苏木酮A的制备和纯化方法与流程
本发明属于制药领域,特别涉及一种苏木酮a的制备和纯化方法。
背景技术:
苏木酮a作为重要中药材苏木中得一种活性成分,近年开始应用于广泛得研究。
北京大学的屠鹏飞教授等在balb/c小鼠炎症模型上验证了苏木酮a的抗神经炎症作用,验证了该化合物可以直接靶向impdh2蛋白酶,有可能进一步开发成具有抗神经炎症的药物。另外,康宁博士等在临床与实验病理学杂志jclinexppathol2018aug;34(8)上也报道了苏木酮a对顺铂所致肾损伤中的抗氧化应激作用,研究发现苏木酮a能显著改善小鼠的肾功能。
苏木酮a制备方面,srivenkatateswarauniversity的vidavalursiddaiah,chundurivenkatarao等报道了采用7-羟基色满-4-酮与原二茶醛在哌啶盐酸盐催化下反应获得,其纯化方法主要采用柱层析法,尚无具体粗品结晶方法。indianinstituteofchemicaltechnology的biswanathdas,ponnaboinathirupathi等在chem.pharm.bull.57(10)1139—1141(2009)上报道采用乙酸乙酯作为反应溶剂,获得的苏木酮a经过柱层析纯化获得,但是其收率低下,制备成本高,不适宜大量生产,其通过植物提取获得的纯品仅7mg左右。采用合成的方法条件苛刻纯化难度大,收率低下,难以纯化
另外一些文献公开的方法均存在获得的产品纯度低,成本高,环境污染大,无法量产等问题。
在此需要说明的是,在背景技术部分中公开的上述信息仅用于增强对本发明构思背景的理解,因此,它可能包含不构成本领域普通技术人员已知的现有技术的信息。
技术实现要素:
本发明合成工艺如下:
上述合成工艺中,步骤i采用市售的间苯二酚与3-氯丙酸在酸催化下酰化反应,催化酸可以选用多聚磷酸,硫酸,甲磺酸等;反应速度快,副产物较少,不用纯化即可进行第二步反应。步骤ii采用水做溶剂,无机碱如氢氧化钠等做碱,快速反应获得关键中间体7-羟基色满酮,调酸萃取干燥以后,直接用于下一步反应。步骤iii工艺采用醇做溶剂,四氢吡咯醋酸盐做催化剂,原儿茶醛与7-羟基色满酮直接脱水缩合获得苏木酮a粗品,粗品溶解在2-4个碳的醇溶液中,降温析晶可获得高纯度苏木酮a。
本发明的目的之一就在于提供一种苏木酮a的制备方法,该制备方法可工业化量产,产生的三废非常少,几乎零污染,反应直接结晶纯化,少去柱层析等繁琐步骤。
技术方案是:一种苏木酮a的制备方法,该方法制备路线为:
作为优选,所述i的酰化反应是在酸催化下反应的。
作为优选,所述酸选自多聚磷酸、硫酸或甲磺酸。
作为优选,所述邻苯二酚与3-氯丙酸mol比为0.2:0.23。
作为优选,所述ii为酰化反应后的反应液经入水溶解,萃取取有机相,除萃取剂后溶于碱液。
作为优选,所述碱为氢氧化钠。本发明中,还可以采用氢氧化钾或其它的碱。
作为优选,所述iii的脱水缩合是在四氢吡咯醋酸盐催化下反应的。
本发明的目的之二在于提供一种苏木酮a的纯化方法,该方法可工业化量产,可获得纯度大于98%的产品,产生的三废非常少。
技术方案是:一种苏木酮a的纯化方法,包括以下步骤:将苏木酮a粗品溶解在2-4个碳的醇溶液中,降温析晶得高纯度苏木酮a。
作为优选,所述方法还包括将溶解有苏木酮a粗品的2-4个碳的醇溶液升温至回流至产物溶解后加入活性炭并经回流后过滤且将滤液加热回流后加入纯净水。
作为优选,所述醇溶液为乙醇、异丙醇或正丁醇。
与现有技术相比,本发明的有益效果在于:
本发明采用乙醇,水等作为生产溶剂,几乎零污染,反应直接结晶纯化,少去柱层析等繁琐步骤,获得纯度大于98%可量产的工艺。
本发明反应条件温和,一般工厂条件能达到,产生的三废非常少,产品纯度高,生产成本低,适合量产。
附图说明
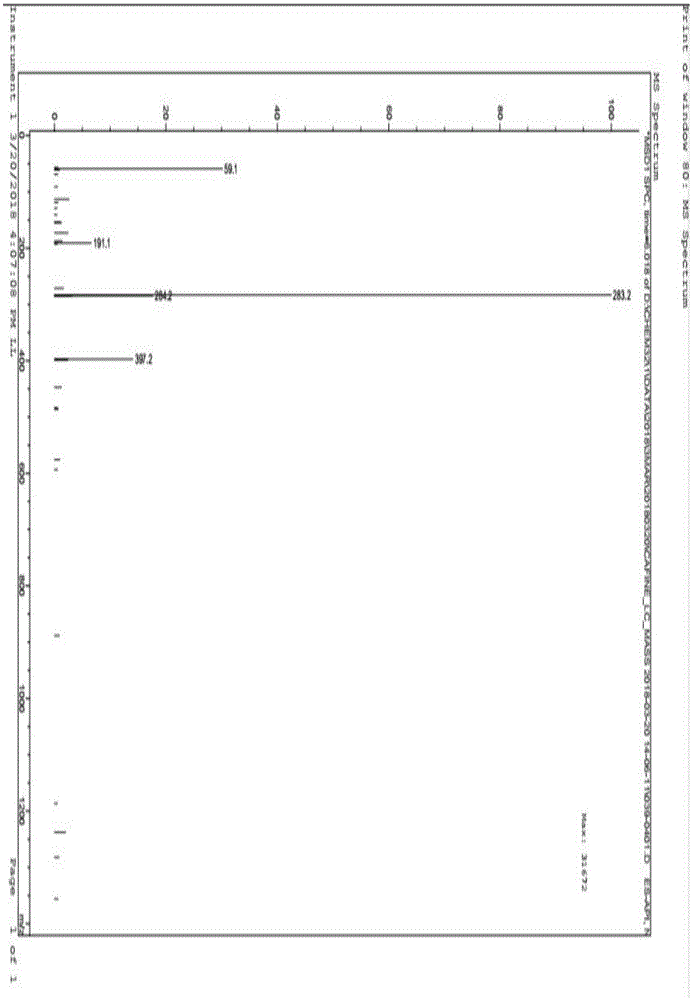
图1是本发明苏木酮a质谱图;
图2是本发明苏木酮a核磁氢谱图;
图3是本发明苏木酮a纯度谱图。
具体实施方式
下面将结合附图对本发明作进一步说明。
实施例17-羟基色满酮的制备
依次向反应瓶中加入22g(0.2mol)间二苯酚,60ml三氟甲磺酸,25g(0.23mol)3-氯丙酸,搅拌均匀后缓慢升温至80℃反应约3h,tlc检测原料无剩余,降温至室温后,将上述反应液直接加入到300ml冰水中,乙酸乙酯萃取三次,合并有机相,减压浓缩获得棕红色固体,将固体溶解在240ml的10%氢氧化钠水溶液中,并在0℃下搅拌1h,缓慢用醋酸调ph至中性,大量浅黄色固体析出,在0℃搅拌约1h后过滤,烘干,获得纯品7-羟基色满酮27g,70%收率,lcmsm/z163.1(m-h)。
实施例2sappanonea的制备
依次向反应瓶中加入7-羟基色满酮20g(0.12mol)(实施例1制备的),乙醇100ml,原儿茶醛18.5g(0.13mol)氮气保护下搅拌均匀,再加入四氢吡咯的醋酸盐3g,升温至89℃后反应约12h,tlc检测无7-羟基色满酮剩余后,降温至室温,浓缩除去大部分溶剂后,直接加入冰水中,大量黄色固体析出,直接过滤,获得sappanonea粗品32g,lcmsm/z283.2(m-h)。
实施例3sappanonea提纯一
依次向反应瓶中加入苏木酮a粗品15g(根据实施例1和2的方法制备出的),乙醇100ml,升温至回流至产物溶解,补加20ml乙醇,再加入1g活性炭,回流30min后热过滤,滤液加热回流后加入12ml纯净水,缓慢降温至室温后搅拌半小时,再降温至10℃左右后,过滤,滤饼用5-10℃的体积比为10:1乙醇水溶液洗涤两次,烘干,获得纯品12.3g,结晶收率82%;hplc纯度98.7%。1hnmr(400mhz,dmso-d6)δ10.69(s,0h),9.52(d,j=143.1hz,1h),7.73(d,j=8.7hz,1h),7.51(d,j=2.0hz,1h),6.84(d,j=8.0hz,2h),6.76(dd,j=8.3,2.1hz,1h),6.54(dd,j=8.7,2.2hz,1h),6.32(d,j=2.2hz,1h),5.35(d,j=1.9hz,2h)。
实施例4sappanonea提纯二
依次向反应瓶中加入苏木酮a粗品5g(根据实施例1和2的方法制备出的),异丙醇75ml,升温至回流至产物溶解,再加入0.5g活性炭,回流30min后热过滤,滤液加热回流后加入7.5ml纯净水,缓慢降温至室温后搅拌半小时,再降温至10℃左右后,过滤,滤饼用冷的10:1异丙醇和水洗涤两次,烘干,获得纯品3.6g,结晶收率72%;hplc纯度96.5%。
实施例5sappanonea提纯三
依次向反应瓶中加入苏木酮a粗品5g(根据实施例1和2的方法制备出的),正丁醇70ml,升温至回流至产物溶解,再加入0.5g活性炭,回流30min后热过滤,滤液加热回流后加入7ml纯净水,缓慢降温至室温后搅拌半小时,再降温至10℃左右后,过滤,滤饼用冷的10:1正丁醇和水洗涤两次,烘干,获得纯品3.2g,结晶收率64%;hplc纯度95.8%。
对比实施例1乙酸乙酯提纯sappanonea
依次向反应瓶中加入苏木酮粗品5g(根据实施例1和2的方法制备出的),乙酸乙酯80ml,加热回流,未完全溶解(加热回流时间足够长),降温至室温后过滤,获得2g产品,收率40%。
上面的实施例采用乙醇,水等作为生产溶剂,几乎零污染,反应直接结晶纯化,少去柱层析等繁琐步骤,获得纯度大于98%。
本发明的制备和提纯方法可工业化量产苏木酮a(由图1-图3可确定)。
本发明反应条件温和,一般工厂条件能达到,产生的三废非常少,产品纯度高,生产成本低。
- 还没有人留言评论。精彩留言会获得点赞!