一种刚性脂环含氟结构的二酐化合物及其制备方法与应用与流程
本发明属于新材料的技术领域,具体涉及一种新型刚性脂环含氟结构的二酐化合物及其合成方法与应用。
背景技术
近年来,随着显示行业更新换代的加速,大尺寸柔性有机发光二极管(oled)的需求将成爆发式的增长,也带动了柔性基板的发展和需求。目前用的较多的柔性基板主要是:聚对苯二甲酸乙二酯(pet),聚碳酸酯(pc),聚萘二甲酸乙二醇酯(pen)等工程塑料。以上几种工程塑料具有较高的透过率(>85%),但是均具有一个致命的缺点:耐热性能差(tg<120℃),耐溶剂性较差。制备大尺寸柔性oled有工序需要使用高温处理,显然上述几种工程塑料不能满足这一条件。因此,具有优异的耐热性能,耐溶剂性能的柔性透明聚酰亚胺薄膜(pi)成了理想的柔性oled基板的最佳选择。
传统的芳香族聚酰亚胺由芳香族具有强吸电子能力的二酐单体与强给电子能力的二胺单体共聚制备,聚合物主链堆积紧密,芳环间存在很强的共轭作用,使得聚酰亚胺的分子链内和分子链间存在很强的电荷转移络合物效应。环间强的分子链间相互作用在赋予pis上述优良性能的同时,也导致了芳香族pis存在溶解加工性差、光学透过性低等缺陷,影响了pis在显示领域的应用。
在题为“colorlesspolyimidesderivedfrom2r,5r,7s,10s-naphthanetetracarboxylicdianhydride”的文献中,作者合成了新型脂环族二酐2r,5r,7s,10s-萘四甲酸二酐(hntda)。他们发现,对比由氢化均苯四甲酸二酐hpmda制备的pis,基于hntda的pis展示出优异的热力学性能,同时,刚性稠环的萘四羧基二酰亚胺部分显著削弱了分子间和分子内相互作用,从而提高薄膜的光学性能,同时维持了良好的热稳定性。但是,薄膜的cte值大于40ppmk-1,cte值大,那么pi薄膜在受热时,会发生翘曲、开裂或者脱层,因此限制了pi薄膜的应用。
在题目“flexibleqledandopvbasedontransparentpolyimidesubstratewithrigidalicyclicasymmetricisomer”的文献中,作者将含有刚性半脂环结构的茚满二胺引入到分子主链上,并与五种不同的商业化二酐聚合得到了一系列pis。性能测试证明,引入这种刚性脂环结构,一方面有效地抑制了ctc效应的形成,提高了薄膜的光学透过率。另一方面,脂环结构直接与苯环相连,避免了传统全脂环结构耐热性差的问题。此外,将cpi-1作为柔性基底,制备了相应的opv与qled器件,表现出较好的器件效率。脂环结构对聚酰亚胺光学性能的改善效果还是非常明显的,但是打断共轭,削弱分子链间相互作用与ctc效应,引起了pis的耐热性能的降低,这一点从上述报道的结构可以看出来。因此,仅仅通过引入脂环结构对聚酰亚胺进行改性还较难达到显示领域对柔性基板性能的要求。
在公开号为cn105131286a的中国专利中,聚酰亚胺是以芳香族二酐化合物3,3',4,4'-联苯四酸二酐(sbpda)或脂环族二酐化合物1,2,3,4-环丁烷四酸二酐(cbda)与芳香族二胺化合物为原料,通过热亚胺化方法制备的。通过调整sbpda和cbda或者1,3-二甲基-1,2,3,4-环丁烷四酸二酐(mcbda)的比例,可以实现对所述的聚酰亚胺薄膜基板耐热稳定性和光学透明性的调控,但是该方法制备的聚酰亚胺薄膜玻璃化转变温度tg小于350℃。
4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)具有sp3杂化季碳结构,这种sp3杂化结构增加了分子扭曲的程度。此外,由cf3基团赋予的空间位阻效应可以很好地破坏分子链的堆砌程度和堆砌密度,从而减少了分子链内和分子链间的相互作用力。因此,聚合物的电荷转移络合物(ctc)的形成起到很大的抑制作用,因此,相关薄膜的颜色可以变得很浅,使得聚酰亚胺更适合于某些特定领域的应用。然而,6fda分子的刚性较弱,导致聚酰亚胺的线性热膨胀系数(cte)值很大。1,2,4,5-环己烷四羧酸二酐(hpmda)作为脂族二酐单体可以显著降低ctc效应并改善pi透射率。然而,hpmda分子的空间异构性会影响聚合反应的活性。另外,基于hpmda的聚酰亚胺显示出良好的热稳定性,弱抗氧化能力,最重要的缺点是在高温下容易发生黄化现象。
在柔性显示领域,理想的基底需要其玻璃化转变温度高于400℃,cte值低于20ppmk-1。本发明综合了6fda和hpmda的优点,设计并合成了含有刚性半脂环,三氟甲基和氟原子取代的二酐9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐(8fda)。
技术实现要素:
本发明解决了现有技术中含有二酐单体的聚合物玻璃化转变温度低、线性热膨胀系数高以及热稳定性差的技术问题,提供了一种刚性脂环含氟结构的二酐化合物及其制备方法与应用。
按照本发明的第一方面,提供了一种刚性脂环含氟结构的二酐化合物,该化合物的结构式如式ⅰ所示:
所述r为f、cl、cf3或
按照本发明的另一方面,提供了一种刚性脂环含氟结构的二酐化合物的制备方法,包含以下步骤:
(1)向2,3,6,7-四甲基蒽-9,10-二酮溶液中加入三甲基(三氟甲基)硅烷或三乙基(三氟甲基)硅烷,冷却后,加入催化剂a,充分混匀后,将反应混合物升温至30℃~50℃,反应6h~24h,使2,3,6,7-四甲基蒽-9,10-二酮发生亲核加成反应,得到((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷);
(2)将步骤(1)得到的((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)溶解后置于酸性环境中,反应时间为0.5h~5h,反应温度为25℃~80℃,使((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)上的三甲基硅氧烷转变成羟基,得到2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇;
将步骤(2)得到的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇溶解后,冰浴条件下,逐滴加入二乙胺基三氟化硫或双(2-甲氧基乙基)氨基三氟化硫,反应12h~15h,得到9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽;
或者步骤(3)为以下技术方案:将步骤(2)得到的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇溶解后,加入乙酰氯,在70℃-80℃的条件下,反应8h-12h,得到9,10-二氯-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽;
或者步骤(3)为以下技术方案:将步骤(2)得到的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇溶解后,加入卤化氢和三氟卤甲烷,反应15h-30h,得到2,3,6,7-四甲基-9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽;
或者步骤(3)为以下技术方案:将步骤(2)得到的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇溶解后,第一步加入三溴化磷,或加入溴化氢和催化剂b,在40℃-60℃的条件下,反应12h-24h,使所述2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇上的羟基转变成溴原子,得到9,10-二溴-2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽;第二步加入苯基溴化镁和催化剂c,或加入苯硼酸和催化剂d,在75℃-90℃条件下,反应8h-15h,使9,10-二溴-2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽上的溴原子被苯基取代,得到2,3,6,7,-四甲基-9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽;
(4)将步骤(3)得到的9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽、9,10-二氯-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽、2,3,6,7-四甲基-9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽或2,3,6,7,-四甲基-9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽溶解后,加入氧化剂,在90℃-110℃条件下,反应12h-15h后,抽滤,旋干滤液,产物溶解后并酸化,得到9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸、9,10-二氯-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸、9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸或9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸;
(5)将步骤(4)得到的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸、9,10-二氯-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸、9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸或9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸脱水成酐,得到9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐、9,10-二氯-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐、9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐或9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐。
优选地,步骤(1)所述催化剂a为氟化铯、四丁基氟化铵或三(二甲氨基甲基)锍二氟三甲基氨酸盐;步骤(3)所述催化剂b为浓硫酸;步骤(3)所述催化剂c为1,3-双(二苯基膦丙烷)二氯化镍,或者为醋酸钯和三苯基膦的混合物;步骤(3)所述催化剂d为四(三苯基膦)钯和碳酸钾的混合物,或者为钯和碳酸钾的混合物,或者为四氯钯酸钠和碳酸钾的混合物;步骤(4)所述氧化剂为高锰酸钾或三氧化铬。
优选地,步骤(3)所述卤化氢为溴化氢、碘化氢或氯化氢;步骤(3)所述三氟卤甲烷为三氟溴甲烷、三氟碘甲烷或三氟氯甲烷。
优选地,步骤(1)所述2,3,6,7-四甲基蒽-9,10-二酮、三甲基(三氟甲基)硅烷与催化剂a的物质的量之比为1:(2~3.5):(0.01~0.05)。
优选地,步骤(3)中所述2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇与二乙胺基三氟化硫,或者与双(2-甲氧基乙基)氨基三氟化硫的物质的量之比为1:(2~3.5)。
优选地,步骤(4)中所述9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽、9,10-二氯-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽、2,3,6,7-四甲基-9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽或2,3,6,7,-四甲基-9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽与氧化剂的物质的量之比为1:(10~12)。
按照本发明的另一方面,提供了所述的刚性脂环含氟结构的二酐化合物应用于制备聚酰亚胺材料的应用。
按照本发明的另一方面,提供了一种聚酰亚胺膜的制备方法,包含以下步骤:
(1)将二胺溶解后,加入所述的刚性脂环含氟结构的二酐化合物,在25℃~35℃条件下反应10h~30h,得到聚酰胺酸溶液;
(2)将步骤(1)得到的聚酰胺酸溶液均匀分散在基底表面,加热使聚酰胺酸溶液中的溶剂挥发;继续加热使所述聚酰胺酸发生脱水环化反应,得到聚酰亚胺膜。
按照本发明的另一方面,提供了由所述方法制备得到的聚酰亚胺膜。
总体而言,通过本发明所构思的以上技术方案与现有技术相比,主要具备以下的技术优点:
(1)本发明提供的刚性脂环含氟结构的二酐化合物中1,4-环己二烯这一半脂环链段可以有效地破坏聚合物链的共轭程度,电荷转移络合物效应得到有效地抑制,其光学性能随之提升。同时含氟基团和脂环结构的空间效应可以提高聚合物链段的自由体积(ffv),提高基于刚性脂环含氟结构的二酐化合物的聚酰亚胺膜的溶解加工性能。此外,刚性脂环结构可以保证分子链的刚性,降低链段的运动能力,从而提高玻璃化转变温度和热稳定性。刚性结构可以降低基于本发明所述的含有刚性脂环含氟结构的二酐化合物的聚酰亚胺的线性热膨胀系数。因此能够用于制备透明耐高温低线性热膨胀系数的聚酰亚胺薄膜。本发明所述的含有刚性脂环含氟结构的二酐化合物结构简单,对透明聚酰亚胺薄膜的开发具有广泛应用价值。
(2)本发明的刚性脂环含氟结构二酐中具有刚性脂环和含氟结构,在制备聚合物材料中可以提高材料的透光率,降低材料的介电常数和吸水率,保持良好的热稳定性与尺寸稳定性,因此适用于制备柔性透明聚酰亚胺薄膜。
(3)本发明制备刚性脂环含氟结构的二酐化合物的方法简单,反应条件温和,反应原料来源方便,成本低,有机溶剂使用种类少,减少环境污染。
(4)本发明综合了4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)和1,2,4,5-环己烷四羧酸二酐(hpmda)的优点,合成了含有刚性半脂环,三氟甲基和氟原子取代的二酐9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐。该物质玻璃化转变温度高,线性热膨胀系数低,是制备聚酰亚胺膜的理想二酐单体材料。
附图说明
图1(a)是实施例1第一步合成的((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)的核磁氢谱图;图1(b)是实施例1第一步合成的((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)的碳谱图。
图2(a)是实施例1第二步合成的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇的核磁氢谱图;图2(b)是实施例1第二步合成的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇的碳谱图。
图3(a)是实施例1第三步合成的9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽的核磁氢谱图;图3(b)是实施例1第三步合成的9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽的碳谱图。
图4(a)是实施例1第四步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸的核磁氢谱图;图4(b)是实施例1第四步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸的碳谱图。
图5(a)是实施例1第五步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的核磁氢谱图;图5(b)是实施例1第五步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的碳谱图。
图6(a)、图6(b)、图6(c)、图6(d)和图6(e)分别是实施例1第一步到第五步合成的各步骤产物的的红外谱图。
图7(a)为实施例1第一步产物的单晶x射线衍射空间结构图;图7(b)为实施例1第二步产物的单晶x射线衍射空间结构图;图7(c)为实施例1第三步产物的单晶x射线衍射空间结构图;图7(d)和图7(e)为实施例1第五步产物升华后得到的单晶x射线衍射空间结构图;图7(f)为实施例1第五步产物在甲苯中重结晶得到的单晶x射线衍射空间结构图。
图8是本发明提供的化合物9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的结构式。
图9为化合物9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐制备过程示意图。
图10为pi膜的dma曲线。
图11为pi膜的tga曲线。
图12为pi膜的透过率曲线。
图13为pi膜的tma曲线。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
实施例1
一种含有刚性脂环含氟结构的二酐化合物9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐,简称为8fda,制备过程示意图如图9所示。
步骤一:将2,3,6,7-四甲基蒽-9,10-二酮(40.00g,151.33mmol,1.00当量)悬浮于thf(400ml)在r.t.并加入三甲基(三氟甲基)硅烷(49.21ml,332.93mmol,2.20当量)。将悬浮液冷却至0℃,加入csf(459.74mg,3.03mmol,0.02当量)。搅拌10分钟后,将反应混合物升温至室温,并搅拌持续1.5小时。过滤反应混合物,剩余的黄色固体用乙醚(25ml)冲洗。粗产物通过柱色谱分离(正戊烷至正戊烷/mtbe15:1)上反应,得到化合物((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)(16.6g,30.27mmol,20%)为白色固体;1hnmr(400mhz,cdcl3)δ7.67(s,1h),2.38(d,j=3.3hz,3h),-0.10(d,j=15.5hz,5h).
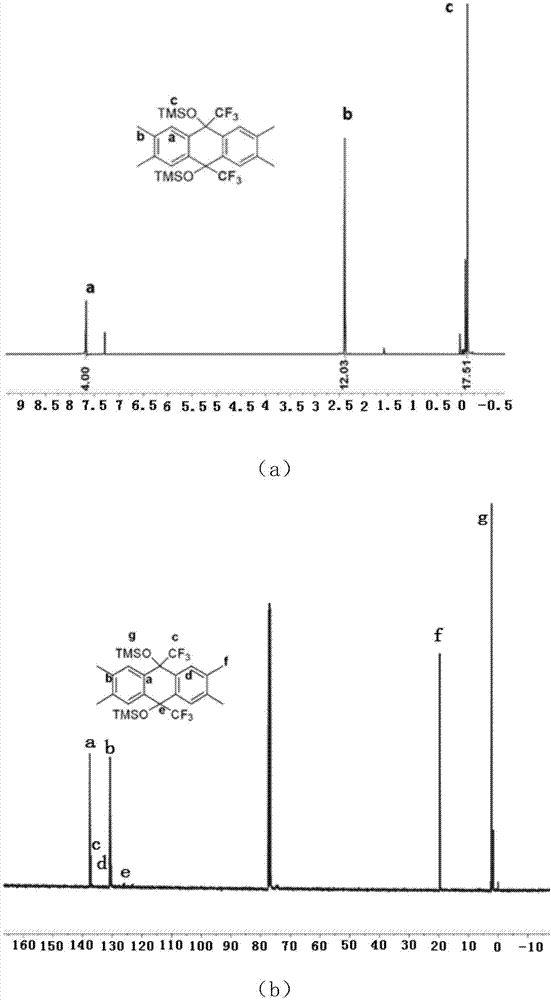
图1(a)是实施例1第一步合成的((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)的核磁氢谱图;图1(b)是实施例1第一步合成的((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)的碳谱图;由图1可以得知,1hnmr(600mhz,cdcl3)δ7.67(s,4h),2.38(d,j=3.3hz,12h),-0.12(d,j=14.4hz,18h)。13cnmr(150mhz,cdcl3)137.56,137.22,131.04-130.69,130.40,125.87,19.67,2.19,1.99-1.54。19fnmr(565mhz,cdcl3)δ-78.39(s,6f),因此步骤一得到的物质是(2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)
步骤二:将((2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二基)双(氧基))双(三甲基硅烷)(双tms)(20.00g,36.4mmol,1.00当量)单体溶于80mlthf中,加热至回流温度后,慢慢加入浓盐酸,立即变浑浊,继续加热反应3小时。监控反应进程,待其完全反应后,冷却至室温,抽滤,滤饼用水洗三次(30ml/次),再用正己烷洗三次(30ml/次)。产物真空干燥。得到无色晶体2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇,产率95%;1hnmr(400mhz,dmso)δ7.68(d,j=1.2hz,1h),7.26(s,1h),2.31(s,3h).
图2(a)是实施例1第二步合成的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇的核磁氢谱图;图2(b)是实施例1第二步合成的2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇的碳谱图;由图2可以得知,1hnmr(600mhz,dmso-d6)δ7.69(d,j=1.2hz,4h),7.32(s,2h),2.32(s,12h)。13cnmr(100mhz,dmso-d6)δ137.26(s),131.90(s),129.46(s),72.11(t,j=26.6hz),19.84(s)。19fnmr(565mhz,dmso-d6)δ-76.69(s,6f),因此步骤二得到的物质是2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇。
步骤三:将2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽-9,10-二醇(10g,24.7mmol,1.00当量)溶于30ml无水thf中,无水无氧条件下冷却至-78℃,慢慢滴加二乙胺基三氟化硫(dast)(7.23ml,54.4mmol,2.20当量),滴加完毕后继续反应12小时。点板监控反应进程。完全反应后,加入碳酸氢钠淬灭过量的dast。淬灭完全后,然后将其过滤,滤液旋干,柱分离。产物用正己烷重结晶。产物真空干燥。得到9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽,产率85%;1hnmr(600mhz,cdcl3)δ7.68(d,j=26.0hz,1h),2.39(s,3h).
图3(a)是实施例1第三步合成的9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽的核磁氢谱图,图3(b)是实施例1第三步合成的9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽的碳谱图;由图3可以得知,1hnmr,cdcl3)δ7.68(d,j=26.0hz,4h),2.39(s,12h)。13cnmr(100mhz,cdcl3)δ139.87,139.61,129.18,128.90,127.44-126.52,
19.90。19fnmr(565mhz,cdcl3)δ-77.80(d,2f),-77.98(m,4f),-157.55(m,2f),因此步骤三得到的物质是9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽。
步骤四:将9,10-二氟-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽(5g,12.2mmol,1.00当量)溶于30ml吡啶和水体积比1:1的溶剂中,加热至回流温度。再称量(19.34g,0.122mmol,10.00当量高锰酸钾),在1小时内分批加入反应瓶中。反应结束后,趁热抽滤,旋干滤液。再用热水溶解产物,加入浓盐酸酸化,抽滤得到四酸产物。乙酸重结晶。产物真空干燥,得到9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸,产率85%;1hnmr(600mhz,dmso)δ13.98(s,1h),8.53–7.94(m,1h).
图4(a)是实施例1第四步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸的核磁氢谱图;图4(b)是实施例1第四步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸的碳谱图;由图4可以得知,1hnmr(600mhz,dmso-d6)δ13.98(s,4h),8.53-7.94(m,4h)。13cnmr(100mhz,dmso-d6)δ167.05,166.97,136.90,136.79-136.53,130.00,129.40。19fnmr(565mhz,dmso-d6)δ-76.31(m,6f),-142.23(m,2f),因此步骤四得到的物质是9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸。
步骤五:将9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸(5g,9.47mmol,1.00当量)溶于30ml乙酸酐溶剂中,加热至回流温度。反应10小时。点板监控反应进程。完全反应后,旋干,真空干燥得到9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐,产率85%。1hnmr(600mhz,dmso)δ8.83–8.63(m,1h),8.34(dd,j=43.5,25.8hz,1h).图8是本发明制备的化合物9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的结构式。
图5(a)是实施例1第五步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的核磁氢谱图;图5(b)是实施例1第五步合成的9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的碳谱图;由图5可以得知,1hnmr(600mhz,dmso-d6)δ8.83-8.63(m,4h)。13cnmr(100mhz,dmso-d6)δ161.87,161.80,135.42,134.13,125.73,125.64。19fnmr(565mhz,dmso-d6)δ-75.64(m,6f),-139.35(s,2f),因此步骤五得到的物质是9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐。
图6(a)、图6(b)、图6(c)、图6(d)和图6(e)分别是本实施例步骤一、步骤二、步骤三、步骤四和步骤五合成的产物的红外谱图。
图7(a)为实施例1第一步产物的单晶x射线衍射空间结构图;图7(b)为实施例1第二步产物的单晶x射线衍射空间结构图;图7(c)为实施例1第三步产物的单晶x射线衍射空间结构图;图7(d)和图7(e)为实施例1第五步产物升华后得到的单晶x射线衍射空间结构图;图7(f)为实施例1第五步产物在甲苯中重结晶得到的单晶x射线衍射空间结构图。从图7中可以得知,各个步骤中得到的是相应的产物。
实施例2
一种含有刚性脂环含氟结构的二酐化合物9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的制备方法,含有以下步骤:
步骤一和步骤二按照实施例1中的方法进行,得到2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽-9,10-二醇。
步骤三:将2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽-9,10-二醇(200mg,0.5mmol,1当量)溶于10ml无水thf中,搅拌使其溶解,再注射pbr3(0.23ml,3.45mg,0.013mmol,0.026当量),避光条件下,室温下搅拌10~30min后升温至50℃,反应24h,冷却。粗产物通过柱色谱分离(正戊烷至正戊烷/mtbe15:1)上反应,得到化合物9,10-二溴-2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽。产率95%。
步骤四:将9,10-二溴-2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽(40mg,0.075mmol,1当量)溶于10ml无水thf中,加入1,3-双(二苯基膦丙烷)二氯化镍(2.03mg,0.0038mmol,0.05当量),再滴加苯基溴化镁(0.2ml,34mg,0.188mmol,2.5当量),在室温下反应一小时后,再升温到回流温度,回流20-24h。冷却至室温,用无水乙醚萃取,再通过柱色谱分离(正己烷)上反应,得到2,3,6,7-四甲基-9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽,产率50%。
步骤五:将2,3,6,7-四甲基-9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽(5g,9.5mmol,1.00当量)溶于30ml吡啶和水体积比1:1的溶剂中,加热至回流温度。再称量(15.01g,95mmol,10.00当量高锰酸钾),在1小时内分批加入反应瓶中。反应结束后,趁热抽滤,旋干滤液。再用热水溶解产物,加入浓盐酸酸化,抽滤得到四酸产物。乙酸重结晶。产物真空干燥,得到9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸,产率85%。
步骤六:将9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸(5g,7.76mmol,1.00当量)溶于30ml乙酸酐溶剂中,加热至回流温度。反应10小时。点板监控反应进程。完全反应后,旋干,真空干燥得到9,10-二苯基-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐,产率85%。
实施例3
一种含有刚性脂环含氟结构的二酐化合物9,10-二氯-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的制备方法,包含以下步骤:
步骤一和步骤二按照实施例1中的方法进行,得到2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽-9,10-二醇。
步骤三:将2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽-9,10-二醇(200mg,0.5mmol,1当量)溶于15ml甲苯中,再加入乙酰氯(98.125mg,1.25mmol,2.5当量),室温下搅拌10min,再升温至75℃,反应8h。冷却至室温,通过柱色谱分离得到产物9,10-二氯-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽,产率65%。
步骤四:将9,10-二氯-2,3,6,7-四甲基-9,10-二(三氟甲基)-9,10-二氢蒽(5g,11.3mmol,1.00当量)溶于30ml吡啶和水体积比1:1的溶剂中,加热至回流温度。再称量(21.4g,135.6mmol,12.00当量高锰酸钾),在1小时内分批加入反应瓶中。反应结束后,趁热抽滤,旋干滤液。再用热水溶解产物,加入浓盐酸酸化,抽滤得到四酸产物。乙酸重结晶。产物真空干燥,得到9,10-二氯-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸,产率85%。
步骤五:将9,10-二氯-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸(5g,8.9mmol,1.00当量)溶于30ml乙酸酐溶剂中,加热至回流温度。反应10小时。点板监控反应进程。完全反应后,旋干,真空干燥得到9,10-二氯-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐,产率85%。
实施例4
一种含有刚性脂环含氟结构的二酐化合物9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐的制备方法,包含以下步骤:
步骤一和步骤二按照实施例1中的方法进行,得到2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽-9,10-二醇。
步骤三:氮气保护下,将2,3,6,7-四甲基-9,10-双(三氟甲基)-9,10-二氢蒽-9,10-二醇(200mg,0.5mmol,1当量)溶于10mlthf中,加入碘化氢(0.17ml,190mg,1.5mmol,3当量),加入三氟碘甲烷(293.85mg,1.5mmol,3当量),在室温下反应24h,通过柱分离,得到2,3,6,7-四甲基-9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽。
步骤四:将2,3,6,7-四甲基-9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽(5g,9.84mmol,1.00当量)溶于30ml吡啶和水体积比1:1的溶剂中,加热至回流温度。再称量(18.66g,118.08mmol,12.00当量高锰酸钾),在1小时内分批加入反应瓶中。反应结束后,趁热抽滤,旋干滤液。再用热水溶解产物,加入浓盐酸酸化,抽滤得到四酸产物。乙酸重结晶。产物真空干燥,得到9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸,产率85%。
步骤五:将9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽-2,3,6,7-四甲酸(5g,7.96mmol,1.00当量)溶于30ml乙酸酐溶剂中,加热至回流温度。反应10小时。点板监控反应进程。完全反应后,旋干,真空干燥得到9,9’,10,10’-四(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐,产率80%。
实施例5
运用实施例1制备的二酐物质9,10-二氟-9,10-双(三氟甲基)-9,10-二氢蒽-2,3,6,7-四羧酸二酐来合成聚酰亚胺,包括以下步骤:
步骤一:将ppda(3mmol)用适量的干燥的nmp溶剂,溶解于50ml烧瓶中的中,然后将8fda,(3mmol)慢慢地,一次性加入到上述溶解了二胺的溶液中。将反应物在25℃条件下搅拌24小时,得到聚酰胺酸(paa)溶液。
步骤二:将聚酰胺酸溶液倒在一块经过ito玻璃清洗剂,去离子水彻底清洗,干燥的洁净的玻璃上面,然后利用流延法将paa均匀的分散在玻璃表面。接下来将流延好的paa玻璃至于预热到80℃的烘箱中。在80℃条件下加热2小时以缓慢释放溶剂,然后按照以下温度程序升温:在100℃下加热1小时,150℃加热1小时,200℃加热1小时,250℃加热1小时,280℃加热1小时。待烘箱温度自然冷却至室温后,在薄膜边缘用手术刀画一条线,将其放入去离子水中浸泡后,从玻璃表面剥离下来。得到热亚胺化的聚酰亚胺薄膜。
实施例6
将实施例5得到的聚酰亚胺薄膜通过热重分析(tga),动态机械分析(dma)和热机械分析(tma)评价pi膜的热性能和机械性能。用perkin-elmertga-2在氮气流下以10℃/分钟的加热速率进行热重分析(tga)。用dmaq800v20.22build41进行动态力学分析,使用拉伸模式,频率为1hz。用tainstrumentq400进行热膨胀系数的测试。氮气流0.05n。升温速率5℃每分钟。
测试结果如表1所示。图10为pi膜的dma曲线;图11为pi膜的tga曲线。图12为pi膜的透过率曲线。图13为pi膜的tma曲线。
根据图10中的dma曲线,基于本发明的刚性脂环含氟结构的二酐化合物(8fda)的pi薄膜的tg值为414℃。而基于4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)的pi薄膜的tg值为332℃。这归因于本发明的刚性脂环含氟结构的二酐化合物(8fda)单体的刚性结构。聚酰亚胺的tg值由分子链的刚性程度,吸电子二酐残基和给电子二胺残基之间的ctc的分子相互作用共同决定。图11为pi薄膜的tga曲线,聚酰亚胺的1%热分解温度(td1)和残炭率的范围是500-517℃和65-66%。而基于本发明的刚性脂环含氟结构的二酐化合物(8fda)的pis具有比相应的4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)膜更高的td1值(分别为517℃和500℃)。本发明的刚性脂环含氟结构的二酐化合物(8fda)基聚酰亚胺的多取代和刚性半脂环1,4-环己二烯,分子链的刚性较4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)强,增强了链间的相互作用和积聚程度,增加了它们的耐热性。与其他薄膜相比,来源于具有本发明的刚性脂环含氟结构的二酐化合物(8fda)的ppda的在400nm处显示出相当低的透射率(t%<30%),如图12所示。由这些测试结果可知通过本发明的刚性脂环含氟结构的二酐化合物(8fda)和ppda聚合得到的pi-1具有优异的热稳定性和尺寸稳定性(tg为414℃,cte为12ppmk-1),t450接近80%。图13为pi薄膜的tma曲线,该方法制备的pi薄膜的cte值为12ppmk-1。而目前已有的基于4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)的薄膜的cte值为47ppmk-1。与4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)相比,基于本发明的刚性脂环含氟结构的二酐化合物(8fda)的薄膜的cte值明显低于基于4,4'-(六氟异亚丙基)二邻苯二甲酸酐(6fda)的薄膜。这也可以归因于本发明的刚性脂环含氟结构的二酐化合物(8fda)的更刚性的结构,可以增加链内取向,更强的分子间相互作用将抑制分子运动,并导致更低的cte产生和更好的尺寸稳定性。
表1基于8fda的pis的热稳定性,机械性能和光学性能数据汇总表
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!