本发明属于合成医药、化工领域,主要涉及一种3-芳基-3’-氨基双季碳双氧化吲哚化合物及其合成方法和应用。
背景技术:
氧化吲哚是一种广泛存在于自然界中的杂环结构,很多具有生物活性的天然产物都含有这一结构。而且此类化合物通常具有一定的生理生物活性,因而受到药物化学届的广泛关注,被认为是一类极具有潜在药物价值的有机小分子母核结构。
但是3,3’-双取代双氧化吲哚结构类骨架合成一直是有机合成中的难点。从传统有机合成方法上来看,该类骨架存在:结构拥挤、化合物空间位阻较大,含有两个连续季碳中心、其中一个为全碳季碳中心,结构中含有多种官能团、化合物分子高度官能团化等合成难点,是一类合成难度较大的化合物骨架。
按照现有方法针对3-芳基-3’-氨基双季碳双氧化吲哚砌块进行合成,都存在合成路线较长,步骤繁琐(5步以上),反应总收率较低(<30%),产生的废料较多,流程耗时较长等缺陷,不利于3-芳基-3’-氨基双季碳双氧化吲哚砌块在药物合成中的应用。
技术实现要素:
本发明人注意到3-芳基氧化吲哚结构在很多药物分子中存在,如式(iii)所示的抗癌药物以及式(iv)所示的非肽类生长激素促分泌素sm-130686:
同时本发明人还注意到3-氨基氧化吲哚结构也在很多药物活性分子中存在,如式(v)所示的胃泌素受体拮抗剂(抗胃癌候选药物)ag-041r,以及式(vi)加压素vib受体拮抗剂(抗抑郁候选药物):
如果将该两种活性分子骨架进行拼接,可以得到一类全新的3-芳基-3’-氨基双季碳双氧化吲哚结构骨架化合物,在本发明之前未有针对该类骨架的合成方法以及生物活性测试评价的相关报道。该骨架化合物可能会产生一系列在结构和活性上有意义的新化合物分子,可能为生物活性筛选提供新的化合物来源。但同时,该类骨架化合物从传统有机合成方法上来看,存在:结构拥挤、化合物空间位阻较大,含有两个连续季碳中心、其中一个为全碳季碳中心,结构中含有多种官能团、化合物分子高度官能团化等合成难点,是一类合成难度较大的化合物骨架。
近几年,胡文浩教授课题组发展了一系列亚胺捕捉离子对中间体的多组分反应策略。本发明基于该研究的基础上,实现了一种新的通过一步反应就可构建3-芳基-3’-氨基双季碳双氧化吲哚骨架分子的合成方法,该方法具有优秀的区域选择性和非对映选择性。
本发明公开一种步骤简短(仅需1步)、反应条件温和(室温25℃)、操作方便、非对映选择性优异(dr>20:1)的3-芳基-3’-氨基双季碳双氧化吲哚的合成方法。
本发明以重氮化合物、n,n-双取代苯胺、靛红亚胺化合物为原料,只经一步反应,即可制备得到3-芳基-3’-氨基双季碳双氧化吲哚。相比于现有技术中的其他相关的类似报道,本方法具有反应条件温和、反应步骤少、反应快、产生的废物少、原子经济性高等特点,因此该方法在全合成及药物合成领域具有很广阔的应用前景。
本发明提出了一种如式(ii)所示的光学活性双季碳双氧化吲哚化合物的合成方法,以重氮化合物、靛红亚胺化合物、n,n-双取代苯胺为原料,以醋酸铑为催化剂,在溶剂中,以分子筛为吸水剂,经过一步反应,得到如式(ii)所示的双季碳双氧化吲哚化合物,该化合物为全新化合物,未有相关报道;
所述合成反应如反应式(i)所示:
式(ii)及反应式(i)中,
r1为烷基、苄基、苄氧羰基、烷氧羰基、卤素取代的苄基、烷基取代的苄基、烷氧基取代的苄基、三氟甲基取代的苄基、硝基取代的苄基、萘甲基、呋喃甲基、烯基、炔基、二苯甲基、二(4-溴苯基)甲基、氢;
r2为烷氧基、卤素、三氟甲基、硝基、烯基、炔基、氢;
r3为烷基、苄基、卤素取代的苄基、烷基取代的苄基、烷氧基取代的苄基、三氟甲基取代的苄基、硝基取代的苄基、萘甲基、呋喃甲基、烯基、炔基、二苯甲基、二(4-溴苯基)甲基、氢;
r4为烷基、卤素、三氟甲基、硝基、烯基、炔基、氢;
z为苄基、甲氧基乙基、卤素取代的苄基、烷基取代的苄基、烷氧基取代的苄基、三氟甲基取代的苄基、硝基取代的苄基、萘甲基、呋喃甲基、烯基、炔基、二苯甲基、二(4-溴苯基)甲基。
优选地,
r1为c1-c6烷基、苄基、苄氧羰基、c1-c6烷氧羰基、卤素取代的苄基、c1-c6烷基取代的苄基、c1-c6烷氧基取代的苄基、三氟甲基取代的苄基、硝基取代的苄基、萘甲基、呋喃甲基、烯丙基、炔丙基、二苯甲基、二(4-溴苯基)甲基、氢;
r2为c1-c6烷氧基、卤素、三氟甲基、硝基、烯丙基、炔丙基、氢;
r3为c1-c6烷基、苄基、卤素取代的苄基、c1-c6烷基取代的苄基、c1-c6烷氧基取代的苄基、三氟甲基取代的苄基、硝基取代的苄基、萘甲基、呋喃甲基、烯丙基、炔丙基、二苯甲基、二(4-溴苯基)甲基、氢;
r4为c1-c6烷基、卤素、三氟甲基、硝基、烯丙基、炔丙基、氢;
z为苄基、甲氧基乙基、卤素取代的苄基、c1-c6烷基取代的苄基、c1-c6烷氧基取代的苄基、三氟甲基取代的苄基、硝基取代的苄基、萘甲基、呋喃甲基、烯丙基、炔丙基、二苯甲基、二(4-溴苯基)甲基。
进一步优选地,
r1为甲基、乙基、异丙基、苄基、苄氧羰基、c1-c6烷氧羰基、2-溴苄基、2-氯苄基、2-氟苄基、2-碘苄基、2-甲基苄基、2-甲氧基苄基、2-三氟甲基苄基、2-硝基苄基、1-萘甲基、4-甲氧基苄基、4-溴苄基、2-萘甲基、2-呋喃甲基、烯丙基、炔丙基、二苯甲基、二(4-溴苯基)甲基、氢;
r2为c1-c6烷氧基、氟、氯、溴、碘、三氟甲基、硝基、烯丙基、炔丙基、氢;
r3为甲基、乙基、异丙基、苄基、2-溴苄基、2-氯苄基、2-氟苄基、2-碘苄基、2-甲基苄基、2-甲氧基苄基、2-三氟甲基苄基、2-硝基苄基、1-萘甲基、4-甲氧基苄基、4-溴苄基、2-萘甲基、2-呋喃甲基、烯丙基、炔丙基、二苯甲基、二(4-溴苯基)甲基、氢;
r4为c1-c6烷基、氟、氯、溴、碘、三氟甲基、硝基、烯丙基、炔丙基、氢;
z为苄基、甲氧基乙基、2-溴苄基、2-氯苄基、2-氟苄基、2-碘苄基、2-甲基苄基、2-甲氧基苄基、2-三氟甲基苄基、2-硝基苄基、1-萘甲基、4-甲氧基苄基、4-溴苄基、2-萘甲基、2-呋喃甲基、乙基、异丙基、烯丙基、炔丙基、二苯甲基、二(4-溴苯基)甲基。
进一步优选地,
r1为甲基、苄基、苄氧羰基、叔丁氧羰基;
r2为5-甲氧基、5-氯、6-溴;
r3为甲基、苄基;
r4为4-氯、5-氟、5-甲基、6-溴、7-氯;
z为苄基或1-甲氧基乙基。
其中,所述重氮化合物:n,n-双取代苯胺:靛红亚胺化合物:醋酸铑的摩尔比为(1~1.5):(1~1.5):(1~1.5):(0.01~0.1);优选地,所述重氮化合物:n,n-双取代苯胺:靛红亚胺化合物:醋酸铑=1.1:1.1:1:0.05。
其中,所述方法包括以下步骤:先将所述靛红亚胺化合物、醋酸铑、n,n-双取代苯胺、分子筛溶于所述溶剂中,在-30℃-100℃下,加入所述重氮化合物的溶剂溶液,反应得到所述式(ii)所示的双季碳双氧化吲哚化合物。其中,所述重氮化合物为靛红重氮类化合物、芳基重氮酮类化合物、烷基重氮酮类化合物、芳基重氮乙酸酯类化合物、烷基重氮乙酸酯类化合物。
其中,所述反应的温度为-30℃-100℃;优选地,为25℃。
其中,所述反应的时间为1小时-48小时;优选地,为2小时。
其中,将反应得到的所述双季碳双氧化吲哚化合物进行分离纯化。所述分离纯化是用体积比为乙酸乙酯:石油醚=1:20~1:5的溶液进行柱层析,然后所有产物可用乙酸乙酯和石油醚进行重结晶。
其中,所述分子筛的投料量以所述靛红亚胺化合物为基准,为500mg/mmol-2000mg/mmol;优选地,所述分子筛投料量以靛红亚胺化合物为基准,为1000mg/mmol。
其中,所述溶剂包括烷烃类、卤代烷烃类、醚类、卤代醚类、取代苯类、醇类、酯类、酮类、卤代苯、杂芳烃类、酰胺类、亚砜类、水等之任意一种或几种的组合。优选地,所述溶剂为甲苯、二氯甲烷。
在一个具体实施方式中,所述合成反应如反应式(i')所示:
本发明有益效果包括:(1)本发明利用了胡文浩教授课题组已报道的亚胺捕捉离子对中间体的反应策略来构建了一类3-芳基-3’-氨基双季碳双氧化吲哚类复杂化合物骨架。(2)本发明以重氮化合物、n,n-双取代苯胺、靛红亚胺化合物为原料,以醋酸铑作为催化剂,通过一步反应构建3-芳基-3’-氨基双季碳双氧化吲哚化合物,具有高非对映选择性(dr>20:1)、高原子经济性(除了释放的一分子氮气以外,所有反应底物的原子都出现在反应产物当中)、高步骤经济性(仅需一步反应)等优点,并且反应条件温和,操作简单安全。(3)本发明所涉及的式(ii)3-芳基-3’-氨基双季碳双氧化吲哚类新型骨架分子,具有潜在的生物药物活性,在药用领域具有较好的应用前景,本发明方法为药物活性分子筛选提供了新的化合物来源。
附图说明
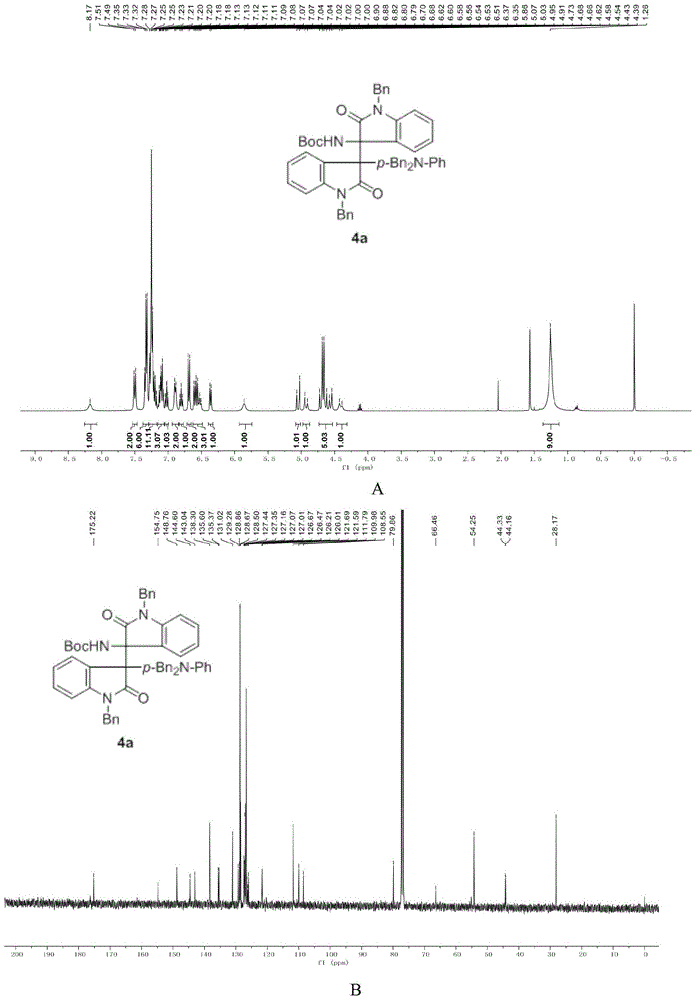
图1为实施例1所得产物4a的1hnmr(图1a)和13cnmr(图1b)示意图。
图2为实施例2所得产物4b的1hnmr(图2a)和13cnmr(图2b)示意图。
图3为实施例3所得产物4c的1hnmr(图3a)和13cnmr(图3b)示意图。
图4为实施例4所得产物4d的1hnmr(图4a)、13cnmr(图4b)和19fnmr(图4c)示意图。
图5为实施例5所得产物4e的1hnmr(图5a)和13cnmr(图5b)示意图。
图6为实施例6所得产物4f的1hnmr(图6a)和13cnmr(图6b)示意图。
图7为实施例7所得产物4g的1hnmr(图7a)和13cnmr(图7b)示意图。
图8为实施例8所得产物4h的1hnmr(图8a)和13cnmr(图8b)示意图。
图9为实施例9所得产物4i的1hnmr(图9a)和13cnmr(图9b)示意图。
图10为实施例10所得产物4j的1hnmr(图10a)和13cnmr(图10b)示意图。
图11为实施例11所得产物4k的1hnmr(图11a)和13cnmr(图11b)示意图。
图12为实施例12所得产物4l的1hnmr(图12a)和13cnmr(图12b)示意图。
图13为实施例13所得产物4m的1hnmr(图13a)和13cnmr(图13b)示意图。
图14为实施例14所得产物4n的1hnmr(图14)和13cnmr(图14b)示意图。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明,本发明的保护内容不局限于以下实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
实施例1:
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4a所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为80%。核磁共振1hnmr、13cnmr图谱如图1所示,1hnmr(400mhz,cdcl3)δ8.17(s,1h),7.50(d,j=8.9hz,2h),7.36–7.30(m,6h),7.29–7.17(m,10h),7.14–7.06(m,3h),7.05–6.99(m,1h),6.89(d,j=6.9hz,2h),6.80(t,j=7.5hz,1h),6.69(d,j=9.2hz,2h),6.63–6.49(m,3h),6.36(d,j=7.3hz,1h),5.86(s,1h),5.05(d,j=16.0hz,1h),4.93(d,j=16.1hz,1h),4.75–4.51(m,5h),4.41(d,j=15.7hz,1h),1.26(s,9h).13cnmr(101mhz,cdcl3)δ175.22,154.75,148.76,144.60,143.04,138.30,135.60,135.37,131.02,129.28,128.86,128.67,128.50,127.44,127.35,127.16,127.07,127.01,126.67,126.47,126.21,126.01,121.69,121.59,111.79,109.98,108.55,79.86,66.46,54.25,44.33,44.16,28.17.
实施例2:
n-甲基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4b所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为66%。1hnmr(400mhz,cdcl3)δ8.12(s,1h),7.48(d,j=8.9hz,2h),7.33(q,j=6.4,6.0hz,5h),7.30–7.21(m,11h),7.09–7.02(m,1h),6.82(t,j=7.5hz,1h),6.74–6.65(m,4h),6.59(d,j=7.8hz,1h),6.29(d,j=6.8hz,1h),5.99(s,1h),5.02(d,j=16.1hz,1h),4.91(d,j=16.0hz,1h),4.76–4.60(m,4h),2.73(s,3h),1.23(s,9h).13cnmr(101mhz,cdcl3)δ174.71,148.74,145.11,143.10,138.31,135.44,130.86,129.36,128.89,128.67,127.35,127.12,127.01,126.67,126.40,126.20,125.96,121.66,121.14,111.81,109.95,107.57,79.84,54.26,44.30,28.14,25.72.
实施例3
n-甲基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4c所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为60%。1hnmr(400mhz,cdcl3)δ7.97(s,1h),7.55(d,j=7.5hz,2h),7.37–7.19(m,16h),7.07(t,j=7.7hz,1h),6.85(d,j=8.1hz,1h),6.74(t,j=7.6hz,1h),6.67–6.57(m,4h),6.39(d,j=7.5hz,1h),5.03(d,j=16.0hz,1h),4.90(d,j=16.0hz,1h),4.74–4.57(m,4h),2.64(s,3h),1.32(s,9h).13cnmr(101mhz,cdcl3)δ174.19,148.90,146.82,142.86,138.29,135.45,131.12,130.10,128.87,128.63,127.38,127.19,126.93,126.77,126.66,126.43,126.33,124.38,120.81,112.23,109.99,79.79,68.98,55.39,54.08,44.34,28.15,25.88.
实施例4
n-甲基-n’-boc-5-氟靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4d所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为75%。1hnmr(400mhz,cdcl3)δ8.23(s,1h),7.47(d,j=8.8hz,2h),7.34(t,j=7.3hz,4h),7.30–7.21(m,11h),7.08(t,j=7.7hz,1h),7.00(s,1h),6.77–6.67(m,3h),6.66–6.58(m,2h),6.02(t,j=12.4hz,2h),4.95(t,j=15.4hz,2h),4.68(q,j=17.2hz,4h),2.72(s,3h),1.27(s,9h).13cnmr(101mhz,cdcl3)δ174.56,157.32,154.62,148.92,143.12,141.11,138.14,135.32,130.75,129.07,128.73,128.69,127.41,127.11,127.01,126.59,126.06,121.18,115.56,115.33,114.30,114.04,111.79,110.13,107.81,107.73,80.10,67.07,54.17,44.35,28.18,25.86.19fnmr(376mhz,cdcl3)δ-120.78.
实施例5
n-甲基-n’-boc-5-甲基靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4e所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为84%。1hnmr(400mhz,cdcl3)δ8.15(s,1h),7.49(d,j=8.7hz,2h),7.37–7.20(m,15h),7.12–7.01(m,2h),6.68(d,j=9.0hz,3h),6.58(d,j=9.6hz,2h),6.00(s,23h),5.02(d,j=16.0hz,1h),4.91(d,j=16.0hz,1h),4.78–4.60(m,4h),2.70(s,3h),2.11(s,3h),1.24(s,9h).13cnmr(101mhz,cdcl3)δ174.66,154.64,148.56,143.04,142.68,138.34,135.46,130.97,130.89,129.40,128.82,128.71,128.66,127.34,127.13,127.01,126.54,126.33,121.06,111.45,109.90,107.18,79.79,54.27,44.30,28.17,25.75,21.14.
实施例6
n-甲基-n’-boc-6-溴靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4f所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为82%。1hnmr(400mhz,cdcl3)δ8.21(s,1h),7.46(d,j=8.7hz,2h),7.39–7.14(m,15h),7.08(t,j=7.4hz,1h),6.94(d,j=8.8hz,1h),6.86(d,j=1.6hz,1h),6.76(t,j=7.4hz,1h),6.68(d,j=9.1hz,2h),6.60(d,j=7.8hz,1h),6.11(d,j=7.9hz,2h),5.10–4.85(m,2h),4.79–4.56(m,4h),2.71(s,3h),1.27(s,9h).13cnmr(101mhz,cdcl3)δ174.66,154.64,148.82,146.45,143.06,138.23,135.29,130.80,129.12,128.71,127.44,127.08,126.66,126.04,125.62,124.60,123.18,121.36,111.91,111.12,110.15,80.16,66.60,54.33,44.35,28.20,25.87.
实施例7
n-甲基-n’-boc-7-氯靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4g所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为93%。1hnmr(400mhz,cdcl3)δ8.12(s,1h),7.51–7.44(m,2h),7.29(dd,j=25.4,9.8hz,16h),7.09(t,j=7.2hz,1h),6.80(t,j=7.1hz,1h),6.75–6.56(m,4h),6.29–6.02(m,2h),4.96(q,j=15.9hz,2h),4.78–4.57(m,4h),3.07(s,3h),1.26(s,9h).13cnmr(101mhz,cdcl3)δ175.95,175.26,154.62,148.81,143.09,140.94,138.25,135.35,131.60,130.94,129.16,128.71,127.44,127.15,127.07,126.68,126.11,125.91,124.39,122.36,119.72,115.10,111.85,110.17,80.18,66.66,54.31,44.38,29.23,28.15.
实施例8
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基-5-氯靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4h所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为92%。1hnmr(400mhz,cdcl3)δ8.02(s,1h),7.45(d,j=8.5hz,2h),7.39–7.20(m,16h),7.12(dt,j=13.9,6.9hz,3h),7.01–6.89(m,3h),6.83(t,j=7.4hz,1h),6.70(d,j=8.0hz,3h),6.39(dd,j=23.0,7.9hz,2h),5.72(s,1h),5.06(d,j=16.0hz,1h),4.84(d,j=16.1hz,1h),4.76–4.56(m,5h),4.40(d,j=14.8hz,1h),1.26(s,9h).13cnmr(101mhz,cdcl3)δ175.86,175.00,154.70,148.89,144.42,141.57,138.18,135.41,134.90,130.89,129.62,128.84,128.78,128.71,128.65,127.69,127.56,127.42,127.21,127.07,126.78,126.64,126.27,126.01,121.96,119.43,111.86,110.77,108.59,80.05,66.35,55.44,54.31,44.45,44.28,28.17.
实施例9
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基-6-溴靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4i所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为96%。1hnmr(400mhz,cdcl3)δ8.04(s,1h),7.56–7.09(m,21h),7.01–6.52(m,8h),6.35(d,j=7.3hz,1h),5.63(s,1h),5.05(d,j=16.0hz,1h),4.92–4.57(m,6h),4.36(d,j=9.7hz,1h),1.26(s,9h).13cnmr(101mhz,cdcl3)δ176.12,175.08,154.64,148.87,144.48,144.33,138.18,135.40,134.73,130.91,129.47,128.83,128.69,128.52,127.61,127.40,127.14,127.05,126.65,126.12,125.21,124.54,122.81,121.83,119.56,113.20,111.82,108.55,80.00,66.20,54.93,54.26,44.32,28.17.
实施例10
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄基-5-甲氧基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4j所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为95%。1hnmr(400mhz,cdcl3)δ8.24(s,1h),7.50(d,j=8.7hz,2h),7.40–7.03(m,6h),6.83(dd,j=16.1,7.8hz,13h),6.67(dd,j=12.9,8.5hz,3h),6.55(dd,j=8.5,2.2hz,3h),6.42(dd,j=19.8,8.0hz,1h),5.41(s,2h),5.07(d,j=16.0hz,1h),4.83(dd,j=22.6,15.8hz,1h),4.74–4.58(m,4h),4.33(d,j=14.9hz,1h),3.12(s,3h),1.27(s,9h).13cnmr(101mhz,cdcl3)δ176.07,175.18,154.79,154.67,148.74,144.69,138.30,136.40,135.62,135.42,130.97,129.24,128.68,128.60,127.47,127.35,127.19,127.15,127.02,126.68,126.07,121.86,115.01,112.52,111.80,110.45,108.49,79.89,66.27,55.03,54.25,44.38,28.20.
实施例11
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-甲基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4k所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为98%。1hnmr(400mhz,cdcl3)δ8.17(s,1h),7.50(d,j=8.9hz,2h),7.37–7.12(m,15h),6.95–6.87(m,2h),6.86–6.76(m,2h),6.66(dd,j=14.4,8.3hz,3h),6.56(d,j=7.8hz,1h),6.32(d,j=7.3hz,1h),5.91(d,j=7.4hz,1h),4.77–4.59(m,4h),4.41(s,2h),3.25(s,3h),1.26(s,9h).13cnmr(101mhz,cdcl3)δ176.09,175.21,154.80,148.74,144.56,144.00,138.28,135.55,130.94,129.28,129.07,128.67,128.49,127.36,127.14,127.01,126.64,126.56,126.14,125.86,121.77,121.64,120.14,111.76,108.80,108.63,79.86,66.68,54.87,54.27,44.09,28.15,26.59.
实施例12
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-叔丁氧羰基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4l所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为99%。1hnmr(400mhz,cdcl3)δ7.93(s,1h),7.87(d,j=8.1hz,1h),7.43(d,j=7.5hz,2h),7.36–7.13(m,15h),6.91(s,2h),6.80(t,j=7.4hz,1h),6.68(d,j=8.4hz,3h),6.56(d,j=7.5hz,1h),6.34(d,j=7.2hz,1h),5.83(s,1h),4.67(q,j=16.3,15.7hz,4h),4.43(s,2h),1.64(s,9h),1.25(s,9h).13cnmr(101mhz,cdcl3)δ174.91,174.85,154.51,148.85,148.75,144.83,140.25,138.17,135.42,130.91,129.47,129.24,128.70,128.50,127.43,127.17,127.04,126.61,126.36,125.70,125.08,123.20,121.92,120.22,115.39,111.86,108.71,84.70,79.98,67.05,54.25,44.10,28.15.
实施例13
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二苄基苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-苄氧羰基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4m所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为99%。1hnmr(400mhz,cdcl3)δ8.18(s,1h),7.50(d,j=8.8hz,2h),7.38–7.29(m,6h),7.23(tt,j=14.1,8.3hz,10h),7.10(dt,j=14.1,6.8hz,3h),7.02(t,j=7.6hz,1h),6.89(d,j=6.8hz,2h),6.81(t,j=7.5hz,1h),6.69(d,j=9.1hz,2h),6.56(ddd,j=21.6,15.0,7.6hz,3h),6.37(d,j=7.3hz,1h),5.86(s,1h),5.05(d,j=16.0hz,1h),4.93(d,j=16.0hz,1h),4.78–4.53(m,5h),4.41(d,j=15.2hz,1h),1.26(s,9h).13cnmr(101mhz,cdcl3)δ176.32,175.24,154.78,148.77,144.60,143.04,138.31,135.59,135.37,131.03,129.30,128.88,128.68,128.52,127.45,127.36,127.17,127.09,127.02,126.68,126.48,126.21,126.03,111.79,110.00,108.57,79.89,66.48,54.26,44.33,44.17,28.19.
实施例14
n-苄基-n’-boc靛红亚胺(0.3mmol),n,n-二(1-甲氧基乙基)苯胺(0.33mmol),rh2(oac)4(0.05mmol)和分子筛(0.2g)溶于二氯甲烷(1.5ml),然后将溶解在二氯甲烷(1ml)中的n-甲基靛红重氮(0.33mmol)用蠕动泵在一小时内加到反应体系中,反应在室温下进行,滴加完毕后,搅拌两小时。其结构如式4n所示,将粗产物进行柱层析(乙酸乙酯:石油醚=1:20~1:5)得到纯产物,产率为97%。1hnmr(400mhz,cdcl3)δ8.16(s,1h),7.56(d,j=8.9hz,2h),7.39–7.17(m,6h),7.17–7.00(m,4h),6.87(dd,j=19.1,7.2hz,3h),6.71–6.53(m,5h),6.44(d,j=7.4hz,1h),5.89(s,1h),5.05(d,j=16.0hz,1h),4.95(d,j=16.0hz,1h),4.59(d,j=15.6hz,1h),4.43(s,1h),3.56(dq,j=9.5,5.3hz,8h),3.36(s,6h),1.26(s,9h).13cnmr(101mhz,cdcl3)δ176.33,175.26,154.73,147.57,144.61,143.06,135.61,135.41,131.16,129.33,128.91,128.71,128.54,127.46,127.38,127.18,127.11,126.80,126.47,126.27,126.06,121.77,121.69,119.58,110.98,110.01,108.59,79.86,70.06,66.45,59.05,55.18,50.83,44.33,44.19,28.19
实施例15抗肿瘤活性测试实验:
1、接种细胞:用含10%胎牛血清、1%青霉素和链霉素的mccoy'5a培养液配成单个细胞悬液,以每孔2500个结直肠癌细胞hct116接种到96孔细胞培养板,每孔体积100μl。
2、给药:分别将本发明前述实施例中合成的式(ii)化合物(表1中所列的化合物4a至4k以及4m)用dmso配制成终浓度为25mm的母液,用完全培养基稀释至100um,作用于细胞,设置三个复孔。
3、培养:5%co2,37℃饱和湿度育孵箱培养72小时。
4、呈色:培养72小时吸出培养基,每孔加入100μl完全1640培养基和10μlcck8,37℃孵育4小时。
5、比色:选择620、450nm波长,在酶标仪上测定各孔光密度(od)值,记录结果。
6、同一孔450nm吸光光度值-620nm吸光光度值(背景吸光光度值)作为最终吸光度,代入以下公式。
7、细胞增殖活力(%)=[a(加药)-a(空白)]/[a(0加药)-a(空白)]×100。
a(加药):具有结直肠癌细胞hct116、cck溶液(cellcountingkit-8,是一种基于wst-8(化学名:2-(2-甲氧基-4-硝苯基)-3-(4-硝苯基)-5-(2,4-二磺基苯)-2h-四唑单钠盐)的细胞增殖和细胞毒性快速高灵敏度检测试剂)和药物溶液(药物溶液即指待测试的化合物样品dmso溶液)的孔的吸光度
a(空白):具有培养基和cck溶液而没有细胞的孔的吸光度
a(0加药):具有细胞、cck溶液而没有药物溶液的孔的吸光度
8、实验结果表明:在浓度为100μm本发明式(ii)化合物的作用下,各化合物(表1中所列的化合物4a至4k以及4m)的结直肠癌细胞hct116细胞抑制率如下表1。
其中,编号4a化合物对结直肠癌细胞hct116的生长抑制率最高,约为43.55%;化合物4m也对结直肠癌细胞hct116的生长表现出一定的抑制效果,抑制率约为30.48%;剩余化合物对结直肠癌细胞hct116的生长抑制作用较弱,抑制率约在12.73%-23.05%之间。
通过抗肿瘤活性筛选,可发现,化合物4a对结直肠癌细胞hct116生长有较好的抑制作用,抑制率约为43.55%。
表1