一种拟穴青蟹高密度遗传图谱的构建方法与流程
本发明属于海洋生物技术领域,特别涉及一种拟穴青蟹高密度遗传图谱的构建方法。
背景技术:
拟穴青蟹(scyllaparamamosain)是我国重要的海洋渔业资源和海水养殖蟹类,具有体型大、生长快、营养丰富和肉质鲜美的特点,深受人们喜爱。近年来,我国拟穴青蟹人工养殖产量连年超过14万吨,并且保持良好的发展势头,具有很大的发展空间和养殖潜力。随着养殖规模的不断扩大,拟穴青蟹良种的需求量也在不断上升。但是,目前为止,人工养殖的苗种还主要来源于海区的自然苗,受海区自然条件的限制,其质量和数量均难以获得保证。此外,传统人工育苗技术的周期长,育苗成功率低。因此,借助于分子标记辅助育种技术,实现人工育苗的产业化和规模化将极大地促进青蟹产业的发展。
高密度遗传连锁图谱是重要经济性状qtl定位和分子标记辅助育种的基础,而且可以用于辅助基因组的组装,具有重要的理论和应用价值。第一个拟穴青蟹的遗传图谱是利用aflp标记构建的(maetal.2016),因其具有标记数量少、分布不均、覆盖密度低等缺陷,严重限制了其在qtl定位和基因组组装中的应用。snp标记是指在基因组上单个核苷酸的变异,因其具有数量多、多态性丰富、遗传稳定等优点,是高密度遗传图谱构建的理想标记。slaf-seq技术是基于高通量测序的大规模snp标记开发与基因分型的新技术,已被成功应用于多个物种的遗传变异研究。
由于一些物种的人工养殖和繁殖技术还不成熟,或者是难以同时获得全同胞家系的父母本,这就限制了遗传图谱的构建。对于青蟹来说,在人工育苗过程中采用的亲蟹大多为海捕野生蟹,其父本难以寻找,因此,建立一种基于单亲本的拟穴青蟹遗传图谱构建技术十分有必要,从而为其经济性状qtl定位、全基因组关联分析和分子辅助育种提供有力支持。
技术实现要素:
本发明的目的在于提供一种拟穴青蟹高密度遗传图谱的构建方法,为样本材料中只有单亲本或难以同时获得双亲本的动植物家系遗传图谱的构建提供了新的思路和方法,有利于动植物遗传图谱的构建,在分子育种领域具有良好的应用前景。
一种拟穴青蟹高密度遗传图谱的构建方法,主要包括以下步骤:
(1)作图群体构建;
(2)简化基因组文库构建及高通量测序:选择最适的酶切方案,对检测合格的基因组dna分别进行酶切;对得到的酶切片段进行3’端加a处理、连接dual-index测序接头、pcr扩增、纯化、混样、切胶选取合适的目的片段,文库质检合格后用illuminahiseqtm2500平台进行测序;
(3)基因分型:过滤掉原始测序数据中低质量的reads,根据dual-index接头识别原始数据,得到每个样品的reads;对过滤后的reads进行质量和数量评估,并通过control测序数据与其基因组的比对效率评估酶切效率;将酶切片段作为一个slaf标签,通过gatk软件对每个slaf标签中的多态性snp标签进行检测和统计分析,并对多态性snp标签进行基因型编码;
(4)高密度遗传图谱构建:为了保证遗传图谱的质量,首先要过滤掉低质量的snp标签,筛选出适合作图的snp标签;利用joinmap软件计算snp标签间的mlod值,过滤掉mlod值低于6的标签,根据mlod值将snp标签分为不同的连锁群,采用highmap软件分析连锁群内marker的线性排列顺序,并估算相邻marker间的遗传距离,然后绘制整合遗传图谱,并根据雌、雄中marker的差异分别构建雌、雄遗传图谱。
进一步的,步骤(1)所述作图群体构建主要包括:选择优质亲蟹并将其培育至抱卵,孵化出幼体,将幼体培育至仔蟹ii期后转移到池塘中养殖,待生长到2-3月龄时,随机取样(雌雄比例接近1:1),连同母本一起作为作图群体;根据腹部形态特征鉴定每只个体的性别,然后使用ctab法分别提取每只个体肌肉组织的基因组dna。
进一步的,步骤(2)所述最适的酶切方案为:最适内切酶为haeiii酶和hpy166ii酶,酶切片段长度为314-414bp。选择最适酶切方案的步骤为:根据青蟹基因组大小以及gc含量等信息,选取中华绒螯蟹的基因组作为参考(因为中华绒螯蟹与拟穴青蟹亲缘关系比较近,而且中华绒螯蟹具有基因组信息,而拟穴青蟹目前没有基因组信息);然后对中华绒螯蟹基因组序列进行电子酶切预测。最适酶切方案的选择原则为:位于重复序列的酶切片段比例尽可能低,酶切片段尽量在基因组上均匀分布,酶切片段长度能够保证序列标签的数量。步骤(2)所述选取合适的目的片段是指最佳酶切方案中的酶切片段,即长度为314-414bp的片段。
进一步的,步骤(3)中所述通过gatk软件对每个slaf标签中的多态性snp标签进行检测和统计分析,步骤为:根据过滤后reads在参考基因组上的定位结果,利用gatk进行局部重比对(localrealignment)、gatk变异检测和samtools变异检测,将gatk和samtools得到的结果一致的变异位点作为多态性snp标签。
进一步的,步骤(3)中所述对多态性snp标签进行基因型编码主要是采用遗传学中通用的二等位编码规则,并根据母本和子代的基因型推测父本的基因型。例如,在某一位点,父本纯合而母本杂合,在二等位编码的8种分型中可以是nn×np和cc×ab,假如是nn×np(父本×母本)这种类型,则子代为nn和np两种基因型,且比例应该是1:1;假如是cc×ab,子代为ac和bc两种杂合基因型,比例也是1:1。因此可以通过子代的基因型及其比例来区分nn×np与cc×ab。同样地,其他的几种分型以此类推。此外,考虑到影响因素很小的测序错误和偏分离导致的缺少一种分型等情况,在基因分型最后确定之前,对子代基因型比例的实际观测值与理论推断值进行卡方检验,p值最大的即认为是最可能的分型。
进一步的,步骤(4)所述筛选出适合作图的snp标签的筛选原则是:母本和子代的测序深度大于4×;单一多态性snp标签在子代中的覆盖率大于90%;过滤偏分离p-value小于0.05的snp标记。
进一步的,步骤(2)还包括以水稻(oryzasativajaponic)作为对照(control)参与建库和测序。
进一步的,步骤(2)所述合格的样品的基因组dna的检测方法为:使用nanodrop仪器检测样品的浓度和纯度,然后通过琼脂糖凝胶电泳来判定样品的完整性。基因组dna在酶切前要进行dna质量和浓度的检测,只有dna纯度和浓度达到要求了才能进行酶切。dna样品检测标准如下:浓度≥18ng/ul,总量≥1.8ug,od260/280介于1.6-2.5,260nm吸收峰正常,点样孔无或有轻微杂质,主带清晰可见,rna污染不严重;dna颜色透明无色且不粘稠,无沉淀物。
与现有技术相比,本发明利用单亲本和子一代家系作为作图群体进行遗传图谱的构建,即利用母本和子代的基因型对父本基因型进行反推,然后进行基因分型,从而构建遗传图谱,为样本材料中只有单亲本或难以同时获得双亲本的动植物家系遗传图谱的构建提供了新的思路和方法,有利于遗传图谱的构建,推进遗传背景的研究进程,在分子育种领域具有良好的应用前景。本发明采用基于snp标记的分型技术,具有标记数量多、多态性丰富、遗传稳定等优点,得到的遗传图谱密度高、质量好,可用于后续重要经济性状的qtl定位和基因组的辅助组装。
附图说明
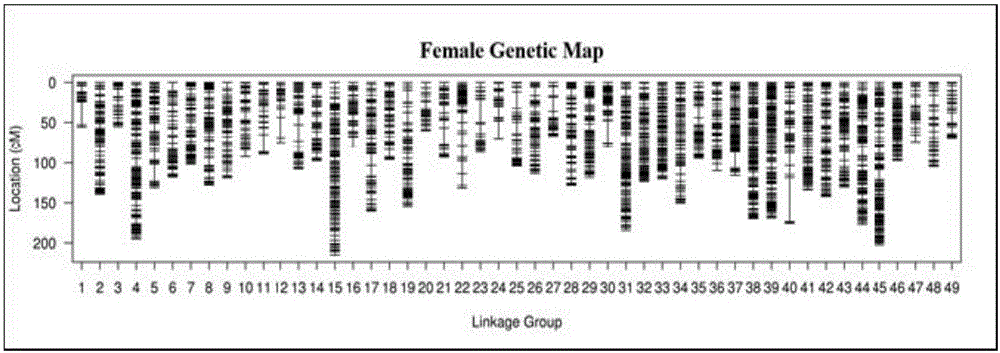
图1是实施例1中构建的雌性连锁图谱;
图2是实施例1中构建的雄性连锁图谱;
图3是实施例1中构建的整合连锁图谱。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明作进一步地详细描述。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的修改和替换,均属于本发明的范围。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
实施例1
一种拟穴青蟹高密度遗传图谱的构建方法,主要包括以下步骤:
1.作图群体构建
选择已经交配过、体型大、体肢完整、卵巢发育饱满、活力强且无寄生虫的雌蟹作为亲蟹,在种蟹池内培育,待其发育至抱卵时将其转移至孵化池中等待孵化。孵化后将幼体转移至育苗池中培育,直至仔蟹ii期。将孵化后的亲蟹样本解剖取大螯内部肌肉,将其存放于-80℃冰箱中,以备后用。然后选择健康的仔ii期蟹苗,转移至池塘中养殖,待其生长到2.5月龄时,随机选择129只个体。根据腹部形态判定雌雄,雄蟹63只,雌蟹66只。将子代个体解剖,取大螯内部肌肉,将其保存于-80℃冰箱中,以备后用。然后利用ctab法提取母本及子代个体的基因组dna,用1.0%琼脂糖电泳检测dna质量,并用紫外分光光度计检测dna浓度。
2.简化基因组文库构建及高通量测序
根据青蟹基因组大小以及gc含量等信息,选取中华绒螯蟹的基因组作为参考;然后对中华绒螯蟹基因组序列进行电子酶切预测。最适酶切方案的选择原则是:位于重复序列的酶切片段比例尽可能低,酶切片段尽量在基因组上均匀分布,酶切片段长度能够保证序列标签的数量。经过酶切预测,确定最适酶切方案:最适内切酶为haeiii酶和hpy166ii酶,酶切片段长度为314-414bp。然后,对检测合格的基因组dna分别进行酶切(酶切的方法跟上述的中华绒螯蟹最适酶切方案一样);对得到的酶切片段进行3’端加a处理、连接dual-index测序接头、pcr扩增(pcr扩增条件如下:98℃预变性3分钟;接着14个循环98℃变性10秒,65℃退火30秒,72℃延伸30秒;最后72℃延伸5分钟)、纯化、混样、切胶选取314-414bp的片段,文库质检合格后用illuminahiseqtm2500平台进行测序。此外,为了评估建库实验的准确性,以水稻(oryzasativajaponic)作为对照(control)参与建库和测序。本发明中,水稻的酶切效率为93.49%,表明酶切效率正常。
基因组dna在酶切前要进行dna质量和浓度的检测,只有dna纯度和浓度达到要求了才能进行酶切。dna检测使用nanodrop仪器检测样品的浓度和纯度,然后通过琼脂糖凝胶电泳来判定样品的完整性。样品检测标准如下:浓度≥18ng/ul,总量≥1.8ug,od260/280介于1.6-2.5,260nm吸收峰正常,点样孔无或有轻微杂质,主带清晰可见,rna污染不严重;dna颜色透明无色且不粘稠,无沉淀物。
3.基因分型
过滤掉原始测序数据中低质量的reads,根据dual-index接头识别原始数据,得到各个样品的reads。对过滤后的reads进行测序质量和数量评估,并通过control测序数据与其基因组的比对效率评估酶切方案实施的有效性和酶切效率(就是用程序对过滤后的数据进行统计,质量评估包括测序数据的q30、gc分布,并画出碱基测序错误率图片和gc分布图;数量评估包括统计过滤后的clean_reads和clean_bases,即得到多少可用reads和可用bases,control数据指的就是实验建库用到的水稻样品测序获得的数据,可以通过水稻测序数据与水稻基因组的比对,来统计实际酶切位点与预测酶切位点的差距,统计切开与未切开酶切位点分别占的比例,统计水稻数据的覆盖度,依此来评估酶切方案实施的有效性和酶切效率);将314-414bp的酶切片段作为一个slaf标签,对每个slaf标签中的多态性snp标签进行检测和统计分析,主要通过gatk软件包实现,其步骤为:根据过滤后reads在参考基因组上的定位结果,利用gatk进行局部重比对(localrealignment)、gatk变异检测和samtools变异检测,将gatk和samtools得到的结果一致的变异位点作为多态性snp标签用于后续分析。
对多态性的snp标签进行基因型编码,采用遗传学中通用的二等位编码规则,并根据母本和子代的基因型推测父本的基因型。例如,在某一位点,父本纯合而母本杂合,在二等位编码的8种分型中可以是nn×np和cc×ab,假如是nn×np(父本×母本)这种类型,则子代为nn和np两种基因型,且比例应该是1:1;假如是cc×ab,子代为ac和bc两种杂合基因型,比例也是1:1。因此可以通过子代的基因型及其比例来区分nn×np与cc×ab。同样地,其他的几种分型以此类推,具体推测方式见表1。此外,考虑到影响因素很小的测序错误和偏分离导致的缺少一种分型等情况,在基因分型最后确定之前,对子代基因型比例的实际观测值与理论推断值进行卡方检验,p值最大的即认为是最可能的分型。
表1:二等位编码规则中父母本基因型、子代基因型及比例
4.高密度遗传图谱构建
为了保证遗传图谱的质量,首先要过滤掉低质量的snp标签,筛选出适合作图的snp标签,筛选原则是:母本和子代的测序深度大于4×;单一多态性snp标签在子代中的覆盖率大于90%;过滤偏分离p-value小于0.05的snp标记。然后利用joinmap软件计算snp标签间的mlod值,过滤掉mlod值低于6标签,根据mlod值将snp标签分为不同的连锁群,采用highmap软件分析连锁群内marker的线性排列顺序,并估算相邻marker间的遗传距离,然后绘制整合遗传图谱,并根据雌、雄中marker的差异分别构建雌、雄遗传图谱。
5.高密度遗传图谱总结
(1)经过测序,共获得731.37mreads数据,平均q30为93.92%,平均gc含量为41.78%,样本gc分布正常。因此,测序数据量和质量均较高。
(2)利用haeiii酶和hpy166ii酶对水稻基因组dna进行酶切,酶切效率为93.49%,slaf建库正常。
(3)经过snp检测,共获得275,876个多态性的snp标签,能成功分型的标签为135,946个,可以用于遗传图谱构建的标记有131,737个。
(4)经过筛选,适合作图的snp标签为17,246个,去除掉mlod值低于6的标签,共得到上图标记16,693个。此外,本图谱中加入了已知的8个性别连锁snp标记,故最后上图snp标记为16,701个。
(5)所有上图标记被分为49个连锁群,构建了雌性连锁图谱、雄性连锁图谱和整合连锁图谱,其总图距分别为5790.08cm、5877.71cm和5996.66cm。三个图谱见图1(雌性图谱)、图2(雄性图谱)和图3(整合图谱)。
- 还没有人留言评论。精彩留言会获得点赞!