重组尘螨II型变应原Derp2和Derf2蛋白的制备与应用的制作方法
本发明属于生物工程基因领域,涉及重组ii型变应原derp2和derf2蛋白的制备方法及其应用。
背景技术:
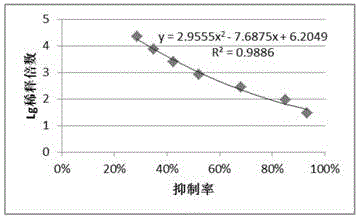
:尘螨种类繁多,广泛存在于人类生活和工作环境中,其排泄物、代谢物及螨体均具有较强的变应原性,据统计全球约10%的人口尘螨过敏,约80%的外源性哮喘由尘螨引起。目前,临床上主要采用尘螨变应原粗提液治疗尘螨引起的变态反应性疾病,例如2006年上市的浙江我武生物的“畅迪”粉尘螨滴剂即为粉尘螨代谢培养物的提取液。尘螨变应原主要存在于排泄物和螨体中,采用提取方法耗时长,过程繁琐,成本较高;此外天然变应原提取液的组成非常复杂,恒定其组分非常困难,且容易受到外源性有毒物质、病原体微生物的污染。长期使用尘螨变应原粗提液易导致红晕、肿胀、硬结、坏死等局部反应和休克、水肿、支气管痉挛、荨麻疹、血管性水肿、全身性红斑等全身反应。此外,采用粗提液进行诊断,无法明确患者对变应原各组分的反应程度,易致误诊。变应原的质量对变态反应疾病的诊断和治疗至关重要,用于免疫诊治的变应原应该是纯品而不宜为粗提取液。重组变应原与粗提液相比具有以下优势:(1)重组变应原具有更高的纯度,与粗提液相比不含非变应原成分、酶类、酶抑制剂和毒性蛋白等;(2)重组蛋白成分单一,具有较好的特异性,而粗提液中成分复杂,患者可能只与其中部分成分反应,特异性差;(3)重组变应原与天然提取液相比减少ige结合的抗原表位,有效降低ige介导的过敏反应,同时保留变应原t细胞识别所必须的结构域,具有较好的免疫原性,减少免疫治疗的危险性,提高脱敏治疗的效果。尘螨变应原组成复杂,约有30余种,其中i型和ii型变应原是最主要的变应原组分。在尘螨过敏患者血清中,70-80%的患者的ige结合derp2(户尘螨ii型变应原,以下简写为dp2)和derf2(粉尘螨ii型变应原,以下简写为df2),并呈现强阳性反应。dp2和df2前体均由146个氨基酸组成,经过去除信号肽加工处理后为129个氨基酸,分子量约为14kd,无糖基化位点。目前利用真核表达系统进行dp2重组表达的研究比较具有代表性的是stallergenes公司2011年的专利(欧洲专利号:ep2388268(a1)),他们利用毕赤酵母表达系统对dp2进行了重组表达和纯化,该专利未针对毕赤酵母系统对dp2基因和分子构建进行优化,产量较低,纯化工艺复杂,收率低,难以满足临床给药的用量。针对df2重组表达比较具有代表性的是胡友莹等2011年在原核表达系统中进行的研究,以及2012年崔玉宝等申请的“一种生产重组粉尘螨变应原derf1和derf2融合蛋白的方法”(中国专利号:cn102676568a),但原核表达系统无翻译后修饰的功能,得到的df2蛋白结构不正确,与血清反应性较弱,且后期分离纯化困难。综合以上研究结果,截止目前为止,针对dp2和df2发酵和纯化的研究较少,且收率,结构,质量等均难以达到临床使用要求;本发明通过对dp2和df2蛋白的发酵工艺和纯化工艺进行优化,使得发酵水平、纯化收率和质量要求均达到了重组脱敏药物临床使用要求。技术实现要素:本申请根据本公司专利申请号201611267033.8(dp2)和201611267247.5(df2)中提供的方法筛选到的基因工程菌株,对dp2和df2蛋白发酵和纯化工艺中的关键质量参数进行了优化;由于dp2和df2基因序列同源性大于90%,因此蛋白的结构和性质有较大的相似性,发明人在研究中发现dp2和df2蛋白在纯化工艺中也有极高的相似性。本发明提供的重组尘螨ii型变应原dp2和df2蛋白的制备方法,包括发酵工艺和纯化工艺,发酵过程包括重组酵母菌株活化、种子液培养、高密度发酵,所用发酵培养基为40~60%bsm培养基,高密度发酵的诱导阶段诱导温度为:20~25℃,ph值为6.0-6.5。作为优选,所用发酵培养基为60%bsm培养基,高密度发酵的诱导阶段诱导温度为:20~22℃,ph值为6.0±0.2。更优选,高密度发酵的诱导阶段甲醇流加速度为30ml/h-1l-1,并维持do不高于40%。更优选,高密度发酵的限速生长阶段开始以30ml/h-1l-1的速率补加50%甘油,后补料速度上调为60ml/h-1l-1。作为优选,重组尘螨ii型变应原蛋白的制备方法的纯化工艺为三步纯化,包括阳离子交换层析、阴离子交换层析、疏水层析。作为优选,阳离子交换层析的纯化填料为spff;优选的ph范围4.0-6.0。更优选的,样品和缓冲液ph为6.0。作为优选,阴离子交换层析的纯化填料为qff;优选的ph范围6.0-9.0;优选的电导值2.0ms/cm-20.0ms/cm。更优选的,样品和缓冲液ph7.5,电导10.0ms/cm。作为优选,疏水层析的纯化填料为phenylff;优选的ph范围6.0-8.0;优选的硫酸铵浓度为1.0-2.0m。更优选的,样品和平衡缓冲液硫酸铵浓度1.0m,ph6.0。本发明采用以上技术方案后,具有以下优点:通过发酵参数优化,制备的发酵液活性更高、产量更高,所采用的发酵培养基成分简单、成本低廉,降低了发酵工艺的成本;发酵工艺中采用特定的培养和发酵控制条件,与未经优化的无机培养基相比,发酵时间缩短20%,活性提高50%,产量提高20%以上;通过对纯化工艺进行优化,收率提高了30%。并且宿主残留蛋白和残留dna已完全符合药典要求,表示经发明人大规模发酵的dp1、df1已完全满足药用级要求。附图说明图1实施例1第一批次重组dp2毕赤酵母菌株中sds-page电泳图以及发酵液od600和菌体湿重变化趋势图。其中,图1-a为实施例1第一批次重组dp2毕赤酵母菌株中sds-page电泳图,泳道m为(10-230kda)宽范围的蛋白上样marker;泳道1为流加甲醇0h发酵上清液,泳道2为流加甲醇8h发酵上清液,泳道3为流加甲醇16h发酵上清液,泳道4为流加甲醇24h发酵上清液,泳道5为流加甲醇32h发酵上清液,泳道6为流加甲醇40h发酵上清液,其中箭头所指即为重组dp2条带。图1-b为实施例1第一批次重组dp2毕赤酵母菌株小规模高密度发酵液od600和菌体湿重变化趋势图。图2为实施例1第二批次重组dp2毕赤酵母菌株中sds-page电泳图以及发酵液od600和菌体湿重变化趋势图。其中,图2-a为实施例1第二批次重组dp2毕赤酵母菌株中sds-page电泳图,泳道m为(10-230kda)宽范围的蛋白上样marker;泳道1为流加甲醇0h发酵上清液,泳道2为流加甲醇6h发酵上清液,泳道3为流加甲醇12h发酵上清液,泳道4为流加甲醇18h发酵上清液,泳道5为流加甲醇24h发酵上清液,泳道6为流加甲醇30h发酵上清液,其中箭头所指即为重组dp2条带。图2-b为实施例1第二批次重组dp2毕赤酵母菌株小规模高密度发酵液od600和菌体湿重变化趋势图。图3为实施例1第三批次重组dp2毕赤酵母菌株sds-page电泳图以及发酵液od600和菌体湿重变化趋势图。其中,图3-a为为实施例1第三批次重组dp2毕赤酵母菌株中sds-page电泳图,泳道m为(10-230kda)宽范围的蛋白上样marker;泳道1为流加甲醇0h发酵上清液,泳道2为流加甲醇6h发酵上清液,泳道3为流加甲醇12h发酵上清液,泳道4为流加甲醇18h发酵上清液,泳道5为流加甲醇24h发酵上清液,泳道6为流加甲醇30h发酵上清液,其中箭头所指即为重组dp2条带。图3-b为实施例1第三批次重组dp2毕赤酵母菌株小规模高密度发酵液od600和菌体湿重变化趋势图。图4为重组dp2毕赤酵母菌株实施例2中sds-page电泳图以及发酵液od600和菌体湿重变化趋势图。其中,图4-a为重组dp2毕赤酵母菌株实施例3中sds-page电泳图,泳道m为(10-230kda)宽范围的蛋白上样marker;泳道1为补料前发酵上清液,泳道2为流加甲醇4h发酵上清液,泳道3为流加甲醇8h发酵上清液,泳道4为流加甲醇12h发酵上清液,泳道5为流加甲醇18h发酵上清液,泳道6为流加甲醇27h发酵上清液,泳道7为流加甲醇30h发酵上清液,,其中箭头所指即为重组dp2条带。图4-b为重组dp2毕赤酵母菌株实施例2中小规模高密度发酵液od600和菌体湿重变化趋势图。图5为重组dp2毕赤酵母菌株实施例3中sds-page电泳图以及发酵液od600和菌体湿重变化趋势图。其中,图5-a为重组dp2毕赤酵母菌株实施例3中sds-page电泳图,泳道m为(10-230kda)宽范围的蛋白上样marker;泳道1为补料前发酵上清液,泳道2为流加甲醇0h发酵上清液,泳道3为流加甲醇6h发酵上清液,泳道4为流加甲醇12h发酵上清液,泳道5为流加甲醇18h发酵上清液,泳道6为流加甲醇24h发酵上清液,泳道7为流加甲醇30h发酵上清液,泳道8为流加甲醇36h发酵上清液,其中箭头所指即为重组dp2条带。图5-b为重组dp2毕赤酵母菌株实施例3中小规模高密度发酵液od600和菌体湿重变化趋势图。图6为dp2发酵液上清ph4.0第一步纯化结果。其中,图6-a为dp2发酵液第一步纯化色谱图;图6-b为dp2发酵液第一步纯化sds-page分析图,泳道1为纯化前样品,泳道2为非预染蛋白marker,泳道3为纯化穿透样品;泳道4-10为纯化洗脱峰各收集管。图7为dp2发酵液上清ph5.0第一步纯化结果。其中,图7-a为dp2发酵液第一步纯化色谱图;图7-b为dp2发酵液第一步纯化sds-page分析图,泳道1为纯化前样品,泳道2为非预染蛋白marker,泳道3为纯化穿透样品;泳道4-10为纯化洗脱峰各收集管。图8为dp2发酵液上清ph6.0第一步纯化结果。其中,图8-a为dp2发酵液第一步纯化色谱图;图8-b为dp2发酵液第一步纯化sds-page分析图,泳道1为纯化前样品,泳道2为非预染蛋白marker,泳道3为纯化穿透样品;泳道4-10为纯化洗脱峰各收集管。图9为dp2蛋白ph6.0第二步纯化结果。其中,图9-a为dp2蛋白第二步纯化色谱图;图9-b为dp2蛋白第二步纯化sds-page分析图,泳道1为纯化前样品,泳道2为纯化穿透样品,泳道3为洗脱样品,泳道4为非预染蛋白marker。图10为dp2蛋白ph7.5第二步纯化结果。其中,图10-a为dp2蛋白第二步纯化色谱图;图10-b为dp2蛋白第二步纯化sds-page分析图,泳道1非预染蛋白marker,泳道2为纯化前样品,泳道3为纯化穿透样品,泳道4为洗脱样品。图11为dp2/df2蛋白ph9.0第二步纯化结果。其中,图11-a为dp2蛋白第二步纯化色谱图;图11-b为dp2蛋白第二步纯化sds-page分析图,泳道1为纯化前样品,泳道2为非预染蛋白marker,泳道3为纯化穿透样品,泳道4为洗脱样品。图12为dp2蛋白ph6.0第三步纯化结果。其中,图12-a为dp2蛋白第三步纯化色谱图;图12-b为dp2蛋白第三步纯化sds-page分析图,泳道1为非预染蛋白marker,泳道2为纯化前样品,泳道3为纯化穿透样品,泳道4-9为洗脱峰各收集管样品。图13为dp2蛋白ph7.0第三步纯化结果。其中,图13-a为dp2蛋白第三步纯化色谱图;图13-b为dp2蛋白第三步纯化sds-page分析图,泳道1为非预染蛋白marker,泳道2为纯化前样品,泳道3为纯化穿透样品,泳道4-9为洗脱峰各收集管样品。图14为dp2蛋白ph8.0第三步纯化结果。其中,图14-a为dp2蛋白第三步纯化色谱图;图14-b为dp2蛋白第三步纯化sds-page分析图,泳道1为纯化前样品,泳道2为非预染蛋白marker,泳道3为纯化穿透样品,泳道4-8为洗脱峰各收集管样品。图15为dp2蛋白纯度hplc分析结果。其中图15-a为dp2蛋白rp-hplc分析结果;图15-b为df2蛋白rp-hplc分析结果。图16为dp2/df2蛋白氨基酸覆盖率分析结果。其中图16-a为dp2蛋白氨基酸覆盖率分析结果;图16-b为df2蛋白氨基酸覆盖率分析结果。图17为dp2与阳性血清库反应抑制曲线。图18为dp2300l发酵液上清第一步纯化结果。其中,图18-a为dp2发酵液第一步纯化色谱图;图18-b为dp2发酵液第一步纯化sds-page分析图,泳道1为11-94kd非预染蛋白marker,泳道2为纯化前样品,泳道3为纯化穿透样品;泳道4为洗脱峰1,泳道5-8为洗脱峰2。图19为dp2蛋白第二步纯化结果。其中,图19-a为dp2蛋白第二步纯化色谱图;图19-b为dp2蛋白第二步纯化sds-page分析图,泳道1为纯化前样品,泳道2为11-100kd非预染蛋白marker,泳道3为纯化后样品,泳道4为洗脱样品。图20为dp2蛋白第三步纯化结果。其中,图20-a为dp2蛋白第三步纯化色谱图;图20-b为dp2蛋白第三步纯化sds-page分析图,泳道1为纯化前样品,泳道2为非预染蛋白marker,泳道3为纯化穿透样品,泳道4-10为洗脱峰各收集管样品。具体实施方式下面结合具体实施例,进一步阐述本发明,应理解,引用实施例仅用于说明本发明而不用于限制本发明的范围。实施例1重组dp2/df2小规模(3l)高密度发酵工艺步骤1:重组菌株活化取冻存于-80℃的dp2工作种子库中甘油菌种子于ypd固体培养基(酵母提取物10g/l,蛋白胨20g/l,葡萄糖20g/l,琼脂糖15g/l)划线,30℃恒温恒湿箱培养3-5天。步骤2:种子液培养挑取固体培养基上单克隆菌落于ypd液体培养基(酵母提取物10g/l,蛋白胨20g/l,葡萄糖20g/l),30℃,220rpm培养至od600≈6.0,并在显微镜下观察无杂菌,即得到发酵用种子液。步骤3:发酵过程洗净赛多利斯b-plus生物反应器,用ph=7.0和ph=4.0的标准液分别校正发酵罐的ph计探头,然后配置bsm培养基作为发酵培养基(不同发酵批次bsm浓度见表1),倒入罐中,121℃高压灭菌20min,待温度降到50℃后,用浓氨水调ph=6.5±0.2。将步骤2中得到的种子液按照1:15(v/v,种子液/发酵培养基)比例接入生物反应器中。初期重组毕赤酵母菌体增殖阶段发酵温度为27℃,ph=5.5±0.2,转速=300rpm培养,do值100%,添加ptm1(2.4ml/l)。此阶段大约22h,期间do持续下降,通过增加搅拌转速、通气量以及通氧气将do维持在20%,当碳源消耗完毕后,溶氧值迅速上升,菌体湿重达到120g/l,此时进入限速生长阶段,此阶段的开始的3h以30ml/h-1l-1的速率补加50%甘油,3h后补料速度上调为60ml/h-1l-1。补加6h,菌体湿重约250g/l,停止补料,do上升,表明碳源耗尽,进入诱导阶段。(不同发酵批次诱导条件见表1)并维持do不高于30%,进行发酵表达,诱导开始后,每隔一段时间开始取样,同时在诱导初期中进行dospike,保证发酵罐罐内甲醇无积累,测发酵液od600吸收值以及菌体湿重。图1-3为三个批次菌体湿重发酵期间变化趋势图。表1重组dp2小规模高密度发酵工艺条件优化其中,bsm发酵培养基即是指life的发酵说明书上确定的标准浓度,即指k2so418.2g/l,mgso414.9g/l,koh0.93g/l,caso44.13g/l,h3po426.7ml/l,甘油40g/l,40%的bsm发酵培养基即是指life的发酵说明书上确定的标准浓度基础上各成分浓度都降为40%,即k2so47.28g/l,mgso45.96g/l,koh0.372g/l,caso41.652g/l,h3po410.68ml/l,甘油16g/l。重组dp2的上述发酵工艺对重组df2同样适用。实施例2重组dp2/df2大规模(30l)高密度发酵验证步骤1:重组菌株活化取冻存于-80℃的工作种子库中甘油菌种子于ypd固体培养基(酵母提取物10g/l,蛋白胨20g/l,葡萄糖20g/l,琼脂糖15g/l)划线,30℃恒温恒湿箱培养3-5天。步骤2:一级种子液培养挑取步骤1中固体培养基上单克隆菌落于ypd液体培养基(酵母提取物10g/l,蛋白胨20g/l,葡萄糖20g/l),30℃,220rpm培养至od600≈6.0,并在显微镜下观察无杂菌,即得到发酵用一级种子液。步骤3:二级种子液培养将步骤2中获得的一级种子液接种至赛多利斯b-plus发酵罐中,并以60%bsm,ph=5.5±0.2,27℃,300rpm值培养,通过通气和转速条件是溶氧保持在25%,最终至od600≈40,湿重达到约80g/l,并在显微镜下观察无杂菌,即得到发酵用二级种子液。步骤4:发酵过程洗净赛多利斯cplus生物反应器,用ph=7.0和ph=4.0的标准液分别校正发酵罐的ph计探头,然后按照实施例1中关键工艺参数的设计区间,确定发酵培养基为60%bsm进行配置发酵培养基20l,并倒入发酵罐中,121℃在线灭菌20min,待温度降到50℃后,用浓氨水调ph=5.5±0.2。将步骤3中得到的种子液按照1:15(v/v,种子液/发酵培养基)比例接入发酵罐中。初期重组毕赤酵母菌体增殖阶段发酵温度为27℃,ph=6.5±0.2,转速=300rpm培养,do值100%,添加ptm1(2ml/l)。此阶段大约20h,当碳源消耗完毕后,溶氧值迅速上升,菌体湿重达到100g/l,此时进入限速生长阶段,此阶段的开始的2h以30ml/h-1l-1的速率补加50%甘油,2h后补料速度上调为60ml/h-1l-1。补加4h,菌体湿重约200g/l,停止补料,do上升,表明碳源耗尽,进入诱导阶段。调节ph=6.5±0.2,以甲醇流加速度为30ml/h-1l-1进行诱导表达重组dp2;维持do不高于40%,诱导温度为20~22℃,ph=6.0±0.2,进行发酵表达,发酵开始后每隔一段时间取样一次,测发酵液od600吸收值以及菌体湿重,图4-b为重组dp2大规模发酵液od600吸收值以及菌体湿重发酵期间变化趋势图,由图可知,在重组毕赤酵母发酵期间,发酵液od600均能达到300以上,且菌体湿重均能达到400g/l。诱导40h发酵结束后,离心收集上清,sds-page电泳鉴定甲醇诱导不同时间的重组dp2产量(图4-a),说明发酵液od600吸收值、菌体湿重变化趋势以及发酵产物均与3l小规模高密度发酵保持一致。申请人因此确定,本发明的发酵工艺,完全能够进行产业化放大生产。重组dp2的上述发酵工艺对重组df2同样适用。实施例3重组dp2/df2中试(300l)高密度发酵验证步骤1:重组菌株活化取冻存于-80℃的工作种子库中甘油菌种子于ypd固体培养基(酵母提取物10g/l,蛋白胨20g/l,葡萄糖20g/l,琼脂糖15g/l)划线,30℃恒温恒湿箱培养3-5天。步骤2:一级种子液培养挑取步骤1中固体培养基上单克隆菌落于ypd液体培养基(酵母提取物10g/l,蛋白胨20g/l,葡萄糖20g/l),30℃,220rpm培养至od600≈6.0,并在显微镜下观察无杂菌,即得到发酵用一级种子液。步骤3:二级种子液培养将步骤2中获得的一级种子液接种至迪沃信bvt-3000型30l种子罐中,并以60%bsm,ph=5.5±0.2,27℃,300rpm值培养,通过通气和转速条件是溶氧保持在25%,最终至od600≈40,湿重达到约80g/l,并在显微镜下观察无杂菌,即得到发酵用二级种子液。步骤4:发酵过程洗净迪沃信bvt-3000型300l发酵罐,用ph=7.0和ph=4.0的标准液分别校正发酵罐的ph计探头,然后按照实施例1中关键工艺参数的设计区间,确定发酵培养基为60%bsm进行配置发酵培养基100l,并倒入发酵罐中,121℃在线灭菌20min,待温度降到50℃后,用浓氨水调ph=5.5±0.2。将步骤3中得到的种子液按照1:10(v/v,种子液/发酵培养基)比例接入发酵罐中。初期重组毕赤酵母菌体增殖阶段发酵温度为27℃,ph=6.5±0.2,转速=300rpm培养,do值100%,添加ptm1(2ml/l)。此阶段大约20h,当碳源消耗完毕后,溶氧值迅速上升,菌体湿重达到100g/l,此时进入限速生长阶段,此阶段的开始的2h以30ml/h-1l-1的速率补加50%甘油,2h后补料速度上调为60ml/h-1l-1。补加4h,菌体湿重约200g/l,停止补料,do上升,表明碳源耗尽,进入诱导阶段。以甲醇流加速度为30ml/h-1l-1进行诱导表达重组dp2;维持do不高于40%,诱导温度为20~22℃,ph=6.0±0.2,进行发酵表达,发酵开始后每隔一段时间取样一次,测发酵液od600吸收值以及菌体湿重,图5-b为重组dp2大规模发酵液od600吸收值以及菌体湿重发酵期间变化趋势图图。诱导40h发酵结束后,离心收集上清,sds-page电泳鉴定甲醇诱导不同时间的重组dp2产量(图5-a),说明发酵液od600吸收值、菌体湿重变化趋势以及发酵产物均与3l小规模、30l大规模高密度发酵保持一致。申请人因此确定,本发明的发酵工艺,完全能够进行产业化放大生产。重组dp2的上述发酵工艺对重组df2同样适用。实施例4重组dp2/df2(30l)发酵液纯化工艺一、重组dp2发酵液上清第一步纯化步骤1、发酵液的预处理按上述实施例中得到的发酵液高速离心,得到上清液;加入硅藻土助滤,过滤得到澄清的发酵液样品;用3kd膜包超滤稀释,降低电导至5ms/cm以下。步骤2、阳离子交换层析将上述处理后的发酵液上清乙酸调节ph4.0,上spff层析柱,柱型为bpg140/100,柱床体积2800ml,平衡缓冲液为50mmnaac,ph4.0,洗脱缓冲液为50mmnaac,1.0mnacl,ph4.0,按照0-100%线性洗脱,dp2/df2目的蛋白主要集中在第二个洗脱峰,图6-a为dp2蛋白第一步纯化色谱图,图6-b为dp2蛋白第一步纯化sds-page分析图。步骤1、2的其余步骤不变,调节样品和缓冲液ph5.0。图7-a为dp2蛋白第一步纯化色谱图,图7-b为dp2蛋白第一步纯化sds-page分析图。步骤1、2的其余步骤不变,调节样品和缓冲液ph6.0。图8-a为dp2蛋白第一步纯化色谱图,图8-b为dp2蛋白第一步纯化sds-page分析图。由图6至图8结果可知,随着ph增加,得到的dp2蛋白纯度更高,杂质更少,利于后续的纯化处理,其中ph6.0为最佳条件。二、重组dp2蛋白第二步纯化步骤1、超滤合并实施例步骤2中第二个洗脱峰,3kd膜包超滤稀释至电导<2.0ms/cm。步骤2、第二步阴离子交换层析将步骤1中样品加入20mmtris,调节ph6.0,上qff阴离子交换层析柱,柱型为hiscale50/40,柱床体积500ml,平衡缓冲液为20mmnah2po4,ph6.0,洗脱缓冲液为20mmnah2po4,1.0mnacl,ph6.0,收集穿透,0-100%线性洗脱,目的蛋白主要集中在穿透。图9-a为dp2蛋白第二步纯化色谱图,图9-b为dp2蛋白第二步纯化sds-page分析图。步骤1、2的其余步骤不变,调节样品和缓冲液ph7.5,电导10.0ms/cm。图10-a为dp2蛋白第二步纯化色谱图,图10-b为dp2蛋白第二步纯化sds-page分析图。步骤1、2的其余步骤不变,调节样品和缓冲液ph9.0,电导20.0ms/cm。图11-a为dp2蛋白第二步纯化色谱图,图11-b为dp2蛋白第二步纯化sds-page分析图。由图9至图11结果可知,图10结果最佳,经优化后目的蛋白与杂质分离度更高,目的蛋白大部分留在穿透中,杂质结合到介质中,实现了完全分离,且大大提高了dp2蛋白在穿透中的收率。三、重组dp2蛋白第三步纯化将第二步纯化后的样品加入硫酸铵至终浓度1.0m,调节ph6.0,上phenylff层析柱,柱型为hiscale50/40,柱床体积500ml,平衡缓冲液为20mmnah2po4,1.0m(nh4)2so4,ph6.0,洗脱缓冲液为20mmnah2po4,ph6.0,按照25,50%,70%,100%等度洗脱。图12-a为dp2蛋白第三步纯化色谱图,图12-b为dp2蛋白第三步纯化sds-page分析图。其余步骤不变,调节样品和平衡缓冲液硫酸铵浓度1.0m,ph7.0。图13-a为dp2蛋白第二步纯化色谱图,图13-b为dp2蛋白第二步纯化sds-page分析图。其余步骤不变,调节样品和平衡缓冲液硫酸铵浓度2.0m,ph8.0。图14-a为dp2蛋白第二步纯化色谱图,图14-b为dp2蛋白第二步纯化sds-page分析图。由图12至图14结果可知,图12结果最佳,经优化后得到的dp2蛋白纯度更高,蛋白量也最多,穿透中无目的蛋白,全部结合到填料中。表2所示经过优化后dp2蛋白纯化收率比优化前提高30%,活性(elisa方法测定)与工艺优化前相比也有所提高。表2.dp2蛋白纯化工艺优化前后收率重组dp2的上述纯化工艺对重组df2同样适用。实施例5重组dp2/df2蛋白鉴定1、浓缩,测定蛋白浓度将实施例3中梯度洗脱峰收集后用vivaflow50切向流超滤膜包(vf05p9-50cm2,sartorius)浓缩,透析袋置换缓冲溶液为ph7.4pbs溶液;piercebca蛋白浓度试剂盒测定蛋白浓度。2、dp2/df2蛋白hplc纯度分析将纯化得到的dp2/df2样品稀释至1mg/ml,0.22um滤膜过滤,即得到样品。流动相为20mmpb,ph7.5,流速1ml/min,进样10ul,rp柱分析纯度,图15-a,15-b为纯度检测结果,经三步纯化的dp2、df2蛋白纯度大于95%。3、dp2/df2蛋白氨基酸覆盖率分析氨基酸覆盖率分析是重组蛋白类药物质量研究中非常重要的指标之一,只有氨基酸一级结构的完全相同,才能保证产品具有与天然蛋白相同的的生物学活性。发明人委托上海中科新生命生物科技有限公司对dp2,df2蛋白进行氨基酸覆盖率分析,图16-a,16-b结果显示氨基酸覆盖率均为100%,符合相关政策法规的要求。4、dp2/df2蛋白活性分析利用竞争抑制elisa方法,对重组dp2,df2蛋白(rdp2,rdf2)生物学活性进行测定,并与天然dp2蛋白(ndp2)进行比较。具体步骤为(以dp2为例):1、包被:分别使用包被液(ph9.60.15m碳酸盐缓冲液)将rdp2、ndp2稀释至2ug/ml,100ul/孔,4℃孵育过夜。2、封闭:pbst(ph7.40.15mpbs+0.05%tween20)洗3次,拍干,每孔加入200ul封闭液(1%bsa/pbst),37℃孵育2h。3、样品稀释:将rdp2、ndp2样品进行30倍稀释,后进行3倍梯度稀释,共7个稀释度。取各个稀释度的样品和适当稀释度的血清库血清(15份以上phadia100检测d1/d2特异性ige值>100kua/l病人血清混合库)等体积混合,即125ul样品+125ul血清,混合后的样品放入4℃孵育过夜。阳性对照:125ul稀释液+125ul血清。4、加样:混合孵育好的样品加入相应的包被孔中,37℃孵育90min。5、二抗孵育:pbst洗4次,拍干,每孔加入100ul二抗稀释液(鼠抗人ige-hrp1:1500稀释),37℃孵育1h。6、显色:pbst洗4次,拍干,每孔加入100ultmbⅰ号显色液,37℃显色10min。7、终止及读数:每孔加入50ul2mh2so4终止反应,450nm波长处读数。8、结果处理:利用excel软件,以抑制率%(阳性值-样品值/阳性值)作为横坐标,以log10稀释倍数为纵坐标,做二次曲线拟合。将50%抑制率代入曲线方程,计算出稀释倍数×100作为生物学活性值(bu/ml)。比活(bu/mg)=活性值/蛋白浓度。实验结果如表3与图17所示,计算得rdp2活性值为125675bu/ml;比活为1.40e+05bu/mg。同理检测ndf2作为对照,实验结果如表4所示,说明酵母表达rdp2,rdf2与天然蛋白相比具有相似的生物学活性。表3:不同稀释倍数rdp2与阳性血清库反应od值od1od2od平均抑制率稀释倍数lg稀释倍数0.1060.1120.10993%301.47712130.2330.2430.23885%901.95424250.4950.5180.506568%2702.43136380.7480.7750.761552%8102.9084850.9210.9070.91442%24303.38560631.0341.0341.03435%72903.86272751.1111.1591.13529%218704.33984881.5871.5911.589阳性对照表4:重组蛋白与天然蛋白活性值比较5、dp2/df2宿主蛋白含量检测宿主残留蛋白(hostcellprotein,简称hcp)是指在生物制品中残留的宿主蛋白。这类蛋白成分复杂,种类繁多,且会因生产过程及纯化工艺的不同而发生变化。基因工程产品中残留的hcp是影响制品纯度的重要因素,反复使用含hcp的基因工程制品会引起机体的过敏反应,具有潜在的“佐剂效应”,也可能使机体对药物产生抗体,进而影响药物的疗效。hcp在生物制品中的残留量反映的不仅是制品批间的一致性,而且还是衡量生物制品质量的一个重要指标。发明人采用毕赤酵母hcp检测试剂盒(f140,cygnus)对制备的样品进行检测,表2显示经三步纯化的dp2,df2蛋白hcp含量均远低于2015版药典规定的重组生物制品(酵母)hcp最高限度。表5.dp2和df2蛋白hcp检测结果6、dp2/df2残留dna含量检测重组蛋白类药物虽然经过多步纯化,但制品中仍有可能残留宿主细胞的dna片段,这些残留dna可能带来传染性或致瘤性风险,可能会造成插入突变、导致抑癌基因失活、癌基因被激活等;此外,微生物来源的基因组dna富含cpg和非甲基化序列,增加了重组蛋白类药物在体内的免疫原性风险,因此who和各国药物注册监管机构对残留dna的限量要求非常严格。目前检测dna残留量的方法主要有dna探针杂交法、荧光染料法和qpcr法,其中前两种方法都存在技术缺陷,很难达到杂质限量检测的灵敏度,fda在最新版usp中规定了qpcr为唯一推荐的残留dna检测方法,因此发明者采用qpcr对样品中的残留dna进行了检测(sk030205p100,湖州申科生物技术有限公司),表3结果显示经三步纯化的样品残留dna含量均远低于2015版药典规定的重组生物制品(酵母)残留dna含量最高限度。表6.dp2和df2蛋白残留dna检测结果以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本
技术领域:
的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。实施例6重组dp2/df2(300l)纯化工艺一、重组dp2发酵液上清第一步纯化步骤1、发酵液的预处理按上述实施例3中得到的发酵液高速离心,得到上清液;加入硅藻土助滤,过滤得到澄清的发酵液样品;用3kd膜包超滤稀释,降低电导至5ms/cm以下。步骤2、阳离子交换层析将上述处理后的发酵液上清乙酸调节ph6.0,上spff层析柱,柱型为bpg140/100,柱床体积2800ml,平衡缓冲液为50mmnaac,ph6.0,洗脱缓冲液为50mmnaac,1.0mnacl,ph6.0,按照0-100%线性洗脱,dp2/df2目的蛋白主要集中在第二个洗脱峰,图18-a为dp2蛋白第一步纯化色谱图,图18-b为dp2蛋白第一步纯化sds-page分析图;可知,300ldp2发酵液第一步纯化工艺放大效果较好,纯化工艺得到很好的放大。二、重组dp2蛋白第二步纯化步骤1、超滤合并实施例4步骤2中第二个洗脱峰,3kd膜包超滤稀释至电导<2.0ms/cm。步骤2、第二步阴离子交换层析将步骤1中样品加入20mmtris,调节ph7.5,上qff阴离子交换层析柱,柱型为hiscale50/40,柱床体积500ml,平衡缓冲液为20mmtris,ph7.5,洗脱缓冲液为20mmtris,1.0mnacl,ph7.5,收集穿透,0-100%线性洗脱,目的蛋白主要集中在穿透。图19-a为dp2蛋白第二步纯化色谱图,图19-b为dp2蛋白第二步纯化sds-page分析图;可知,dp2蛋白第二步纯化工艺放大效果较好,纯化工艺得到很好的放大。三、重组dp2蛋白第三步纯化将第二步纯化后的样品加入硫酸铵至终浓度1.0m,调节ph6.0,上phenylff层析柱,柱型为hiscale50/40,柱床体积500ml,平衡缓冲液为20mmnah2po4,1.0m(nh4)2so4,ph6.0,洗脱缓冲液为20mmnah2po4,ph6.0,按照25,50%,70%,100%等度洗脱。图20-a为dp2蛋白第三步纯化色谱图,图20-b为dp2蛋白第三步纯化sds-page分析图;可知,dp1蛋白第三步纯化工艺放大效果较好,三步纯化得到的dp1蛋白纯度>95%。重组dp2的上述纯化工艺对重组df2同样适用。实施例7重组dp2/df2(300l发酵液纯化样品)蛋白活性分析按照实施例5步骤4中活性检测方法对300l发酵液制备得到的dp2和df2蛋白进行活性检测,以30l发酵液制备得到的样品和天然蛋白作为比较,表6显示300l发酵及纯化样品与30l及天然蛋白基本一致,处于相同水平;说明发酵及纯化工艺得到了较好的放大。表6:300l发酵纯化样品与30l发酵纯化及天然蛋白活性值比较当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1