一种枣根提取物、提取分离方法及其应用与流程
本发明属于植物提取分析技术领域,特别涉及一种枣根提取物、提取分离方法及其应用。
背景技术
枣,别名为干枣、红枣、枣子、大枣、良枣、刺枣、贯枣,是植物枣(ziziphusjujubamill.)的干燥成熟果实,归属于鼠李科(rhamnaceae)枣属(ziziphusmill.)植物。始载于第一部中药学专著《神农本草经》,且被列为诸药之上品。大枣的历史悠久,被列为我国的“五果”(桃、李、栗、杏、枣)之一,既是普通食品,又是传统中药,其味甘、平、无毒,归脾、胃、心经。
枣树,高达10余米,为落叶小乔木,稀灌木。树皮为褐色或灰褐色,枝平滑无毛,分长枝(枣头)和短枝(枣股),长枝弯曲呈“之”字形。据《中国果树志枣卷》记载,枣树在我国有700个品种,栽培范围极广,树种分布集中于河北、山西、山东、陕西、山西、河南以及新疆维吾尔自治区等新兴枣产区。在《本经》中有记载,大枣有养血安神、补中益气、缓和药性的功效,现代临床应用上,大枣可提高人体免疫力,抑制癌细胞,治疗胆结石,预防骨质疏松,防治高血压,治疗溃疡病,现代药理学研究表明大枣具有免疫调节、抗氧化、抗肿瘤、护肝、降血糖等活性。
近年来对枣属植物化学成分和药理作用研究较多的是果实和枝叶部分,而对枣根的化学成分及药理活性方面尚未报道。本发明提供了一种枣根的提取分离方法。
技术实现要素:
本发明利用乙醇提取、乙酸乙酯萃取,结合运用硅胶、chp-20p、大孔树脂、sephadexlh-20等柱层析技术对枣根提取物进行了分离,可应用于延缓阿尔兹海默症和治疗糖尿病药物的研究。
本发明提供了一种枣根提取物,包含以下组分:式①所示的熊果酸、式②所示的ceanothenicacid、式③所示的丁香酚-β-d-葡萄糖苷、式④所示的二氢查尔酮-4′-β-d-吡喃葡萄糖苷、式⑤所示的齐墩果酸、式⑥所示的3-氧代齐墩果酸、式⑦所示的儿茶素、式⑧所示的桦木酸、式⑨所示的大枣萘醌、式⑩所示的无刺枣苄苷ⅱ、式
一种上述枣根提取物的提取分离方法,包括如下步骤:
s1,将枣根粉碎过40目筛,用体积浓度为90%的乙醇溶液渗漏提取2次,每次48h后再开始渗漏,合并两次提取液并减压浓缩,得浸膏;
s2,加水溶解s1中的浸膏,得浸膏溶液,用相当于浸膏溶液2倍体积的乙酸乙酯萃取2次,合并2次乙酸乙酯萃取层,减压浓缩,得乙酸乙酯萃取浸膏;
s3,用甲醇将s2中乙酸乙酯萃取浸膏溶解,加入100~200目硅胶拌样,然后加热使溶剂完全挥发,将得到的固体干燥后研磨,过80~100目筛,采用干法上样在100~200目硅胶柱上,用二氯甲烷-甲醇溶液进行梯度洗脱,同时进行薄层液相色谱tlc跟踪监测,结合hplc谱图分析,将乙酸乙酯萃取层浸膏划分为极性不同的8份馏分,然后运用硅胶、chp-20p、大孔树脂、sephadexlh-20柱层析技术中的一种或多种反复层析分离,得式①-
优选地,二氯甲烷-甲醇溶液的体积比为(200~1)∶1。
一种上述枣根提取物在制备延缓阿尔兹海默症和治疗糖尿病药物中的应用。
优选地,所述枣根提取物中大枣萘醌、齐墩果酸、儿茶素、熊果酸、ceanothenicacid、无刺枣苄苷ⅱ在抑制α-葡萄糖苷酶活性、乙酰胆碱酯酶活性中的应用。
与现有技术相比,本发明的有益效果:
本发明利用乙醇提取、乙酸乙酯萃取,结合运用硅胶、chp-20p、大孔树脂、sephadexlh-20等柱层析技术对枣根提取物进行了分离,其中部分提取物具有良好的乙酰胆碱酯酶和α-葡萄糖苷酶抑制活性,可应用于延缓阿尔兹海默症和治疗糖尿病药物的研究,极具应用前景。
附图说明
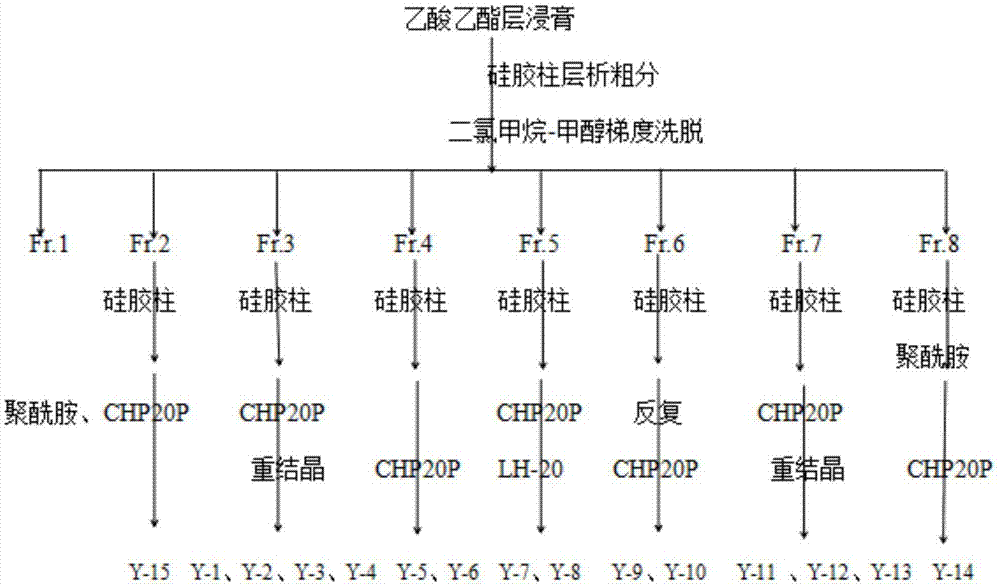
图1为本发明实施例1中乙酸乙酯萃取浸膏的分离过程图;
图2为本发明枣根提取物中6种化合物分析时的高效液相色谱图;
图3为本发明枣根提取物中6种化合物的混合标准品的高效液相色谱图。
其中,图2-3中1-二氢查尔酮-4′-β-d-葡萄糖苷,2-丁香酚-β-d-葡萄糖苷,3-无刺枣苄苷ⅱ,4-对羟基苯甲酸,5-儿茶素,6-槲皮素。
具体实施方式
下面结合附图1-3对本发明的几个具体实施方式进行详细描述,但应当理解本发明的保护范围并不受具体实施方式的限制。
实施例1
一种枣根提取物的提取分离方法,包括以下步骤:
s1,将枣根粉碎过40目筛,用体积浓度为90%的乙醇溶液渗漏提取2次,每次浸泡48h后再开始渗漏,合并两次提取液并减压浓缩,得浸膏,具体如下:
4.7kg枣根粉碎过40目筛,用60l体积浓度为90%的乙醇溶液渗漏法提取,每次浸泡48h,然后开始渗透,分别收集一次滤液和一次滤渣,一次滤液作为得到提取液,一次滤渣再按照上述步骤渗漏提取1次,合并两次提取液并45℃、0.09mpa下减压浓缩,得浸膏(293.60g)。
s2,加水溶解s1中的浸膏,得浸膏溶液,用相当于2倍体积浸膏溶液的乙酸乙酯萃取2次,合并2次萃取时得到的乙酸乙酯萃取层,在45℃、0.09mpa下减压浓缩,得乙酸乙酯萃取浸膏(107g)。
s3,用甲醇将s2中乙酸乙酯萃取浸膏溶解,加入100~200目硅胶拌样,然后加热使溶剂完全挥发,将得到的固体干燥后研磨,过80~100目筛,采用干法上样在100~200目硅胶柱上,用体积比为(200~1)∶1的二氯甲烷-甲醇溶液进行梯度洗脱,同时进行薄层液相色谱tlc跟踪监测,结合hplc谱图分析,按洗脱溶剂的体积比大致将乙酸乙酯萃取层浸膏划分为极性不同的8种馏分,分别命名为fr1(洗脱液体积比200:1~190:1)、fr2(洗脱液体积比180:1~150:1)、fr3(洗脱液体积比140:1~110:1)、fr4(洗脱液体积比100:1~80:1)、fr5(洗脱液体积比70:1~60:1)、fr6(洗脱液体积比50:1~30:1)、fr7(洗脱液体积比30:1~20:1)、fr8(洗脱液体积比10:1~1:1)。
fr3部分采用硅胶柱层析,干法上样,用ch2cl2-meoh(50:1~1:1)梯度洗脱,tlc跟踪监测结合hplc谱图分析又大致划分为5小段(洗脱液体积比:50:1~40:1、35:1~30:1、20:1~15:1、10:1~5:1、5:1~1:1),发现在第2段样品中有细小针状晶体出现,过滤将晶体分离出来后,取少量溶解,点板发现只有一个紫色斑点且呈点性较好,进而进行hplc分析,得到的谱图为单峰且峰形较好,得到式①,编号y-1。将其余几段馏分分别进行硅胶柱分离,梯度洗脱,洗脱溶剂为甲醇-二氯甲烷(70:1~1:1),最后用纯甲醇冲洗,在过柱过程中结合薄层液相色谱分析(tlc)跟踪检测,合并成分相同或相似的馏分并用旋转蒸发仪将溶剂蒸干,反复进行硅胶柱层析,最终分离得到只有两到三个点的样品以及只需除杂的样品,尝试chp、lh-20等多种反相填料,最终确定用填料chp-20p继续分离,得到式②(甲醇:水体积比为10:90)、式③(甲醇:水体积比为20:80)、式④(甲醇:水体积比为10:90)三种化合物,分别编号为y-2、y-3、y-4。
比较fr4部分与fr3部分的峰形谱图发现两部分有一些重合的峰,因此先进行大致划段,将重合部分大致划分开来后,重点对化合物不同的部分进行分离。粗分划段,用粒径为200-300目的硅胶粉处理样品,ch2cl2-meoh(60:1~1:1)梯度洗脱,结合薄层液相色谱(tlc)跟踪检测、高效液相色谱(hplc)分析以及反复硅胶柱层析,最终得到一小段馏分,其中主要有两个点,离得很近,轻微的拖尾现象,可知填料硅胶并不适用于该段样品,于是收集该段馏分,将洗脱液回收并蒸发掉溶剂后,采用反相填料chp-20p根据分子量不同的原理对该段样品进行分离,得到式⑤(甲醇:水体积比为30:70)、式⑥(甲醇:水体积比为60:40)两个化合物,编号为y-5、y-6。
fr5部分样品量较少,但它的峰形谱图显示里面的峰响应面积大,响应值高,出峰状况良好,且各个峰分布比较分散,因此很具有分离的价值。经过多次尝试发现,样品在单一溶剂(ch2cl2、meoh、etoh、pe、乙酸乙酯等)里的溶解性不是很好,却能很好的溶解于甲醇-二氯甲烷混合溶剂,考虑到样品量较少,因此采用湿法上样。硅胶装柱,将溶解好的样品平整的铺于硅胶上层,ch2cl2-meoh进行梯度洗脱,梯度变化为40:1~1:1,多次反复的硅胶柱层析,tlc跟踪检测,hplc图谱分析,得到式⑦(ch2cl2-meoh10:1),编号为y-7。将量少点多且色素带又严重的馏分合并浓缩,暂时搁置。选取其他的有分离意义的馏分继续进行反复硅胶柱层析分离,反复chp-20p、lh-20分离,得到化合物式⑧(甲醇:水体积比为20:80),编号为y-8。
fr6部分在处理样品时,样品溶解性不好,用单一溶剂甲醇、二氯甲烷、正丁醇等溶解效果都不是很好,经过多次尝试,样品最终在混合溶剂甲醇与二氯甲烷中(2:1)溶解。采用多次硅胶柱层析分离、反复chp-20p分离,分离过程中一直不断调整展开剂种类及比例,保证每个点都能以最好的状态呈现出来,以减少实验中出现不必要的误差,最终分离得到式⑨(甲醇:水体积比为10:90)、式⑩(甲醇:水体积比为30:70)两种化合物,编号为y-9、y-10。
fr7部分与fr6部分hplc谱图有重叠峰,因此采用与fr4部分相同的方法先进行粗分划段,对峰形不同的部分重点分离。第一次硅胶柱层析之后得到的第三小段fr6-3(洗脱液体积比30:1~20:1)点多量大,但仔细观察发现里面有两个主要峰,且分的较开,呈点性较好,因此我们对该段继续进行多次硅胶柱层析分离将两个主要点分开,利用反相填料将两个点分离分别得到式
fr8部分极性较大,先用硅胶柱色谱分离,ch2cl2-meoh(20:1~1:1)梯度洗脱,结果发现化合物基本没有被分开,这时我们考虑换一种填料尝试,因此选用聚酰胺大孔吸附树脂柱层析。采用梯度洗脱方法,流动相为水和乙醇,洗脱梯度25%乙醇-水,50%乙醇-水,75%乙醇-水,90%乙醇-水,100%乙醇,将得到的第三小段(75%乙醇-水)馏分接着用300~400目硅胶反复柱层析分离,采用三元展开剂石油醚:甲醇:醋酸,比例为10:2:0.2,展开后,从展开缸中取出,用吹风机将溶剂吹干,显色剂(5%浓硫酸-无水乙醇)加热显色,在硅胶板上发现一个疑似白点,当继续用吹风机高温吹该点时,此斑点没有变黑,结合高效液相图谱分析,确定该斑点为一纯净斑点,得到式
接着分离fr2部分,因其极性较小,用石油醚-乙酸乙酯(30:1~1:1)进行梯度洗脱,得到5小段馏分(30:1~25:1、20:1~15:1、13:1~10:1、8:1~5:1、3:1~1:1),点均分布集中,所以选用反相填料聚酰胺、chp-20p,经反复柱层析,结合薄层色谱tlc检测,出点相似的馏分合并浓缩,跟踪hplc分析图谱,得到式
为鉴定15种化合物结构,对提取的15种化合物分别通过核磁共振技术(1hnmr、l3cnmr、1h-1hcosy、dept、hmbc、hmqc),紫外光谱技术(uv),质谱技术(ms)等现代波谱技术确定了具体结构,这15中化合物分别为:熊果酸、ceanothenicacid、丁香酚-β-d-葡萄糖苷、二氢查尔酮-4′-β-d-吡喃葡萄糖苷、齐墩果酸、3-氧代齐墩果酸、儿茶素、桦木酸、大枣萘醌、无刺枣苄苷ⅱ、β-谷甾醇、豆甾醇、胡萝卜苷、对羟基苯甲酸、槲皮素。结构式如下:
实施例2
一种上述枣根提取物中6种物质的分析方法,包括如下步骤:
1、购买各物质的标准品:二氢查尔酮-4′-β-d-葡萄糖苷(1),丁香酚-β-d-葡萄糖苷(2),无刺枣苄苷ⅱ(3),对羟基苯甲酸(4),儿茶素(5),槲皮素(6),用甲醇配制成浓度为10mg/l、40mg/l、80mg/l、150mg/l、200mg/l的系列标准溶液,4℃冰箱中保存待用。
2、用高效液相色谱仪分别对6种标准品溶液进行检测,选择不同的流动相梯度洗脱方法,根据检测结构不断调整色谱条件,确定最佳色谱条件为:色谱柱:lunac-18柱(5μm,250×4.60mm,phenomenex公司,托伦斯,加利福尼亚州,美国);流动相a:0.2%磷酸-水,流动相b:乙腈;梯度洗脱程序为:0~10min,100%~85%a,0%~15%b;10~30min,85%~75%a,15%~25%b;30~40min,75%~65%a,25%~35%b;40~55min,65%~5%a,35%~95%b;55~56min,5%~100%a,95%~0%b;56~60min,100%~100%a,0%~0%b。柱温:25℃;柱压:19.11mpa;流速:1ml/min;进样量:15μl;检测波长:210nm,254nm,280nm。
用高效液相色谱仪分析上述配制的各化合物的系列标准溶液,分别记录各化合物的峰面积,以对照品质量浓度(μg/ml)为横坐标(x),其对应的峰面积响应值(mau)为纵坐标(y),绘制标准曲线,计算回归方程,标准曲线、相关系数和线性范围,6个成分均呈良好线性关系(r2>0.99)。
如图2为6种化合物进行hplc分析后所得的高效液相色谱图,图3为6种化合物的混合标准品的高效液相色谱图。
各化合物标准曲线及其参数见表1:
表1枣根提取物的标准曲线公式
3、称取枣根粉5.0g,放入100ml带塞锥形瓶中,加入90%的乙醇溶液40ml,称重。放置过夜后,超声提取1h,待提取液冷却至室温后,将上清夜经0.45μm微孔滤膜过滤,取1ml注入hplc样品瓶中,4℃下保存待用。
分别吸取25μl各对照品溶液和供试品溶液,用高效液相色谱仪进行检测,根据检测结果,找出各对照品化合物对应的峰,记录其峰面积并用标准曲线法计算各化合物的含量,结果见表2。计算公式为:
表2各化合物含量
实施例3
本发明枣根提取物在制备延缓阿尔兹海默症和治疗糖尿病药物上的应用。为验证其功效,作如下实验:
1、乙酰胆碱酯酶活性抑制实验
测定方法:将实施例1提取物溶于dmso配成浓度为1.0mg/ml的溶液,在96孔板中,1、2、3列24孔中均加入140μl0.1mol/l磷酸缓冲液(ph为7.4)、20μl待测样品和20μl乙酰胆碱酯酶,作为样品组;4、5、6列24孔中均加入160μl0.1mol/l磷酸缓冲液(ph为7.4)和20μl待测样品,作为控制组;7、8、9列3孔中均加入160μl0.1mol/l磷酸缓冲液(ph为7.4)和20μl乙酰胆碱酯酶,作为空白组;在4℃下培养20min后,每孔加入10μl15mmacti和10μl2mmdtnb,再在37℃下培养20min后,用酶标仪测定在412nm波长处的吸光值大小。用石杉碱甲作为阳性对照,抑制率%=[1-(od样品-od控制)/od空白]×100%。结果如表3:
表3乙酰胆碱酯酶活性抑制率
乙酰胆碱酯酶是生物神经传导中的一种关键性酶,在胆碱能突触间,该酶能降解乙酰胆碱,终止神经递质对突触后膜的兴奋作用,保证神经信号在生物体内的正常传递。乙酰胆碱酯酶参与细胞的发育和成熟,能促进神经元发育和神经再生,具有益智健脑的作用。
从活性实验结果来看,枣根中提取分离得到的部分化合物大枣萘醌、齐墩果酸、儿茶素、熊果酸、ceanothenicacid、无刺枣苄苷ⅱ对乙酰胆碱酯酶活性具有明显的抑制作用,能应用于延缓阿尔兹海默症药物的研究。
2、α-葡萄糖苷酶活性抑制实验
测定方法:将实施例1提取物溶于dmso配成浓度为1.0mg/ml的溶液,在96孔板中,1、2、3列24孔中均加入20μl0.1mol/l磷酸缓冲液(ph为6.8)、20μl待测样品和20μlα-葡萄糖苷酶,作为样品组;4、5、6列24孔中均加入40μl0.1mol/l磷酸缓冲液(ph为6.8)和20μl待测样品,作为控制组。
7、8、9列3孔中均加入40μl0.1mol/l磷酸缓冲液(ph为6.8)和20μlα-葡萄糖苷酶作为空白组;在37℃下培养15min后,每孔加入20μl2.5mmol/lpnpg糖苷,再在37℃下培养15min后,每孔加入80μl0.2mol/lna2co3溶液终止反应。用酶标仪测定在405nm波长处的吸光值大小。用阿卡波糖作为阳性对照。
抑制率%=[1-(od样品-od控制)/od空白]×100%。结果如表4:
表4α-葡萄糖苷酶活性抑制率
葡萄糖苷酶是生物体内糖代谢途径中的重要成员,α-葡萄糖苷酶更是直接参与淀粉及糖原的代谢途径。通过抑制α-葡萄糖苷酶,可以降低唐化学代谢,从而达到降血糖的功效。
从表4来看,枣根中提取分离得到的部分化合物大枣萘醌、齐墩果酸、儿茶素、熊果酸、ceanothenicacid、无刺枣苄苷ⅱ对α-葡萄糖苷酶活性具有明显的抑制作用,能应用于延缓阿尔兹海默症和治疗糖尿病药物的研究。
实施例4
重复性实验:
平行称取6份5.0g枣根样品粉末,按分析方法处理,进行液相分析,进样量均为10μl,准确记录各个标准品化合物的峰面积响应值,计算其相对应的标准品化合物的含量,rsd值均低于1..5%,说明本实验所使用的分析方法重复性良好。结果如表5所示:
稳定性实验
依次取1.0mg/ml的6个标准品溶液,于0、2、4、6、8、10、12、24和48h进行高效液相色谱分析,记录各个标准品化合物的峰面积响应值,计算其相对应的标准品化合物的含量,rsd值均低于1.5%,说明标准品溶液的稳定性好。结果如表5所示:
表5分析方法的重复性和稳定性
加样回收率实验
称取6份5.0g粉碎后的枣根药材粉末,按分析方法进行处理,制成供试液,分别向其中准确加入相对应的一定量的各标准品溶液进行高效液相色谱(hplc)分析,准确记录各个标准品化合物的峰面积响应值,计算其相对应的各个标准品化合物的含量,然后计算各个标准品化合物所对应的回收率,rsd值均低于1.5%,回收率均大于90.0%。结果如表6所示,回收率(n=6)。
表6回收率
需要说明的是,本发明权利要求书中采用的步骤方法与上述实施例相同,为了防止赘述,本发明的描述了优选的实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例做出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!