N,N-二甲基环己烷叔胺衍生物作为制备聚氨酯和/或聚异氰脲酸酯泡沫催化剂的用途的制作方法
本发明涉及一种聚氨酯和/或聚异氰脲酸酯泡沫催化剂,尤其适用于反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))发泡体系的一种新的凝胶型催化剂n,n-二甲基环己烷叔胺衍生物的制备方法和应用。
背景技术:
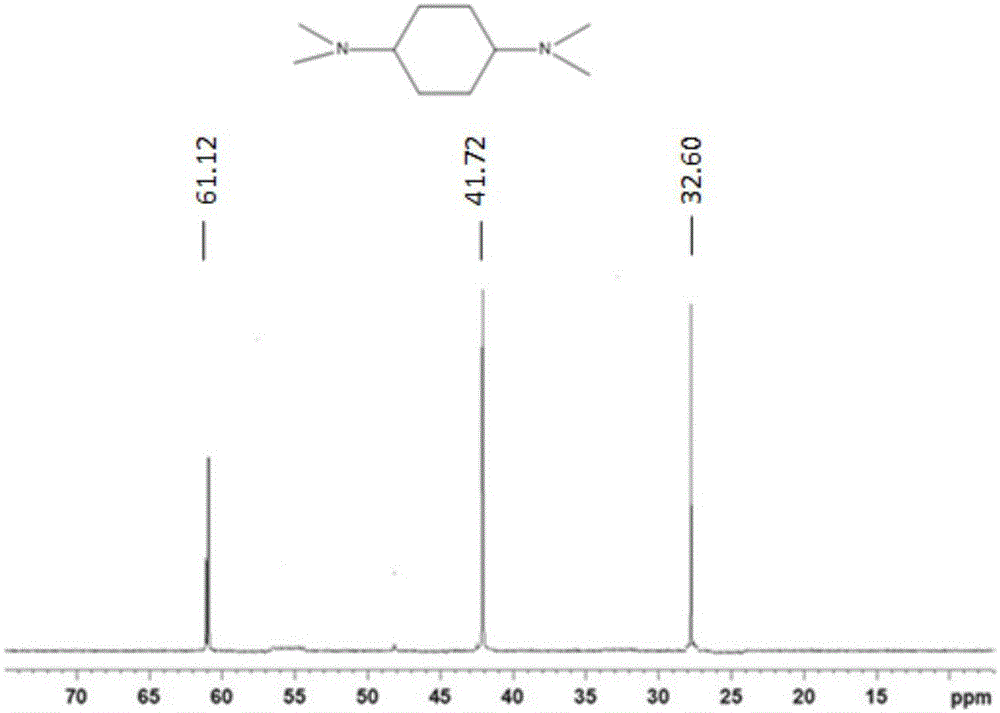
:近年来,低沸点化合物发泡剂的替代成为了聚氨酯泡沫领域的热点。目前已经经历里了四代发泡剂的变迁。其中第一代发泡剂以cfc-11为代表,其化学性质稳定、毒性低、沸点适中、发泡效率高、导热系数小等优良特性,几十年来一直被广泛使用,然而,该类化合物是破环地球臭氧层的元凶。cfcs进入平流层后,受到强烈紫外线照射释放出氯原子,与o3分子发生链反应,即使进入平流层的cfcs极少,也能导致臭氧层的破坏,形成臭氧空洞,使到达地球的紫外线uv-b辐射量增加,危及人类和其他生物的生存。为此43国在1987年签订了《对关于消耗臭氧层物质的蒙特利尔议定书》,对cfcs的使用提出控制。发达国家在1996年以前全面禁用,而发展中国家则可使用到2010年。替代cfcs的理想发泡剂应具备以下条件:(1)不含氯原子,不会对大气臭氧层造成破坏,即odp(臭氧消耗潜值)为零;(2)不会引起温室效应,即gwp(地球变暖潜值)为零;(3)安全、不易燃、无毒;(4)产品原料易得、生产简单、价格低廉;(5)在配方原料组分中化学稳定性好,并具有良好的互溶性;(6)沸点、潜热适中;(7)分子量低,导热系数变化率要小。经过实验研究,人们筛选出比较理想的cfcs的替代品,主要有:氢氟氯烃(hcfcs)、氢氟烃(hfcs)、戊烷系列及液态co2。综合多种因素,多数厂家倾向采用hcfc类化合物作为cfc-11的替代品,1994年前后,一氟二氯乙烷(hcfc-141b)已普遍被聚氨酯泡沫塑料行业承认为主要的第二代发泡剂。由于hcfc-141b的odp值并不为零,对臭氧层仍有损耗,它只是一种过渡发泡剂,根据蒙特利尔协议,hcfc-141b的使用期限为2020年。根据科技发展情况,禁用期限有可能提前,如欧美等国现已在pu硬泡生产中全面禁用hcfc-141b。目前世界范围内都在进行hcfc发泡剂在聚氨酯硬泡领域的替代工作,以减少发泡剂对大气臭氧层的破坏。市场上家电发泡剂的替代产品主要是第三代发泡剂:烷烃(环戊烷)及hfc类(245fa及365mfc),但这几种产品均有不足之处,环戊烷具有易燃的特性,因此在生产和应用过程中都存在安全隐患,而且泡沫隔热性能相对较差,难以满足日益提高的家电能效标准。hfc类产品虽然无臭氧层消耗(odp)问题,但gwp(全球变暖潜值)较高,还是不能满足全球日益严格的对温室效应气体的限制,而且365mfc本身也具有一定的可燃性。从冰箱生产实践中我们发现,245fa从工艺角度看,具有不可燃的特性,从而无需防爆设备的投资,并且泡沫制品具有更低的导热系数、更好的流动性、更好的强度和尺寸稳定性。但是,245fagwp值较高,这在将来全球日趋严格的环保要求下,必将被逐渐取代,而反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))正是针对这个缺点开发出的理想的新一代发泡剂。评估结果表明反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))的gwp值小于5。据估计,此发泡剂如能全球使用,将等效于每年降低6000万吨co2的排放。同时,由于反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))低mir值(乙烷的mir值为0.19±0.03,而反式-1-氯-3,3,3-三氟丙烯的mir值为0.16±0.02),将不会像烃类一样被定义为voc。研究发现,由霍尼韦尔公司开发出的低gwp发泡剂反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))是适用于聚氨酯发泡行业的一种能够同时满足各种工艺及环保要求的新一代发泡剂,具有高效节能、不燃、不含可挥发性有机物(voc)、低gwp、安全环保等特点。经过不断的配方及工艺参数的优化后,以反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))新一代高效节能环保发泡剂制得的聚氨酯泡沫和现有发泡剂体系(245fa和环戊烷)相比具有更为优异的导热系数和整机能耗水平,分别比相同型号的245fa以及环戊烷体系冰箱在导热系数方面降低7%(和245fa体系相比)和12%(和环戊烷体系相比),并且在整机能耗方面降低了3%(245fa)和7%(环戊烷)。反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))体系泡沫具有优良的力学强度,而且和冰箱内胆及钢板外壳粘结性能良好。反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))体系泡沫和环戊烷体系相比具有优异的流动性及密度分布,因此可以在环戊烷体系的基础上进一步降低注料量。反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))发泡剂对hips材料没有侵蚀性,相容性良好。总之,反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))体系泡沫很好地解决了现有环戊烷发泡体系冰箱的绝热性能较差、能耗高、易燃易爆、含可挥发性有机物(voc)等问题,在提升聚氨酯泡沫节能环保性能的同时,提升了泡沫流动性、密度分布和内胆之间相容性等工艺性能参数,并显著提高了冰箱生产和使用过程中的安全性。可以预见,反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))发泡剂对现有发泡体系绝热性能的升级换代具有建设性的推动作用。但反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))在碱性较强的叔胺催化剂作用下容易分解,导致组合料储存稳定性和发泡性能受到影响,其因此组合料的储存稳定性大大降低。在国内,反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))已经开始在冰箱组合料中广泛使用;在欧美,反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))除了在冰箱组合料中的应用外,在喷涂组合料中也得到了广发使用。而五甲基二乙烯三胺、三乙烯二胺、1,3,5-三(二甲氨基丙基)六氢三嗪是硬泡组合料中用量较大的胺类催化剂,多家研究单位的实验研究表明五甲基二乙烯三胺、1,3,5-三(二甲氨基丙基)六氢三嗪、三乙烯二胺等传统胺类催化剂容易与反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))亲核反应,生成具有氮正离子和碳负离子的中间体,但是这样结构的物质非常不稳定,一定条件下容易分解成结构相对稳定的物质同时产生部分的f-和cl-。进一步导致组合料体系的稳定性和反应活性都出现了问题,不能作为正常体系使用。原因是:活性高、位阻低的叔胺类催化剂会和分子结构中同时含有氯离子和碳碳双键的hcfo-1233zd(e)发生亲核反应,生成具有氮正离子和碳负离子的中间体,但是这样结构的物质非常不稳定,一定条件下容易分解成结构相对稳定的物质同时产生部分的f-和cl-。而n,n-二甲基环己胺和n,n-二甲基苄胺等空间位阻较大的催化剂与hcfo-1233zd(e)的反应性较弱,较为稳定,不易导致组合聚醚体系不稳定。因此针对现有胺类催化剂与反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))之间不匹配的问题,需要寻求一种新的催化剂体系,克服反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))组合聚醚体系稳定性差的缺陷。技术实现要素:本发明的一个目的在于提供一种n,n-二甲基环己烷叔胺衍生物作为制备聚氨酯和/或聚异氰脲酸酯泡沫催化剂的用途,新催化剂n,n-二甲基环己烷叔胺衍生物的结构与n,n-二甲基环己胺的结构很相似,但多了一个叔胺基团。其催化效果比n,n-二甲基环己胺强烈,且偏凝胶作用。该新型催化剂是活性较高的胺类催化剂,用于冰箱硬泡、板材、喷涂、现场灌注聚氨酯硬泡。该类催化剂对凝胶和发泡都有催化作用,但偏凝胶作用,它对水与异氰酸酯的反应(发泡反应)有催化作用,但对多元醇与异氰酸酯的反应也较强的催化性,可以在反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))组合料体系中替代三乙烯二胺的凝胶作用,且具有一定的发泡催化作用,可以替代部分n,n,n’,n〞,n〞-五甲基二亚乙基三胺,主要用于反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))的喷涂体系,且能降低施工过程的胺臭味。除用于硬泡,也可用于模塑软泡及半硬泡等的辅助催化剂。本发明的另一个目的在于提供一种发泡组合物,将新的叔胺催化剂用到反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))组合聚醚体系中,替代三乙烯二胺或n,n,n,n,n-五甲基三乙烯二胺等现有催化剂,从而克服了反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))组合聚醚体系稳定性差的弱点。且催化剂的合成原料易得,工艺条件易于控制。为实现以上发明目的,本发明的技术方案如下:n,n-二甲基环己烷叔胺衍生物作为制备聚氨酯和/或聚异氰脲酸酯泡沫催化剂的用途,所述的n,n-二甲基环己烷叔胺衍生物结构式如式(1)所示:其中,r基团代表叔胺基类衍生物,如-n(ch3)2,或c1~c10烷基-n(ch3)2,或芳香基-n(ch3)2,优选式(3)~(6)结构:这类叔胺化合物的位阻大、电位低,不易与hcfo-1233zd(e)发生亲核反应,也就不会导致其分解,因此hcfo-1233zd(e)的组合聚醚体系相对稳定。为了得到n,n-二甲基环己烷叔胺衍生物,所采用的原料是如下结构式(2)所示:r1基团代表伯胺基类衍生物,如-nh2,或c1~c10烷基-nh2,或芳香基-nh2等,可以被加氢胺化得到叔胺基团。环状结构的胺类催化剂有一定的选择性,偏向于凝胶反应,因此这类催化剂可用来替代三乙烯二胺的凝胶作用。本发明中,所述的n,n-二甲基环己烷叔胺衍生物的制备方法如下:将原料结构式(2)所示的伯胺化合物与甲醛、氢气在催化剂的作用下进行反应,得到油水两相的反应液,其中油相主要含有叔胺化合物,水相主要含甲醛;将反应液分相(例如经碱洗),油相再进行精馏提纯得到n,n-二甲基环己烷叔胺衍生物。其化学反应方程式通式如下:其中,所述的甲醛采用甲醛水溶液和/或多聚甲醛的粗解聚水溶液,例如10-40wt%,优选约37wt%甲醛的水溶液;甲醛与脱氨轻组分的摩尔比为2-10:1,优选2-4:1。其中,所述反应的催化剂选自负载型钯系催化剂和/或雷尼型催化剂,优选负载型钯系催化剂,所述负载型钯系催化剂包含钯、助剂及载体,其中,钯的含量为0.1-50wt%,优选为2-10wt%,所述助剂选自钌、铑、铂、钴、铜中的一种或多种,助剂的含量是0.02-3wt%,优选0.05-2wt%,所述助剂优选钌和铑,其中钌的含量为0.5-2wt%,铑的含量为0.05-1wt%,以负载型钯系催化剂的总质量计算,载体选自氧化铝、二氧化硅、硅胶、硅藻土、沸石分子筛、活性炭、二氧化钛、铝酸锂和氧化锆中的一种或两种或多种。进一步地,反应温度为40-200℃,优选60-160℃;反应压力(表压)为0.5-10mpa,优选1-5mpa。进一步地,氢气的引入压力可以为0.5-3mpa,优选约1-2.5mpa。催化剂的用量相对于原料分可以为1-5%(质量分数),优选2-3%。本发明中,所述负载型钯系催化剂可以按常规浸渍法制备,例如所述负载型钯系催化剂的制备步骤包括:按照比例,将钯的金属盐和选自钌、铑、铂、钴、铜中的一种或多种的金属盐溶于去离子水中形成浓度约10-40wt%的均一溶液;然后加入载体,旋转浸渍后蒸干水分,烘干和焙烧,冷却后得到负载型钯系催化剂。在一个更具体的实施方式中,按照比例,将钯的金属盐和选自钌、铑、铂、钴、铜中的一种或多种的金属盐溶于60-80℃去离子水中形成浓度约10-40wt%的均一溶液;然后加入载体,在60-80℃下旋转浸渍4-6h后逐渐蒸干水分;于100-120℃烘箱中烘10-16小时;最后移至马弗炉,在空气气氛中以2-3℃/min升温至500-600℃焙烧6-8小时,自然冷却后即可得到所述负载型钯系催化剂。本发明所述的精馏提纯采用常压、减压精馏操作,优选减压精馏。精馏提纯压力(绝压)可以为0.1-2kpa,优选0.5-1.2kpa,精馏柱理论塔板数为20-50块,优选约30-45块,回流比为3-20:1,优选约5-15:1的条件下进行。本发明所述的方法制备的叔胺化合物作为制备聚氨酯和/或聚异氰脲酸酯泡沫的催化剂。本发明所述的用途,其中:将叔胺化合物可以单独使用也可以与制备聚氨酯和/或聚异氰脲酸酯泡沫所常用叔胺类、有机锡类以及金属盐类催化剂组合使用。优选将n,n-二甲基环己烷叔胺衍生物与n,n,n’,n〞,n〞-五甲基二亚乙基三胺、n,n,n′,n〞,n〞-五甲基二亚丙基三胺、双(2-二甲基氨基乙基)醚、2,4,6-三(二甲氨基甲基)苯酚、n-甲基吗啉、n-乙基吗啉、2,2-双吗啉基二乙基醚、n-甲基咪唑、1,2-二甲基咪唑、二氮杂二环、1,4-二甲基哌嗪、n,n,n-三甲基氨乙基哌嗪、三(二甲氨丙基)胺、1,3,5-三(二甲基胺丙基)-1,3,5-六氢化三嗪、n,n-二甲基环己胺、n,n-二甲基苄胺、辛酸亚锡、异辛酸钾、二(十二烷基硫)二丁基锡、二醋酸二丁基锡、二丁基锡二月桂酸酯、醋酸钾等中的一种或两种或多种组合使用。催化剂或催化剂组合物用量以每100重量份多元醇为基准(phr)计可为1~15份。优选的用量为2~12份。本发明所述的制备聚氨酯和/或聚异氰脲酸酯泡沫是将聚合多异氰酸酯与至少一种含活泼氢的化合物在催化剂及发泡剂的作用下反应,制得聚氨酯和/或聚异氰脲酸酯泡沫的过程。含活泼氢的化合物是至少一种聚醚多元醇、至少一种聚酯多元醇,或其任意组合。合适多元醇的例子是聚亚烷基醚型和聚酯型多元醇。聚亚烷基醚型多元醇包括聚环氧烷聚合物,例如聚环氧乙烷和聚环氧丙烷聚合物和共聚物,它们的端羟基是由包括二元醇和三元醇在内的多元醇化合物衍生而来的;例如其中包括乙二醇、丙二醇、1,3-丁二醇、1,4-丁二醇、1,6-己二醇、新戊二醇、一缩二乙二醇、一缩二丙二醇、季戊四醇、甘油、一缩二甘油、三羟甲基丙烷以及类似的低分子量多元醇。本发明所述的聚合多异氰酸酯是采用的本领域熟知的pm-200或者其他类似的多亚甲基多苯基异氰酸酯,俗称聚合mdi或粗mdi。还有合适的是这些多异氰酸酯的“预聚物”,包括多异氰酸酯与聚醚或聚酯型多元醇的部分预反应的混合物。本发明所述的配方中,还可加入在制备聚氨酯泡沫塑料配方中所常见的其他类型的助剂:包括链增长剂例如乙二醇、丁二醇、二甘醇等;交联剂例如二乙醇胺、二异丙醇胺、三乙醇胺和三丙醇胺等;发泡剂例如水、戊烷、141b、甲酸甲酯等;泡沫稳定剂如硅氧烷等。一种发泡组合物,基于组合物总重,包含如下组分:a,反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))发泡剂,用量2~20%,b,一种或多种多元醇,优选包括聚醚多元醇和聚酯多元醇,用量50~70%,c,一种或多种硅油,1~2.5%,d,水及其他助剂,1~30%,e,其他催化剂和n,n-二甲基环己烷叔胺衍生物,催化剂总用量约1~10%。(跟权利要求书同样修改)所述的n,n-二甲基环己烷叔胺衍生物结构式所述的n,n-二甲基环己烷叔胺衍生物结构式如式(1)所示:其中,r基团代表叔胺基类衍生物,如-n(ch3)2,或c1~c10烷基-n(ch3)2,或芳香基-n(ch3)2,优选式(3)~(6)结构:其中,所述的多元醇包括聚醚多元醇和聚酯多元醇,优选包括蔗糖聚醚、山梨醇聚醚、甘油聚醚、丙二醇聚醚、脂肪族聚酯、芳香族聚酯中的一种或多种。其中,所述的其他催化剂包含叔胺类、季铵盐类、有机锡类以及金属盐类催化剂中的一种或多种组合使用;优选n,n-二甲基环己胺、n,n-二甲基苄胺、2-羟基-n,n,n-三甲基-1-1丙胺甲酸盐、n-甲基吗啉、n-乙基吗啉、2,2-双吗啉基二乙基醚、n-甲基咪唑、1,2-二甲基咪唑、二氮杂二环、1,4-二甲基哌嗪、n,n,n-三甲基氨乙基哌嗪、三(二甲氨丙基)胺、辛酸亚锡、异辛酸钾、二(十二烷基硫)二丁基锡、二醋酸二丁基锡、二丁基锡二月桂酸酯、醋酸钾中的一种或多种。其中,所述的其他助剂包括包括交联剂例如二乙醇胺、二异丙醇胺、三乙醇胺和三丙醇胺等;阻燃剂包含tcpp等。上述的发泡组合物可以与多异氰酸酯反应制备聚氨酯和/或聚异氰脲酸酯泡沫。本发明的积极效果是:将具有类似n,n-二甲基环己胺结构的n,n-二甲基环己烷叔胺衍生物作为一类用作制备聚异氰脲酸酯/聚氨酯的催化剂,与三乙烯二胺相比,虽然催化活性稍弱,但其在反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))组合聚醚体系中,不与发泡剂反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))反应,不会导致其分解,体系稳定,且具有很强的凝胶效果,可以替代三乙烯二胺在喷涂泡沫体系中的使用。也可以应用在家电组合聚醚或其他浇注型组合聚醚中,替代n,n-二甲基环己胺等偏凝胶的催化剂。且其具有较低的胺臭味,对操作人员的毒害性小,催化活性高,可以减少臭味大的n,n-二甲基环己胺的使用。其与聚氨酯泡沫最常用的催化剂三乙烯二胺相比,克服了与反式-1-氯-3,3,3-三氟丙烯(hcfo-1233zd(e))反应的弊端,且能够提供良好的凝胶催化效果;与n,n-二甲基环己胺相比,用量更少,发泡过程气味更小,凝胶效果更好。附图说明图1为实施例3制备的产品的核磁共振谱图。图2为实施例4制备的产品的核磁共振谱图。图3为实施例5制备的产品的核磁共振谱图。图4为实施例6制备的产品的核磁共振谱图。具体实施方式本发明通过以下实施对本发明进行进一步的说明,但本发明并不限于所列出的实施例。以下实施例中进行气相色谱分析的条件为:安捷伦db-5色谱柱,进样口温度280℃,fid检测器温度300℃,柱流速1.5ml/min,氢气流速35ml/min,空气流速350ml/min,程序升温方式为60℃保持1min,以20℃/min升温至280℃,保持10min。1hnmr分析仪器型号:avanceⅲ400mhz,厂家:bruker。实施例1制备钯系催化剂1:将25.04g二水合硝酸钯、0.14g硝酸铑、2.76g醋酸钌溶于100ml去离子水中,加热至60℃形成均一溶液,再加入88.95g氧化铝(平均粒径50μm,比表面积180m2/g,孔容0.30cc/g),在70℃的水浴中旋转浸渍5h后逐渐蒸干水分,于100℃烘箱中烘16小时;最后移至马弗炉,在空气气氛中以3℃/min升温至550℃焙烧8小时,自然冷却后即可得到催化剂。催化剂的组成为:pd为10wt%,rh为0.05wt%,ru为1wt%,其余为氧化铝,以相应金属元素占催化剂总质量计,金属以氧化态形式存在,使用时需要还原。实施例2制备钯系催化剂2:将5.01g二水合硝酸钯、1.40g硝酸铑、5.51g醋酸钌溶于100ml去离子水中,加热至70℃形成均一溶液,再加入95.5g二氧化硅(平均粒径60μm,比表面积240m2/g,孔容0.38cc/g),在60℃的水浴中旋转浸渍6h后逐渐蒸干水分,于120℃烘箱中烘12小时;最后移至马弗炉,在空气气氛中以2℃/min升温至600℃焙烧6小时,自然冷却后即可得到催化剂。催化剂的组成为:pd为2wt%,rh为0.5wt%,ru为2wt%,其余为二氧化硅,以相应金属元素占催化剂总质量计,金属以氧化态形式存在,使用时需要还原。实施例3制备反式n,n,n,n-四甲基-1,4-环己烷二胺:向1l的反应釜中加入4g的实施例1中的钯系催化剂,加入甲醇溶剂打底,密封反应釜,分别用氮气和氢气置换三遍,在温度为240℃,氢气压力为5mpa下活化催化剂6小时。之后降温、泄压、氮气置换三遍,将溶剂从反应釜中过滤出来。之后加入100g的反式1,4-环己烷二胺,依次用氮气和氢气置换三遍,起始氢气压力为2mpa,开启搅拌为700转/分钟,等反应温度升至160℃,将氢气压力调整到3mpa并持续通氢,开始用平流泵以2g/min的速度往反应釜中通37%的甲醛水溶液170g,待流量计的瞬时流量低于50sccm,关闭氢气阀门,再继续反应半小时停止反应。之后降温、泄压、用氮气置换三遍、过滤得到油水两相反应液。往反应液中加入5g市售分析纯的氢氧化钠,在60℃下搅拌2h,用梨形分液漏斗进行分相,取油相进行气相色谱分析,对反式1,4-环己基二胺的含量为0.4%,n,n,n,n-四甲基-1,4-环己烷二胺的含量为97.8%。再将上述操作得到的油相进行减压精馏,在压力为1kpa,精馏柱理论塔板数为30块,回流比为5:1下减压精馏得到反式n,n,n,n-四甲基-1,4-环己烷二胺纯度为99.5%的产品。由1hnmr分析表明,该产物为所需的结构,谱图如图1所示。实施例4制备n,n,n,n-四甲基-1-亚甲基-环己胺:向1l的反应釜中加入4g的实施例1中的钯系催化剂,加入甲醇溶剂打底,密封反应釜,分别用氮气和氢气置换三遍,在温度为240℃,氢气压力为5mpa下活化催化剂6小时。之后降温、泄压、氮气置换三遍,将溶剂从反应釜中过滤出来。之后加入110g的4-氨基亚甲基-环己胺,依次用氮气和氢气置换三遍,起始氢气压力为2mpa,开启搅拌为700转/分钟,等反应温度升至160℃,将氢气压力调整到3mpa并持续通氢,开始用平流泵以2g/min的速度往反应釜中通37%的甲醛水溶液200g,待流量计的瞬时流量低于50sccm,关闭氢气阀门,再继续反应半小时停止反应。之后降温、泄压、用氮气置换三遍、过滤得到油水两相反应液。往反应液中加入5g市售分析纯的氢氧化钠,在60℃下搅拌2h,用梨形分液漏斗进行分相,取油相进行气相色谱分析,4-氨基亚甲基-环己胺的含量为0.5%,n,n,n,n-四甲基-1-亚甲基-环己胺的含量为98.9%。再将上述操作得到的油相进行减压精馏,在压力为1kpa,精馏柱理论塔板数为30块,回流比为5:1下减压精馏得到n,n,n,n-四甲基-1-亚甲基-环己胺纯度为99.3%的产品。由1hnmr分析表明,该产物为所需的结构,谱图如图2所示。实施例5制备4,4′-亚甲基双(n,n-二甲基环己胺):向1l的反应釜中加入4g的实施例2中的钯系催化剂,加入甲醇溶剂打底,密封反应釜,分别用氮气和氢气置换三遍,在温度为240℃,氢气压力为5mpa下活化催化剂6小时。之后降温、泄压、氮气置换三遍,将溶剂从反应釜中过滤出来。之后加入200g的二氨基二环己基甲烷(简称hmda)的反应产物,依次用氮气和氢气置换三遍,起始氢气压力为1mpa,开启搅拌为700转/分钟,等反应温度升至160℃,将氢气压力调整到5mpa并持续通氢,开始用平流泵以2g/min的速度往反应釜中通37%的甲醛水溶液200g,待流量计的瞬时流量低于50sccm,关闭氢气阀门,再继续反应半小时停止反应。之后降温、泄压、用氮气置换三遍、过滤得到油水两相反应液。往反应液中加入5g市售分析纯的氢氧化钠,在60℃下搅拌2h,用梨形分液漏斗进行分相,取油相进行气相色谱分析,二氨基二环己基甲烷(简称hmda)反应产物的含量为0.2%,反式4,4′-亚甲基双(n,n-二甲基环己胺)的含量为97.4%。再将上述操作得到的油相进行减压精馏,在压力为1kpa,精馏柱理论塔板数为30块,回流比为5:1下减压精馏得到反式4,4′-亚甲基双(n,n-二甲基环己胺)纯度为99.6%的产品。由1hnmr分析表明,该产物为所需的结构,谱图如图3所示。实施例6制备n,n,n′,n′-四甲基-1,4-二亚甲基-二环己烷:向1l的反应釜中加入4g的实施例2中的钯系催化剂,加入甲醇溶剂打底,密封反应釜,分别用氮气和氢气置换三遍,在温度为240℃,氢气压力为5mpa下活化催化剂6小时。之后降温、泄压、氮气置换三遍,将溶剂从反应釜中过滤出来。之后加入200g的1,4′-二氨基-1,4-二亚甲基-二环己烷,依次用氮气和氢气置换三遍,起始氢气压力为2mpa,开启搅拌为700转/分钟,等反应温度升至160℃,将氢气压力调整到3mpa并持续通氢,开始用平流泵以2g/min的速度往反应釜中通37%的甲醛水溶液200g,待流量计的瞬时流量低于50sccm,关闭氢气阀门,再继续反应半小时停止反应。之后降温、泄压、用氮气置换三遍、过滤得到油水两相反应液。往反应液中加入5g市售分析纯的氢氧化钠,在60℃下搅拌2h,用梨形分液漏斗进行分相,取油相进行气相色谱分析,对1,4′-二氨基-1,4-二亚甲基-二环己烷的含量为0.8%,n,n,n,n-四甲基-1,4-二亚甲基-二环己胺的含量为97.5%。再将上述操作得到的油相进行减压精馏,在压力为1kpa,精馏柱理论塔板数为35块,回流比为5:1下减压精馏得到n,n,n′,n′-四甲基-1,4-二亚甲基-二环己烷纯度为99.5%的产品。由1hnmr分析表明,该产物为所需的结构,谱图如图4所示。实施例7采用表1所列的配方制备预混配料:表2:组合聚醚在老化前后的的外观、酸值、f-和cl-的变化配方1(对比例)配方2配方3老化前颜色微黄微黄微黄45℃老化后颜色深橘微黄浅黄老化前酸值0.1760.1450.12945℃老化后酸值16.221.9572.011老化前f-/ppm20182245℃老化后f-/ppm5500202545℃老化前cl-/ppm00045℃老化后cl-/ppm25296从表2中可以看出,45℃老化后,配方1(对比例)的外观、酸值、f-和cl-的含量均发生了明显的变化,说明组合聚醚中的发泡剂反式-1-氯-3,3,3-三氟丙烯发生了分解。而配方2~3的外观、酸值、f-和cl-的含量变化很小,基本可以视为不变,可见组合聚醚中的发泡剂反式-1-氯-3,3,3-三氟丙烯没有发生了分解。三乙烯二胺对反式-1-氯-3,3,3-三氟丙烯的影响较大,导致反式-1-氯-3,3,3-三氟丙烯分解。而反式n,n,n,n-四甲基-1,4-环己烷二胺和反式-1-氯-3,3,3-三氟丙烯影响较小。表3:拉丝时间对比将组合聚醚与pm-200分别在22℃下恒温3小时,然后将两者混合,迅速以3000rpm的速度搅拌7s,然后观察其起拉丝时间。本发明中,主要以拉丝时间为特征参数表征催化剂的催化活性、聚氨酯泡沫制备过程的反应时间。其中,拉丝时间(又称纤维时间,凝胶时间)是指从物料混合开始至泡沫中开始可以抽出3厘米丝状纤维的时间。配方1(对比例)和配方3在刚配好时的拉丝时间相同,但用量不同;而配方1(对比例)和配方2的催化剂用量相同时,拉丝时间不同,说明两种催化剂的催化活性不同;有拉丝时间比较,可以看出催化剂的催化活性是:三乙烯二胺>反式n,n,n,n-四甲基-1,4-环己烷二胺。将3款配方的组合聚醚放在45℃下劣化三周后,配方1的组合聚醚发生了明显的变化,不仅组合聚醚的颜色发深,状态变浑,拉丝时间也已经测不出,体系已经不能发泡。说明配方1(对比例)的组合聚醚已经被破坏了。而配方2~3的颜色和;拉丝时间均没有发生明显的变化。说明配方2~3在45℃下放置三周后,体系相对稳定。实施例8表4:预混配料的配方比例表5:实验结果将组合聚醚与pm-200分别在22℃下恒温3小时,然后将两者混合,迅速以3000rpm的速度搅拌2s,然后观察其起拉丝时间。本发明中,主要以拉丝时间为特征参数表征催化剂的催化活性、聚氨酯泡沫制备过程的反应时间。其中,拉丝时间(又称纤维时间,凝胶时间)是指从物料混合开始至泡沫中开始可以抽出3厘米丝状纤维的时间。配方4(对比例)和配方6在刚配好时的拉丝时间相同,但用量不同;而配方4(对比例)和配方5的催化剂用量相同时,拉丝时间不同,说明三乙烯二胺>n,n,n,n-四甲基-1-亚甲基-环己胺的催化活性不同;由拉丝时间比较,可以看出催化剂的催化活性分别是:三乙烯二胺>n,n,n,n-四甲基-1-亚甲基-环己胺,与实施案例7的结论相同。将配方4~6的组合聚醚放在45℃下劣化三周后,配方4的组合聚醚发生了明显的变化,不仅组合聚醚的颜色发深,状态变浑,拉丝时间也已经测不出,体系已经不能发泡。说明配方4(对比例)的组合聚醚已经被破坏了。而配方5和6均没有发生明显的变化。说明配方5~6在45℃下放置三周后,虽然拉丝时间稍有变化,但变化不大,体系相对稳定。由此可以看出,n,n,n,n-四甲基-1-亚甲基-环己胺的组合聚醚比三乙烯二胺的组合聚醚更稳定,且n,n,n,n-四甲基-1-亚甲基-环己胺可以替代三乙烯二胺的作用。实施例9表6:预混配料的配方比例表7:实验结果将组合聚醚与pm-200分别在22℃下恒温3小时,然后将两者混合,迅速以3000rpm的速度搅拌2s,然后观察其起拉丝时间。本发明中,主要以拉丝时间为特征参数表征催化剂的催化活性、聚氨酯泡沫制备过程的反应时间。其中,拉丝时间(又称纤维时间,凝胶时间)是指从物料混合开始至泡沫中开始可以抽出3厘米丝状纤维的时间。配方7(对比例)和配方9在刚配好时的拉丝时间相同,但用量不同;而配方7(对比例)和配方8的催化剂用量相同时,拉丝时间不同,说明三乙烯二胺>4,4′-亚甲基双(n,n-二甲基环己胺)的催化活性不同;由拉丝时间比较,可以看出催化剂的催化活性分别是:三乙烯二胺>4,4′-亚甲基双(n,n-二甲基环己胺),与实施案例7的结论相同。将配方7~9的组合聚醚放在45℃下劣化三周后,配方7(对比例)的组合聚醚发生了明显的变化,不仅组合聚醚的颜色发深,状态变浑,拉丝时间也已经测不出,体系已经不能发泡。说明配方7(对比例)的组合聚醚已经被破坏了。而配方8和9均没有发生明显的变化。说明配方8和9在45℃下放置三周后,虽然拉丝时间稍有变化,但变化不大,体系相对稳定。由此可以看出,4,4′-亚甲基双(n,n-二甲基环己胺)的组合聚醚比三乙烯二胺的组合聚醚更稳定,且4,4′-亚甲基双(n,n-二甲基环己胺)可以替代三乙烯二胺的作用。实施例10表8:预混配料的配方比例表9:实验结果将组合聚醚与pm-200分别在22℃下恒温3小时,然后将两者混合,迅速以3000rpm的速度搅拌2s,然后观察其起拉丝时间。本发明中,主要以拉丝时间为特征参数表征催化剂的催化活性、聚氨酯泡沫制备过程的反应时间。其中,拉丝时间(又称纤维时间,凝胶时间)是指从物料混合开始至泡沫中开始可以抽出3厘米丝状纤维的时间。配方10(对比例)和配方11在刚配好时的拉丝时间相同,但用量不同;而配方10(对比例)和配方11的催化剂用量相同时,拉丝时间不同,说明三乙烯二胺>n,n,n′,n′-四甲基-1,4-二亚甲基-二环己烷的催化活性不同;由拉丝时间比较,可以看出催化剂的催化活性分别是:三乙烯二胺>n,n,n′,n′-四甲基-1,4-二亚甲基-二环己烷,与实施案例7的结论相同。将配方10~12的组合聚醚放在45℃下劣化三周后,配方10(对比例)的组合聚醚发生了明显的变化,不仅组合聚醚的颜色发深,状态变浑,拉丝时间也已经测不出,体系已经不能发泡。说明配方10(对比例)的组合聚醚已经被破坏了。而配方11和12均没有发生明显的变化。说明配方11、12在45℃下放置三周后,虽然拉丝时间稍有变化,但变化不大,体系相对稳定。由此可以看出,n,n,n′,n′-四甲基-1,4-二亚甲基-二环己烷的组合聚醚比三乙烯二胺的组合聚醚更稳定,且n,n,n′,n′-四甲基-1,4-二亚甲基-二环己烷可以替代三乙烯二胺的作用。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1