一种以蒽结构为中心的三胺单体及其制备方法和应用与流程
本发明涉及材料科学技术领域,更具体地,涉及一种以蒽结构为中心的三胺单体及其制备方法和应用。
技术背景
聚酰亚胺是一类耐热性极佳的高分子材料,由于具有优异的综合性能,在航空、航天、电气、机械、化工、微电子等各个领域都有非常广泛的应用。超支化聚合物由于具有粘度低、大量末端官能团等独特的优点,近年来被广大学者研究应用到各大高分子材料上。
超支化聚酰亚胺(hbpis)因综合了超支化聚合物和聚酰亚胺两者的优点而具有一系列独特的理化性能,近年来受到广大科研工作者的关注。目前超支化聚酰亚胺主要应用于气体分离膜和渗透膜等膜材料,以及其它如光波导、光敏、液晶、介电材料和传感器(检测电极)等高新技术领域。相对于线性聚酰亚胺,超支化聚酰亚胺的溶解性得到显著改善,但是其热性能相对下降。因此,在保持优异溶解性的同时,提高超支化聚酰亚胺的热性能,这对于超支化聚酰亚胺的应用具有重要意义。但是目前三胺单体品种相对较少,而且现有三胺的热性能不稳定,合成率低,已工业化的单体种类就更为有限,较少的单体种类严重制约了超支化聚酰亚胺的市场化、大规模发展。聚酰亚胺具有优异的结构可设计性,可通过共聚单体的结构设计,引入功能性基团,赋予其特定的性能。因此,通过分子结构设计开发合成新型单体并用于超支化聚酰亚胺的制备,这对于超支化聚酰亚胺材料应用领域的拓展具有重要意义。
蒽环作为稠环芳烃的一类重要的化合物,具有强的刚性,大的共扼结构,活跃的性质,将蒽环引入到聚合物材料中是近几年才发展起来的新兴领域。amitavam.等人制备了含蒽环结构的聚芳砜类聚合物,该聚合物具有优异的热性能,但是溶解性能差(majumdara,biswasm.synthesisandthermalstability,dielectric,andconductivitycharacteristicsofpolymerswithpendantandbackboneanthracenesulfonylgroups[j].journalofappliedpolymerscience,1991,42(9):2489-2498.)。本发明设计合成以蒽结构为中心的三胺单体,刚性蒽结构的引入提高了单体的热性能,采用此类三胺单体制备超支化聚酰亚胺,不但可以提高聚合物的耐热性,而且通过三胺把刚性大体积的蒽结构引入到超支化聚酰亚胺主链,可以扩大聚合物链之间的距离,增大聚合物的自由体积,进一步改善其溶解性和加工性能,提高其气体渗透性能。由本发明三胺单体制备的超支化聚酰亚胺同时具有高热稳定性和优异的溶解性能,其在耐高温领域以及气体渗透分离膜等材料领域具有较好的应用前景。
技术实现要素:
本发明要解决的技术问题是针对现有技术中三胺的热性能不稳定,制备方法复杂,质量不稳定,合成率低,制备的聚酰亚胺不能兼得优异的溶解性和热性能等技术上的不足,提供一种以蒽结构为中心的三胺单体,可用于合成超支化和功能化的聚酰胺、聚酰亚胺、聚酰胺酰亚胺和聚酯酰亚胺等聚合物。
本发明要解决的另一技术问题是提供上述以蒽结构为中心的三胺单体的制备方法。
本发明还一要解决的技术问题是提供上述以蒽结构为中心的三胺单体的应用。
本发明提供的一种以蒽结构为中心的三胺单体结构如通式(i)所示:
其中ar1选自下列结构式中的任何一种:
优选地,所述ar1选自
其中ar2选自下列结构式中的任何一种:
优选地,所述ar2选自
本发明的以蒽结构为中心的三胺单体通过利用含有三个卤原子取代的ar1单体通过经suzuki反应一步反应或经suzuki反应、还原反应两步反应制备如结构通式所示的功能三胺单体。具体包括以下步骤:
将三个卤原子取代的ar1单体和含有一个硼酸和一个氨基取代的ar2单体加入溶剂,通过suzuki反应即得如结构通式(i)所示的以蒽结构为中心的三胺单体;
或s1.将三个卤原子取代的ar1单体和含有一个硼酸和一个硝基取代的ar2单体加入溶剂,通过suzuki反应得到单体1;
s2.将步骤s1中单体1加入到溶剂中,通过还原反应即得如结构通式i所示的以蒽结构为中心的三胺单体;
进一步地,步骤s1中单体1具有如下结构特征:
进一步地,所述suzuki反应过程中需要加入碱,搅拌并通保护气体,加热,加入催化剂,回流反应后提纯、干燥;所述还原反应过程需要搅拌并通保护气体,加热,加入催化剂和还原剂,回流反应后提纯、干燥。
进一步地,所述suzuki反应中ar1单体与ar2单体的投料物质的量比为:1︰2.5~1︰6;所述碱与ar2单体的投料物质的量之比为1︰1~1︰6。
优选地,所述suzuki反应中ar1单体与ar2单体的投料物质的量比为1︰3~1︰5;所述碱与ar2单体的投料物质的量之比为1︰1.5~1︰4。
进一步地,所述还原反应中单体1与还原剂的投料物质的量比为1︰2~1︰32。
优选地,所述还原反应中单体1与还原剂的投料物质的量比为1︰15~1︰25。
进一步地,所述保护气体为氮气,氦气、氖气、氩气、氪气、氙气、氡气中的一种或几种;
进一步地,所述碱为氢化钠、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、氟化铯、正丁基锂、叔丁醇钾、叔丁醇纳、六甲基二硅基胺基锂中的一种或几种;
进一步地,所述suzuki反应中的溶剂为二甲基亚砜、n,n-二甲基甲酰胺、四氢呋喃、1,4二氧六环、甲苯、二甲苯、丙酮、乙腈、水中的一种或几种;步骤s2中所述溶剂为乙醇、甲醇、正丙醇、叔丁醇、叔戊醇、乙醇、己醇、四氢呋喃、1,4二氧六环、二甲基亚砜、n,n-二甲基甲酰胺、乙酸乙酯、甲苯中的一种或几种;
进一步地,所述催化剂为pd[pph3]4、pd(dppf)cl2、pdcl2(ch3cn)2、pdcl2、pd(oac)2、pd(pph3)2cl2、钯碳中的一种或几种;
进一步地,所述还原剂为水合肼、甲酸铵、硼氢化钠、维生素c、柠檬酸钠、铁粉、锌粉中的一种或几种。
进一步地,所述加热温度为50℃~170℃;所述回流反应时间为10~48h;所述干燥温度40℃~120℃;所述干燥时间为6~30h。
优选地,所述加热温度为70℃~90℃;所述回流反应时间为20~30h;所述干燥温度80℃~100℃;所述干燥时间为18~28h。
与现有技术相比,本发明具有以下有益效果:
本发明创造性的将蒽环引入三胺单体中,所制备的三胺单体耐热性优异,因而能够更进一步提高由其制备的聚合物的耐热性和热稳定性;同时还能增强聚合物的机械性能,显著改善其溶解性和加工性能,还可以改善其在电及磁等方面的性能。
本发明所提出的含蒽结构的三胺单体制备所需原材料组分简单,合成工艺反应条件温和,简单易控,绿色环保。通过精确的制备工艺控制,使得制备的产品易纯化,杂质较少,产率较高,因而适于大规模工业生产。本发明研发的三胺单体的耐热性和热稳定性高,可用于合成超支化和功能化的聚酰胺、聚酰亚胺、聚酰胺酰亚胺和聚酯酰亚胺等聚合物。
附图说明
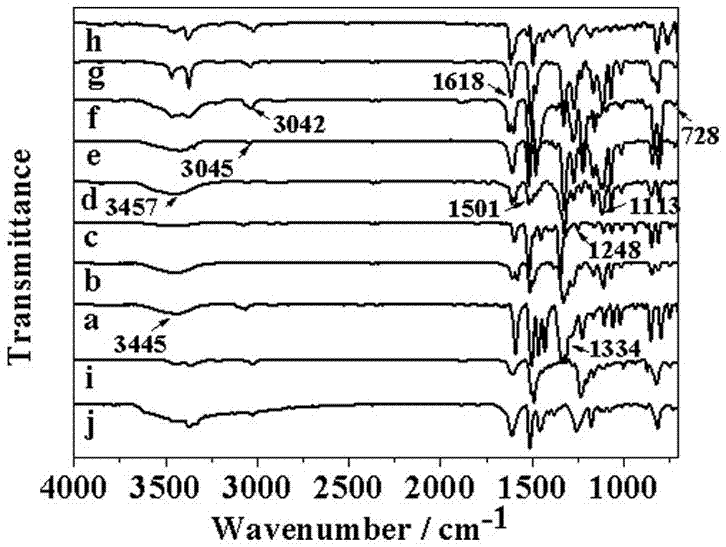
图1是实施例1~10所得单体的红外光谱图,其中:
a对应4,4',4”-(anthracene-1,9,10-triyl)trianiline;
b对应4,4',4”-(anthracene-1,8,9-triyl)trianiline;
c对应4,4',4”-(anthracene-2,9,10-triyl)trianiline;
d对应4,4',4”-(anthracene-2,6,9-triyl)trianiline;
e对应4,4',4”-(anthracene-1,2,4-triyl)trianiline;
f对应5,5',5”-(anthracene-2,6,9-triyl)tris(thiophen-2-amine);
g对应6,6',6”-(anthracene-2,6,9-triyl)tris(naphthalen-2-amine);
h对应5,5',5”-(anthracene-2,6,9-triyl)tris(pyridin-2-amine);
i对应7,7',7”-(anthracene-2,6,9-triyl)tris(2-amino-9h-fluoren-9-one);
j对应n,n',n”-(anthracene-2,6,9-triyltris(benzene-4,1-diyl))tris(4-aminobenzamide)。
从红外光谱图中可以看到,在3500-3340cm-1的范围内出现了-nh2的特征吸收峰,1618cm-1为n-h键的弯曲振动吸收峰;1334和1248cm-1处出现c-n伸缩振动的特征吸收峰;3042cm-1处出现了ar-h的特征吸收频率,1501cm-1处出现了特征的苯环骨架伸缩振动吸收峰,1113~728cm-1为ar–h伸缩振动的特征吸收峰,这些都说明实施例1~10所合成的产物都具有含蒽三胺的特征结构。
具体实施方式
下面结合附图和具体实施例进一步详细说明本发明。除非特别说明,本发明实施例使用的各种原料均可以通过常规市购得到,或根据本领域的常规方法制备得到,所用设备为实验常用设备。除非另有定义或说明,本文中所使用的所有专业与科学用语与本领域技术熟练人员所熟悉的意义相同。
实施例1
4,4',4”-(anthracene-1,9,10-triyl)trianiline的合成:
将4.149(0.01mol)1,9,10-tribromoanthracene和8.671g(0.05mol)对氨基苯硼酸盐酸盐加入到500ml三口瓶中,加入400ml四氢呋喃(thf),再加入2mol/l的碳酸钾溶液45ml,并加入0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在90℃真空中干燥24h,得目标产物2.935g,产率为65%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
将均苯四甲酸二酐(pmda)0.4362g(2mmol)和n,n-二甲基甲酰胺36ml加入三口烧瓶中,通入氩气,升温至30℃,将三胺单体4,4',4”-(anthracene-1,9,10-triyl)trianiline0.4516g(1mmol)溶解到40mln,n-二甲基甲酰胺用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应12h,然后加入6ml乙酸酐和2ml三乙胺,升温至45℃继续反应10h,反应结束冷却到室温后出料在乙醇中,过滤,洗涤,重复2~3次,最后置于80℃真空干燥箱中干燥24h,得到红棕色的超支化聚酰亚胺聚合物,其结构式如下:
实施例2
4,4',4”-(anthracene-1,8,9-triyl)trianiline的合成:
将4.149(0.01mol)1,8,9-tribromoanthracene和8.671g(0.05mol)对氨基苯硼酸盐酸盐加入到500ml三口瓶中,加入400ml四氢呋喃(thf),再加入2mol/l的碳酸钾溶液45ml,并加入0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在80℃真空中干燥28h,得目标产物2.484g,产率为55%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
4,4',4”-(anthracene-1,8,9-triyl)trianiline0.4516g(1mmol)和n,n-二甲基甲酰胺50ml加入三口烧瓶中,通入氩气,升温至30℃,将环丁烷四甲酸二酐(cbda)0.1628g(0.83mmol)溶解到50mln,n-二甲基甲酰胺中用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应20h,然后加入6ml乙酸酐和2ml三乙胺,升温至40℃继续反应10h,反应结束冷却到室温后出料在乙醇中,过滤,洗涤,重复2~3次,最后置于80℃真空干燥箱中干燥24h,得到棕色的超支化聚酰亚胺聚合物,其结构式如下:
实施例3
4,4',4”-(anthracene-2,9,10-triyl)trianiline的合成:
将4.149(0.01mol)2,9,10-tribromoanthracene和8.671g(0.05mol)对氨基苯硼酸盐酸盐加入到500ml三口瓶中,加入400ml四氢呋喃(thf),再加入2mol/l的碳酸钾溶液45ml,并加入0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在85℃真空中干燥24h,得目标产物3.387g,产率为75%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
将均苯四甲酸二酐(pmda)0.4515g(2.07mmol)和n,n-二甲基乙酰胺15ml加入三口烧瓶中,通入氩气,升温至30℃,将三胺单体4,4',4”-(anthracene-2,9,10-triyl)trianiline0.4516g(1mmol)溶解到16mln,n-二甲基乙酰胺用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应14h,然后加入6.2ml乙酸酐和2.1ml三乙胺,升温至45℃继续反应14h,反应结束冷却到室温后出料在甲醇中,过滤,洗涤,重复2~3次,最后置于80℃真空干燥箱中干燥24h,得到浅褐色的超支化聚酰亚胺聚合物,其结构式如下:
实施例4
4,4',4”-(anthracene-2,6,9-triyl)trianiline的合成:
将4.149(0.01mol)1,3,5-tribromobenzene和8.671g(0.05mol)对氨基苯硼酸盐酸盐加入到500ml三口瓶中,加入400ml四氢呋喃(thf),再加入2mol/l的碳酸钾溶液45ml,并加入0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在100℃真空中干燥28h,得目标产物3.161g,产率为70%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
4,4',4”-(anthracene-2,6,9-triyl)trianiline0.9032g(2mmol)和n,n-二甲基甲酰胺3ml加入三口烧瓶中,通入氩气,升温至30℃,将二苯醚四甲酸二酐(odpa)0.7446g(2.4mmol)溶解到2mln,n-二甲基甲酰胺中用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应20h。将所得超支化聚酰亚胺酸胶液刮涂在干燥洁净的玻璃板上,再将玻璃板置于真空烘箱中,抽真空,80下干燥3h,随后升温至120℃后恒温整个过程2h,从120℃升温至200℃后恒温整个过程2h,从200℃升温至350℃恒温整个过程1h,冷却、取出超支化聚酰亚胺膜,其结构式如下:
实施例5
4,4',4”-(anthracene-1,2,4-triyl)trianiline的合成:
将4.149(0.01mol)1,2,4-tribromoanthracene和8.671g(0.05mol)对氨基苯硼酸盐酸盐加入到500ml三口瓶中,加入400ml四氢呋喃(thf),再加入2mol/l的碳酸钾溶液75ml,并加入0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在100℃真空中干燥30h,得目标产物2.484g,产率为55%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
4,4',4”-(anthracene-1,2,4-triyl)trianiline0.4968g(1.1mmol)和四氢呋喃8ml加入三口烧瓶中,通入氩气,升温至30℃,将3,3',4,4'--联苯四甲酸二酐(bpda)0.2942g(1mmol)溶解到8ml四氢呋喃中用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应18h。将所得超支化聚酰亚胺酸胶液刮涂在干燥洁净的玻璃板上,再将玻璃板置于真空烘箱中,抽真空,80下干燥3h,随后升温至120℃后恒温整个过程2h,从120℃升温至200℃后恒温整个过程2h,从200℃升温至350℃恒温整个过程2h,冷却、取出超支化聚酰亚胺膜,其结构式如下:
实施例6
5,5',5”-(anthracene-2,6,9-triyl)tris(thiophen-2-amine)的合成:
将4.149(0.01mol)2,6,9-tribromoanthracene和7.149g(0.05mol)(5-aminothiophen-2-yl)boronicacid加入到500ml三口瓶中,加入400ml四氢呋喃(thf),再加入2mol/l的碳酸钾溶液75ml,并加入0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在100℃真空中干燥28h,得目标产物2.395g,产率为51%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
将3,3',4,4'--联苯四甲酸二酐(bpda)0.4413g(1.5mmol)和n-甲基吡咯烷酮10ml加入三口烧瓶中,通入氩气,升温至30℃,将三胺单体5,5',5”-(anthracene-2,6,9-triyl)tris(thiophen-2-amine)0.4696g(1mmol)溶解到8mln-甲基吡咯烷酮用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应20h,然后加入12ml乙酸酐和3ml三乙胺,升温至45℃继续反应10h,反应结束冷却到室温后出料在甲醇中,过滤,洗涤,重复2~3次,最后置于80℃真空干燥箱中干燥24h,得到棕色的超支化聚酰亚胺聚合物,其结构式如下:
实施例7
6,6',6”-(anthracene-2,6,9-triyl)tris(naphthalen-2-amine)的合成:
6,6',6"-(anthracene-2,6,9-triyl)tris(naphthalen-2-amine)
601.74
将4.149(0.01mol)2,6,9-tribromoanthracene和9.350g(0.05mol)(6-aminonaphthalen-2-yl)boronicacid加入到500ml三口瓶中,加入400ml四氢呋喃(thf),再加入2mol/l的碳酸钾溶液75ml,并加入0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在100℃真空中干燥28h,得目标产物3.009g,产率为50%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
将6,6',6”-(anthracene-2,6,9-triyl)tris(naphthalen-2-amine)1.2035g(2mmol)和n,n-二甲基甲酰胺8ml加入三口烧瓶中,通入氩气,升温至30℃,将3,3',4,4'-二苯酮四酸二酐(btda)0.6444g(2mmol)溶解到8mln,n-二甲基甲酰胺中用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应15h,然后加入6ml乙酸酐和2ml三乙胺,升温至45℃继续反应12h,反应结束冷却到室温后出料在甲醇中,过滤,洗涤,重复2~3次,最后置于80℃真空干燥箱中干燥24h,得到浅黄色的超支化聚酰亚胺聚合物,其结构式如下:
实施例8
5,5',5”-(anthracene-2,6,9-triyl)tris(pyridin-2-amine)的合成:
s1.合成中间体5,5',5”-(anthracene-2,6,9-triyl)tris(2-nitropyridine):
将4.149(0.01mol)2,6,9-tribromoanthracene和6.297g(0.0375mol)(6-nitropyridin-3-yl)boronicacid加入到500ml三口瓶中,四氢呋喃(thf)为溶剂,再加入2mol/l的碳酸钾溶液56.25ml和0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h后,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=2∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在90℃真空中干燥24h,得到黄色固体,产率为72%。该中间体结构如下:
s2.合成5,5',5”-(anthracene-2,6,9-triyl)tris(pyridin-2-amine):
将5.4447g(0.01mol)5,5',5”-(anthracene-2,6,9-triyl)tris(2-nitropyridine)加入到500ml三口瓶中,加入450ml无水乙醇,磁力搅拌并通氩气,油浴加热至80℃后,加入10%wt的钯碳0.1g,并加入15ml水合肼,回流反应30h后,将反应液抽滤,将滤液冷结晶,抽滤所得滤饼80℃真空干燥24h,得到产物4.0908g,产率为90%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
5,5',5”-(anthracene-2,6,9-triyl)tris(pyridin-2-amine)0.9091g(2mmol)和n,n-二甲基乙酰胺5ml加入三口烧瓶中,通入氩气,升温至30℃,将六氟二酐(6fda)0.8618g(1.94mmol)溶解到6mln,n-二甲基乙酰胺中用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应14h,然后加入6ml乙酸酐和2ml三乙胺升温至45℃继续反应12h,反应结束冷却到室温后出料在甲醇中,过滤,洗涤,重复2~3次,最后置于80℃真空干燥箱中干燥24h,得到棕褐色的超支化聚酰亚胺聚合物,其结构式如下:
实施例9
7,7',7”-(anthracene-2,6,9-triyl)tris(2-amino-9h-fluoren-9-one)的合成:
s1.合成中间体7,7',7”-(anthracene-2,6,9-triyl)tris(2-nitro-9h-fluoren-9-one):
将4.149(0.01mol)2,6,9-tribromoanthracene和10.0883g(0.0375mol)(7-nitro-9-oxo-9h-fluoren-2-yl)boronicacid加入到500ml三口瓶中,四氢呋喃(thf)为溶剂,再加入2mol/l的碳酸钾溶液56.25ml和0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h后,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=2∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在90℃真空中干燥24h,得到黄色固体,产率为80%。该中间体结构如下:
s2.合成7,7',7”-(anthracene-2,6,9-triyl)tris(2-amino-9h-fluoren-9-one)
将8.4778g(0.01mol)7,7',7”-(anthracene-2,6,9-triyl)tris(2-nitro-9h-fluoren-9-one)加入到500ml三口瓶中,加入450ml无水乙醇,磁力搅拌并通氩气,油浴加热至80℃后,加入10%wt的钯碳0.1g,并加入10ml水合肼,回流反应36h后,将反应液抽滤,将滤液冷结晶,抽滤所得滤饼80℃真空干燥24h,得到产物6.9720g,产率为92%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
将均苯四甲酸二酐(pmda)0.5453g(2.5mmol)和n,n-二甲基乙酰胺30ml加入三口烧瓶中,通入氩气,升温至30℃,将三胺单体7,7',7”-(anthracene-2,6,9-triyl)tris(2-amino-9h-fluoren-9-one)0.7579g(1mmol)溶解到40mln,n-二甲基乙酰胺用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应14h,然后加入6.2ml乙酸酐和2.1ml三乙胺,升温至45℃继续反应12h,反应结束冷却到室温后出料在乙醇中,过滤,洗涤,重复2~3次,最后置于80℃真空干燥箱中干燥24h,得到深红棕色的超支化聚酰亚胺聚合物,其结构式如下:
实施例10
n,n',n”-(anthracene-2,6,9-triyltris(benzene-4,1-diyl))tris(4-aminobenzamide)的合成:
s1.合成中间体n,n',n”-(anthracene-2,6,9-triyltris(benzene-4,1-diyl))tris(4-nitrobenzamide):
将4.149(0.01mol)2,6,9-tribromoanthracene和10.7269g(0.0375mol)(4-(4-nitrobenzamido)phenyl)boronicacid加入到500ml三口瓶中,四氢呋喃(thf)为溶剂,再加入2mol/l的碳酸钾溶液56.25ml和0.5ml的aliquat336,磁力搅拌并通氩气,油浴加热至75℃后,加入0.020g四三苯基膦钯,回流反应24h后,将反应液倒入水中,有大量沉淀析出。用漏斗抽滤,减压蒸去溶剂。产物以二氯甲烷∶正己烷=4∶1(体积比)为流动相硅胶为固定相作柱色谱提纯,收集产物并旋干,在90℃真空中干燥24h,得到黄色固体,产率为75%。该中间体结构如下:
s2.合成n,n',n”-(anthracene-2,6,9-triyltris(benzene-4,1-diyl))tris(4-aminobenzamide)
将8.9887g(0.01mol)n,n',n”-(anthracene-2,6,9-triyltris(benzene-4,1-diyl))tris(4-nitrobenzamide)加入到500ml三口瓶中,加入450ml无水乙醇,磁力搅拌并通氩气,油浴加热至80℃后,加入10%wt的钯碳0.1g,并加入11ml水合肼,回流反应30h后,将反应液抽滤,将滤液冷结晶,抽滤所得滤饼80℃真空干燥24h,得到产物7.2803g,产率为90%。合成产物的红外光谱图如图1所示。
聚酰亚胺的合成:
n,n',n”-(anthracene-2,6,9-triyltris(benzene-4,1-diyl))tris(4-aminobenzamide)0.8089g(1mmol)和四氢呋喃15ml加入三口烧瓶中,通入氩气,升温至30℃,将均苯四甲酸二酐(pmda)0.2181g(1mmol)溶解到15ml四氢呋喃中用恒压滴液漏斗在1~2h均匀滴加入三口烧瓶中,然后继续反应15h。将所得超支化聚酰亚胺酸胶液刮涂在干燥洁净的玻璃板上,再将玻璃板置于真空烘箱中,抽真空,80下干燥3h,随后升温至120℃后恒温整个过程2h,从120℃升温至200℃后恒温整个过程2h,从200℃升温至350℃恒温整个过程1h,冷却、取出超支化聚酰亚胺膜,其结构式如下:
采用耐驰公司的差示扫描量热仪(dsc204)和ta公司的热重分析仪(q50)分别对实施例1~10所制备的超支化聚酰亚胺进行玻璃化转变温度(tg)和5%热失重温度(t5%)测试,测试结果如表1所示,超支化聚酰亚胺的溶解性能数据如表2所示。
表1超支化聚酰亚胺的热性能
表2超支化聚酰亚胺的溶解性
注:++代表室温可以完全溶解
从表1和表2可以看出,由本发明蒽结构为中心的三胺单体制备的超支化聚酰亚胺具有高玻璃化转变温度和热稳定性,优异的溶解性。
本发明通过上述实施例来说明本发明的详细工艺设备和工艺流程,但本发明并不局限于上述详细工艺设备和工艺流程,即不意味着本发明必须依赖上述详细工艺设备和工艺流程才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!