应液中加入水125ml,使上述偕胺肟化合物的结晶析出。接下来,对得到的浆液进行减压过滤,分取析出的结晶,在50℃下干燥,得到4.2g的上述偕胺肟化合物的结晶(上述偕胺肟化合物纯度:85.7%、上述酰胺体:5.1%、上述腈化合物:0.5%、上述偕胺肟脱乙基体:5.9%、上述酰胺脱乙基体:0.4%、上述腈脱乙基体:0.1%)(收率:78.8%)。将结果汇总于表2、3。
【表2】
表2
*每1g腈化合物的使用量(ml)
**每1mol腈化合物的使用量(mol)
【表3】
表3
b.第二本发明涉及的实施例和比较例
[制造例1](含有酯保护基的化合物a;r1:甲基、r2:乙基的合成)
在直径15cm的具备2片搅拌叶片的2l四口烧瓶中量取上述偕胺肟化合物120g(270.0mmol),加入二氯甲烷840ml、三乙胺33.0g(324.0mmol),边搅拌边冷却到0℃。在得到的溶液中缓慢地滴加将氯甲酸乙酯35.4g(324.0mmol)用二氯甲烷360ml稀释而成的溶液。将全部量滴入后,在0℃下边搅拌2小时边反应。将反应后的溶液升温到20℃,加入水480ml,将有机层抽出。对得到的有机层减压浓缩后,在残渣中加入1-丙醇600ml,在20℃下搅拌3小时。将得到的浆料溶液通过减压过滤固液分离后,在40℃下减压干燥,得到含有酯保护基的化合物a122.86g(含有酯保护基的化合物a纯度:96.1%)(收率88.1%)。
[制造例2](含有酯保护基的化合物b;r1:甲基、r2:2-乙基己基的合成)
在直径7.5cm的具备2片搅拌叶片的500ml四口烧瓶中量取上述偕胺肟化合物30g(67.5mmol),加入二氯甲烷210ml、三乙胺8.2g(81.0mmol),边搅拌边冷却到0℃。在得到的溶液中缓慢地滴加将氯甲酸-2-乙基己酯15.6g(81.0mmol)用二氯甲烷90ml稀释的溶液。全部量滴入后,在0℃下边搅拌2小时边反应。将反应后的溶液升温到20℃,加入水120ml,将有机层抽出。将得到的有机层减压浓缩后,在残渣中加入1-丙醇150ml,在20℃下搅拌3小时。将得到的浆料溶液通过减压过滤固液分离后,在40℃下减压干燥,得到含有酯保护基的化合物b31.9g(含有酯保护基的化合物b纯度:94.5%)(收率78.7%)。
[实施例10]
在直径3.5cm的具有2片搅拌叶片的100ml三口烧瓶中量取制造例1中得到的含有酯保护基的化合物a5g(9.7mmol),加入1-丙醇45ml,加热到回流温度(约95℃)后,在该温度下进行16小时反应。为上述阿齐沙坦甲酯的纯度:91.5%、上述含有酯保护基的化合物a:1.8%。将反应后的反应溶液以20℃/小时的速度冷却到0℃,在0℃下搅拌14小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到3.7g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦烷基酯的纯度:97.3%)(收率:81.1%)。将结果汇总于表4中。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,将该阿齐沙坦甲酯作为试样,测定xrd,得到图1中所示的x射线衍射图。可知该结晶为具有在2θ=9.9°、10.9°、13.6°、17.2°、23.2°给予特征峰的新型结晶结构的化合物。
[实施例11]
在直径5cm的具有2片搅拌叶片的200ml三口烧瓶中量取制造例1中得到的含有酯保护基的化合物a10g(19.4mmol),加入1-丙醇90ml、三乙胺0.4g(3.9mmol),加热到回流温度(约94℃)后,在该温度下进行9小时反应。为上述阿齐沙坦甲酯的纯度:93.0%、上述含有酯保护基的化合物a:0.7%。将反应后的反应溶液以20℃/小时的速度冷却到0℃,在0℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到7.6g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦甲酯的纯度:97.8%)(收率:83.4%)。将结果汇总于表4中。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,将该阿齐沙坦甲酯作为试样,测定xrd,可知其为具有在2θ=9.8°、10.9°、13.6°、17.2°、23.2°给予特征峰的新型结晶结构的化合物。
[实施例12]
在直径3.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取制造例2中得到的含有酯保护基的化合物b5g(8.3mmol),加入1-丙醇45ml、三乙胺0.2g(1.7mmol),加热到回流温度(约94℃)后,在该温度下进行10小时反应。为上述阿齐沙坦甲酯的纯度:91.7%、上述含有酯保护基的化合物b:0.6%。将反应后的反应溶液以20℃/小时的速度冷却到0℃,在0℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到3.2g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦甲酯的纯度:97.5%)(收率:81.8%)。将结果汇总于表4中。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,xrd的结果也与实施例10、11的阿齐沙坦甲酯没有变化。
[实施例13]
在实施例11中,除了将三乙胺的使用量从0.4g(3.9mmol)变为1.96g(19.4mmol)以外,进行同样的操作。用6小时使反应完结。
得到5.9g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦甲酯的纯度:98.6%)(收率:64.4%)。将结果汇总于表4中。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,xrd的结果也与实施例10、11的阿齐沙坦甲酯没有变化。
[实施例14]
在实施例10中,除了将反应溶剂从1-丙醇变为1-丁醇以外,进行同样的操作。
得到3.7g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦甲酯的纯度:97.1%)(收率:80.9%)。将结果汇总于表4。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,xrd的结果也与实施例10、11的阿齐沙坦甲酯没有变化。
[实施例15]
在实施例10中,除了将1-丙醇的使用量从45ml变为125ml以外,进行同样的操作。
得到2.3g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦甲酯的纯度:98.2%)(收率:50.9%)。将结果汇总于表4。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,xrd的结果也与实施例10、11的阿齐沙坦甲酯没有变化。
[实施例16]
在实施例11中,除了将使用的碱从三乙胺变为吡啶以外,进行同样的操作。
得到7.6g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦甲酯的纯度:97.7%)(收率:83.8%)。将结果汇总于表4。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,xrd的结果也与实施例10、11的阿齐沙坦甲酯没有变化。
[实施例17](阿齐沙坦的合成)
在直径10cm的具备2片搅拌叶片的1l三口烧瓶中量取实施例11中得到的阿齐沙坦甲酯5g,加入1.25m氢氧化钠水溶液50ml,加热到50℃后,在该温度下进行3小时反应。将反应液冷却到45℃后,在该温度下加入丙酮25ml、醋酸17ml、水17ml,使阿齐沙坦的结晶析出。将反应液以20℃/小时的速度冷却到20℃后,在该温度下搅拌6小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下干燥,得到4.2g的阿齐沙坦的结晶(阿齐沙坦的纯度:99.0%)。
[比较例3](采用专利文献1记载的方法的上述阿齐沙坦甲酯的制造)
在直径5cm的具备2片搅拌叶片的200ml三口烧瓶中量取制造例1中得到的含有酯保护基的化合物a5g,加入二甲苯50ml,加热到回流温度(约130℃)后,在该温度下进行1.5小时反应。为上述阿齐沙坦甲酯的纯度:70.1%、上述含有酯保护基的化合物a:未检测出。将反应后的溶液减压浓缩,在残渣中加入醋酸乙酯100ml,但残渣中的结晶未溶解。因此,加入异丙醚50ml,在20℃下搅拌12小时。将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到1.4g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦烷基酯的纯度:79.8%)(收率:30.4%)。分子量比阿齐沙坦甲酯大10的杂质为12.1%。将结果汇总于表4。
[比较例4](采用非专利文献1记载的方法的上述阿齐沙坦甲酯的制造)
在直径5cm的具备2片搅拌叶片的200ml三口烧瓶中量取制造例2中得到的含有酯保护基的化合物b5g,加入二甲苯50ml,加热到回流温度(约130℃)后,在该温度下进行2小时反应。为上述阿齐沙坦甲酯的纯度:72.8%、上述含有酯保护基的化合物b:未检测出。将反应后的溶液减压浓缩,在残渣中加入醋酸乙酯50ml,升温到回流温度(约80℃),使浓缩残渣的结晶完全地溶解。将得到的溶液冷却到20℃,在20℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到2.0g的上述阿齐沙坦甲酯的结晶(上述阿齐沙坦甲酯的纯度:88.4%)(收率:50.2%)。分子量比阿齐沙坦甲酯大10的杂质为10.8%。将结果汇总于表4中。另外,将该阿齐沙坦甲酯作为试样,测定xrd,得到图2中所示的x射线衍射图,可知该结晶是在2θ=8.0°、10.4°、12.0°、15.9°、21.4°给予特征峰的化合物。
【表4】
表4
*每1g含有酯保护基的化合物的使用量(ml)
**每lmol含有酯保护基的化合物的使用量(mol)
c.第三本发明涉及的实施例和比较例
[制造例3](对象阿齐沙坦甲酯的合成)
在直径10cm的具备2片搅拌叶片的1000ml四口烧瓶中量取含有酯保护基的化合物(上述式(3)中r1为甲基、r2为乙基的化合物)50g(96.80mmol),加入1-丙醇400ml、三乙胺1.96g(19.36mmol),加热到回流温度(约95℃)后,在该温度下进行14小时反应。将反应后的溶液以20℃/小时的速度冷却到0℃,在0℃下搅拌14小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到38.3g的对象阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:97.3%、上述脱乙基体:0.10%、上述二聚体:0.16%)(收率:84.2%)。另外,未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,将该对象阿齐沙坦甲酯作为试样,测定xrd,得到在2θ=9.9°、10.9°、13.6°、17.2°、23.2°给予特征峰的x射线衍射图。进而,进行dsc测定,能够确认在90.9℃(熔解热量15.790j/g)和186.6℃(熔解热量58.886j/g)具有熔点。
[实施例18](阿齐沙坦甲酯的结晶化)
在直径5cm的具备2片搅拌叶片的200ml三口烧瓶中量取制造例3中得到的对象阿齐沙坦甲酯10g,加入丙酮100ml,加热到回流温度(约57℃),将对象阿齐沙坦甲酯溶解。溶解后,以20℃/小时的速度冷却到0℃,在0℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到8.7g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:99.1%、上述脱乙基体:未检测出、上述二聚体:0.04%)(收率:87.2%)。将结果汇总于表5。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,将该阿齐沙坦甲酯作为试样,测定xrd,获知是如图3中所示那样在2θ=9.2°、15.8°、18.2°、22.1°、25.1°、27.4°给予特征峰的阿齐沙坦甲酯的准结晶。将结果汇总于表6。进而,进行dsc测定,能够确认如图5中所示那样在160.6℃(熔解热量9.635j/g)和189.9℃(熔解热量51.766j/g)具有熔点。
[实施例19](阿齐沙坦甲酯的重结晶)
在直径3.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取制造例3中得到的对象阿齐沙坦甲酯5g,加入丙酮30ml、1-丙醇20ml,加热到回流温度,将对象阿齐沙坦甲酯溶解。溶解后,以20℃/小时的速度冷却到0℃,在0℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到4.3g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:99.0%、上述脱乙基体:未检测出、上述二聚体:0.04%)(收率:86.0%)。将结果汇总于表5。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,将该阿齐沙坦甲酯作为试样,测定xrd,获知是在2θ=9.2°、15.7°、18.2°、22.1°、25.2°、27.4°给予特征峰的阿齐沙坦甲酯的准结晶。将结果汇总于表6。进而,进行dsc测定,能够确认在157.2℃(熔解热量5.249j/g)和189.7℃(熔解热量57.266j/g)具有熔点。
[实施例20](阿齐沙坦甲酯的结晶化)
在实施例19中,除了将1-丙醇变为1-丁醇以外,进行同样的操作。
得到4.2g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:99.1%、上述脱乙基体:未检测出、上述二聚体:0.04%)(收率:85.4%)。将结果汇总于表5。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,xrd的结果也与实施例19的阿齐沙坦甲酯相同。将结果汇总于表6。进而,进行dsc测定,能够确认在158.1℃(熔解热量4.191j/g)和189.5℃(熔解热量58.734j/g)具有熔点。
[实施例21](阿齐沙坦甲酯的结晶化)
在直径3.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取采用下述的比较例5的方法制造的对象阿齐沙坦甲酯4g,加入丙酮40ml,加热到回流温度,将对象阿齐沙坦甲酯溶解。溶解后,以20℃/小时的速度冷却到0℃,在0℃下搅拌14小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到2.8g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:96.6%、上述脱乙基体:未检测出、上述二聚体:0.07%)(收率:70.2%)。将结果汇总于表5。分子量比阿齐沙坦甲酯大10的杂质为1.64%。另外,将该阿齐沙坦甲酯作为试样,测定xrd,获知是在2θ=9.2°、15.7°、18.1°、22.1°、25.3°、27.4°给予特征峰的阿齐沙坦甲酯的准结晶。将结果汇总于表6。进而,进行dsc测定,能够确认在159.4℃(熔解热量7.921j/g)和190.2℃(熔解热量55.015j/g)具有熔点。
[实施例22](阿齐沙坦甲酯的重结晶)
将实施例21中制造的阿齐沙坦甲酯作为对象阿齐沙坦甲酯。
使用实施例21中制造的阿齐沙坦甲酯2g,反复进行与实施例21同样的操作。
得到1.7g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:99.3%、上述脱乙基体:未检测出、上述二聚体:0.02%)(收率:85.9%)。将结果汇总于表5。未能确认分子量比阿齐沙坦甲酯大10的杂质。另外,xrd的结果也与实施例18的阿齐沙坦甲酯相同。将结果汇总于表5。进而,进行dsc测定,能够确认在160.2℃(熔解热量6.424j/g)和190.1℃(熔解热量56.971j/g)具有熔点。
[实施例23](阿齐沙坦的合成)
在直径3.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取实施例19中得到的阿齐沙坦甲酯5g,加入1.25m氢氧化钠水溶液50ml,加热到50℃后,在该温度下进行3小时反应。将反应液冷却到45℃后,在该温度下加入丙酮25ml、醋酸17ml、水17ml,使阿齐沙坦的结晶析出。将反应液以20℃/小时的速度冷却到20℃后,在该温度下搅拌6小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下干燥,得到4.3g的阿齐沙坦的结晶(阿齐沙坦的纯度:99.7%)。
[比较例5](采用非专利文献1记载的方法的上述阿齐沙坦甲酯的制造)
在直径5cm的具备2片搅拌叶片的200ml三口烧瓶中量取含有酯保护基的化合物(上述式(3)中r1为甲基、r2为2-乙基己基的化合物)10g,加入二甲苯100ml,加热到回流温度(约130℃)后,在该温度下进行2小时反应。将反应后的溶液减压浓缩,在残渣中加入醋酸乙酯100ml,升温到回流温度(约80℃),使浓缩残渣的结晶完全地溶解。将得到的溶液冷却到20℃,在20℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到4.1g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:88.3%、上述脱乙基体:0.34%、上述二聚体:0.27%))(收率:50.2%)。分子量比阿齐沙坦甲酯大10的杂质为10.8%。另外,将该阿齐沙坦甲酯作为试样,测定xrd,得到图4中所示的x射线衍射图,获知该结晶为在2θ=8.0°、10.4°、12.0°、15.9°、21.4°给予特征峰的化合物。将结果汇总于表6。进而,进行dsc测定,如图6中所示那样,能够确认在193.6℃(熔解热量76.619j/g)具有熔点。
【表5】
表5
*括弧内的数值表示溶剂的容积比率。
**每1g对象阿齐沙坦烷基酯的使用量(ml)
***分子量比阿齐沙坦甲酯大10的杂质
【表6】
表6
d.其他实施例
[制造例4](阿齐沙坦甲酯的制造)
(酯保护反应)
在直径15cm的具备2片搅拌叶片的2l四口烧瓶中量取上述偕胺肟化合物(r1为甲基的化合物)120g(270.0mmol),加入二氯甲烷840ml、三乙胺33.0g(324.0mmol),边搅拌边冷却到0℃。在得到的溶液中慢慢地滴加将氯甲酸乙酯35.4g(324.0mmol)用二氯甲烷360ml稀释而成的溶液。将全部量滴入后,在0℃下边搅拌2小时边反应。将反应后的溶液升温到20℃,加入水480ml,将有机层抽出。将得到的有机层减压浓缩,作为残渣得到上述含有酯保护基的化合物(r1为甲基、r2为乙基的化合物)。为上述含有酯基的化合物的纯度:96.1%、上述偕胺肟化合物:0.14%。
(环化反应)
在上述减压浓缩后的残渣中加入1-丙醇960ml,加热到回流温度(约95℃)后,在该温度下进行12小时反应。为上述阿齐沙坦甲酯的纯度:91.5%、上述含有酯保护基的化合物:1.8%。将反应后的反应溶液以20℃/小时的速度冷却到0℃,在0℃下搅拌14小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到107.6g的上述阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:97.3%、上述阿齐沙坦甲酯脱乙基体:0.14%、上述阿齐沙坦甲酯二聚体:0.20%)(收率:84.7%)。另外,未能确认分子量比阿齐沙坦甲酯大10的杂质。
[制造例5](阿齐沙坦甲酯的重结晶)
在直径10cm的具备2片搅拌叶片的1000ml三口烧瓶中量取制造例4中得到的阿齐沙坦甲酯90g,加入丙酮900ml,加热到回流温度(约57℃),将阿齐沙坦甲酯溶解。溶解后,以20℃/小时的速度冷却到0℃,在0℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到78.9g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:99.1%、上述脱乙基体:0.02%、上述阿齐沙坦甲酯二聚体:0.07%)(收率:87.7%)。另外,未能确认分子量比阿齐沙坦甲酯大10的杂质。
[制造例6](采用非专利文献1记载的方法的阿齐沙坦甲酯的制造)
(酯保护反应)
在直径7.5cm的具备2片搅拌叶片的500ml四口烧瓶中量取上述偕胺肟化合物(r1为甲基的化合物)30g(67.5mmol),加入二甲基甲酰胺120ml、吡啶5.9g(74.3mmol),边搅拌边冷却到0℃。在得到的溶液中慢慢地滴加氯甲酸-2-乙基己酯13.0g(67.5mmol)。将全部量滴入后,在0℃下边搅拌1小时边反应。将反应后的溶液升温到20℃,加入醋酸乙酯270ml、水60ml,将有机层抽出。将得到的有机层进一步用水60ml清洗后,将有机层减压浓缩,作为残渣得到上述含有酯保护基的化合物(r1为甲基、r2为2-乙基己基的化合物)。为上述含有酯基的化合物的纯度:94.5%、上述偕胺肟化合物:1.26%。
(环化反应)
在上述减压浓缩后的残渣中加入二甲苯420ml,加热到回流温度(约130℃)后,在该温度下进行2小时反应。将反应后的溶液减压浓缩,在残渣中加入醋酸乙酯420ml,升温到回流温度(约80℃),将浓缩残渣的结晶完全地溶解。将得到的溶液冷却到20℃,在20℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到15.9g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:88.3%、上述脱乙基体:0.36%、上述阿齐沙坦甲酯二聚体:0.27%))(收率:50.1%)。分子量比阿齐沙坦甲酯大10的杂质为10.8%。
[实施例24](阿齐沙坦的制造;有活性炭处理)
(水解)
在直径3.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取制造例5中得到的阿齐沙坦甲酯5g,加入1.25m氢氧化钠水溶液40ml,加热到70℃后,在该温度下进行2小时反应。为反应后的粗阿齐沙坦溶液的阿齐沙坦纯度:99.61%、阿齐沙坦脱乙基体:0.06%、阿齐沙坦二聚体:0.08%。将反应后的粗阿齐沙坦溶液的阿齐沙坦纯度和杂质量的结果示于表7中。
(活性炭处理)
将水解反应完成后的溶液冷却到30℃后,加入精制白鹭(大阪瓦斯化学制造、比表面积:1430m2/g、累计细孔容积:1.17ml/g)0.24g,在20~30℃下进行1小时搅拌。为活性炭处理后的溶液的阿齐沙坦纯度:99.85%、阿齐沙坦脱乙基体:0.05%、阿齐沙坦二聚体:0.01%。
(活性炭的除去和精制)
接下来,减压过滤,将精制白鹭除去,将得到的滤液加热到40℃后,在该温度下加入丙酮25ml、醋酸17ml、水17ml,使阿齐沙坦的结晶析出。将反应液以20℃/小时的速度冷却到20℃后,在该温度下搅拌6小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下干燥,得到4.6g的阿齐沙坦的结晶(收率:95.6%)。为上述阿齐沙坦的纯度:99.89%、阿齐沙坦脱乙基体:0.03%、阿齐沙坦二聚体:未检测出、不明杂质:未检测出。将结果示于表8中。
[实施例25~26]
(水解)
除了使用表7中所示的制造例的阿齐沙坦烷基酯作为原料以外,与实施例24同样地进行水解反应。将水解反应后的粗阿齐沙坦溶液的纯度和杂质量的测定结果示于表7中。
((活性炭处理)、(活性炭的除去和精制))
另外,对水解后的溶液,采用与实施例24同样的方法进行(活性炭处理)、(活性炭的除去和精制),得到阿齐沙坦的结晶。对于得到的阿齐沙坦的结晶,同样地进行纯度和杂质量的测定。将其结果示于表8中。
[实施例27~28]
(水解)
与实施例24同样地进行水解反应。将反应后的粗阿齐沙坦溶液的纯度和杂质量的测定结果示于表7中。
(活性炭处理)
另外,除了如表8中所示那样改变活性炭处理时的活性炭的使用量以外,与实施例24同样地进行处理。
(活性炭的除去和精制)
关于活性炭的除去和精制,进行与实施例24同样的操作。对于得到的阿齐沙坦的结晶进行纯度和杂质量的测定。将其结果示于表8中。
[实施例29~32]
(水解)
与实施例24同样地进行水解反应。将反应后的粗阿齐沙坦溶液的纯度和杂质量的测定结果示于表7中。
(活性炭处理)
另外,除了如表8中所示那样改变活性炭处理时的活性炭的种类、使用量以外,与实施例24同样地进行处理。在表9中汇总了实施例中使用的活性炭的特性(比表面积、累计细孔容积)。
(活性炭的除去和精制)
关于活性炭的除去和精制,进行与实施例24同样的操作。对于得到的阿齐沙坦的结晶进行纯度和杂质量的测定。将其结果示于表8中。
[参考例1](阿齐沙坦的制造;无活性炭处理)
在直径3.5cm的具备2片搅拌叶片的100ml三口烧瓶中量取制造例4中得到的阿齐沙坦甲酯5g,加入1.25m氢氧化钠水溶液40ml,加热到70℃后,在该温度下进行2小时反应。为反应后的粗阿齐沙坦溶液的阿齐沙坦纯度:98.98%、阿齐沙坦脱乙基体:0.20%、阿齐沙坦二聚体:0.22%。将反应后的粗阿齐沙坦溶液的阿齐沙坦纯度和杂质量的结果示于表7中。
接下来,将得到的反应液冷却到45℃后,在该温度下加入丙酮25ml、醋酸17ml、水17ml,使阿齐沙坦的结晶析出。将反应液以20℃/小时的速度冷却到20℃后,在该温度下搅拌6小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下干燥,得到4.7g的阿齐沙坦的结晶(收率:96.5%)。为上述阿齐沙坦的纯度:99.17%、阿齐沙坦脱乙基体:0.15%、阿齐沙坦二聚体:0.20%、不明杂质:未检测出。将结果示于表8中。
[参考例2~3](阿齐沙坦的制造;无活性炭处理)
除了使用表7中所示的制造例的阿齐沙坦烷基酯作为原料以外,与参考例1同样地进行水解反应。将反应后的粗阿齐沙坦溶液的纯度和杂质量的测定结果示于表8中。
另外,从采用与参考例1同样的方法得到的反应液中将阿齐沙坦的结晶取出。对于得到的阿齐沙坦的结晶,同样地进行纯度和杂质量的测定。将其结果示于表8中。
【表7】
表7
*分子量比阿齐沙坦甲酯大10的杂质
【表8】
表8
*分子量比阿齐沙坦甲酯大10的杂质
**每1g阿齐沙坦的使用量(g)
【表9】
表9
e.其他实施例
[实施例33]
(第一工序:羟基脒基化)
在直径10cm的具备2片搅拌叶片的1l四口烧瓶中量取上述腈化合物70g(170.1mmol),加入1-丙醇700ml、三乙胺5.16g(51.0mmol)、市售的50质量%羟基胺水溶液56.2g(850.5mmol),加热到回流温度(约92℃)后,在该温度下进行13小时反应。为上述偕胺肟化合物的纯度:83.5%、上述酰胺体:2.5%、上述腈化合物:2.4%、上述偕胺肟脱乙基体:7.6%、上述酰胺脱乙基体:0.6%、上述腈脱乙基体:0.01%。
将反应后的溶液以20℃/小时的速度冷却到20℃,在20℃下搅拌13小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在50℃下干燥,得到60.9g的上述偕胺肟化合物的结晶(上述偕胺肟化合物的纯度:96.6%、上述酰胺体:0.6%、上述腈化合物:0.1%、上述偕胺肟脱乙基体:0.6%、上述酰胺脱乙基体:0.1%、上述腈脱乙基体:未检测出)(收率:80.6%)。
(第二工序:环化)
在直径10cm的具备2片搅拌叶片的1l四口烧瓶中量取上述偕胺肟化合物60g(135.0mmol),加入二氯甲烷420ml、三乙胺16.4g(162.0mmol),边搅拌边冷却到0℃。在得到的溶液中缓慢地滴加将氯甲酸乙酯17.6g(162.0mmol)用二氯甲烷180ml稀释而成的溶液。将全部量滴入后,在0℃下边搅拌2小时边反应。将反应后的溶液升温到20℃,加入水240ml,将有机层抽出。将得到的有机层用10%食盐水240ml清洗后,将有机层中的溶剂在减压下浓缩,作为残渣得到含有酯保护基的化合物(含有酯保护基的化合物纯度:96.4%)。
在得到的残渣中加入1-丙醇480ml,加热到回流温度(约92℃)后,在该温度下进行12小时反应。为上述阿齐沙坦甲酯的纯度:91.7%、上述含有酯保护基的化合物:1.7%。将反应后的反应溶液以20℃/小时的速度冷却到4℃,在4℃下搅拌12小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到54.0g的对象阿齐沙坦甲酯的结晶(对象阿齐沙坦烷基酯的纯度:97.5%、上述阿齐沙坦甲酯脱乙基体:0.09%、上述阿齐沙坦甲酯二聚体:0.15%)(收率:85.0%)。未能确认分子量比阿齐沙坦甲酯大10的杂质。
(第三工序:al-02精制)
在直径10cm的具备2片搅拌叶片的1l四口烧瓶中量取对象阿齐沙坦甲酯50g,加入丙酮500ml,加热到回流温度(约57℃),将对象阿齐沙坦甲酯溶解。溶解后,以20℃/小时的速度冷却到0℃,在0℃下搅拌16小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下减压干燥,得到44.0g的阿齐沙坦甲酯的结晶(阿齐沙坦甲酯的纯度:98.9%、上述阿齐沙坦甲酯脱乙基体:未检测出、上述阿齐沙坦甲酯二聚体:0.04%)(收率:88.1%)。
(第四工序:水解)
在直径10cm的具备2片搅拌叶片的1l四口烧瓶中量取上述阿齐沙坦甲酯40g,加入1.25m氢氧化钠水溶液260ml,加热到70℃后,在该温度下进行2小时反应。为反应后的粗阿齐沙坦溶液的阿齐沙坦纯度:99.69%、阿齐沙坦脱乙基体:0.05%、阿齐沙坦二聚体:0.04%。
将水解反应完成后的溶液冷却到30℃后,加入精制白鹭(大阪瓦斯化学制造、比表面积:1430m2/g、累计细孔容积:1.17ml/g)2.0g,在20~30℃下进行1小时搅拌。为活性炭处理后的溶液的阿齐沙坦纯度:99.85%、阿齐沙坦脱乙基体:0.04%、阿齐沙坦二聚体:未检测出。
接下来,减压过滤,将精制白鹭除去,将得到的滤液加热到40℃后,在该温度下加入甲醇260ml、醋酸29.2ml,使阿齐沙坦的结晶析出。将反应液以20℃/小时的速度冷却到20℃后,在该温度下搅拌6小时。接下来,将得到的浆液减压过滤,分取析出的结晶,在40℃下干燥,得到38.2g的阿齐沙坦(上述阿齐沙坦的纯度:99.88%、阿齐沙坦脱乙基体:0.02%、阿齐沙坦二聚体:未检测出)的结晶(收率:95.5%)。
(第五工序:azl精制)
在直径10cm的具备2片搅拌叶片的1l四口烧瓶中量取上述阿齐沙坦35g,装入二甲基甲酰胺70ml,在30℃下加热溶解。在得到的阿齐沙坦溶液中加入醋酸乙酯350ml后,冷却到5℃,搅拌15小时。接下来,减压过滤,分取析出的结晶,在50℃下干燥,得到34.7g的阿齐沙坦的结晶(收率:99.2%)。将该阿齐沙坦作为试样,测定xrd,得到图7中所示的x射线衍射图,获知该结晶为具有在2θ=9.41°、11.52°、13.33°、14.81°、26.01°给予特征峰的新型结晶结构的化合物。另外,采用dsc测定得到的熔点为127℃。
- 还没有人留言评论。精彩留言会获得点赞!