一种基于同源重组的变棕溶杆菌OH23基因敲除系统的构建方法与应用与流程
本发明涉及微生物基因工程领域,具体涉及一种基于同源重组的变棕溶杆菌oh23基因敲除系统的构建方法与应用。
背景技术:
溶杆菌广泛存在自然界中,分类地位上属于黄单胞科(xanthomonadaceae),溶杆菌属(lysobacter),能够产生多种胞外水解酶和小分子抗菌物质,对农作物产生病害的细菌、真菌、卵菌及线虫产生显著的拮抗作用。
变棕溶杆菌oh23(lysobacterbrunescens)为本实验室分离于植物根际土壤一株革兰氏阴性细菌。oh23能够特异性的拮抗水稻白叶枯病菌(xanthomonasoryzaepv.oryzaepxo99a)、水稻细菌性条斑病菌(xanthomonasoryzaepv.oryzaers105)、水稻白叶枯病菌(xanthomonasoryzaepv.oryzaekacc)、野油菜黄单胞菌(xanthomonascampestrispathovars)、地毯草黄单胞菌大豆致病变种(xanthomonasaxonopodispv.glycines)等黄单胞菌。由于溶杆菌对许多抗生素有抗性,且敏感的抗生素种类也不相同,导致溶杆菌的基因敲除系统不易构建,而且不同溶杆菌甚至同一个种的不同菌株其有效的基因敲除系统也不相同,不能共用。因此,针对变棕溶杆菌oh23基因敲除系统的构建,有助于对oh23进行分子操作,特别是可为其小分子代谢产物生物合成、遗传调控和作用机制研究奠定基础。
技术实现要素:
针对现有技术中变棕溶杆菌oh23的基因敲除系统不能共用的问题,本发明的目的在于提供一种基于同源重组的变棕溶杆菌oh23基因敲除系统的构建方法与应用,构建方法简单,有助于对oh23进行分子操作,尤其为其小分子代谢产物生物合成、遗传调控和作用机制研究奠定基础。
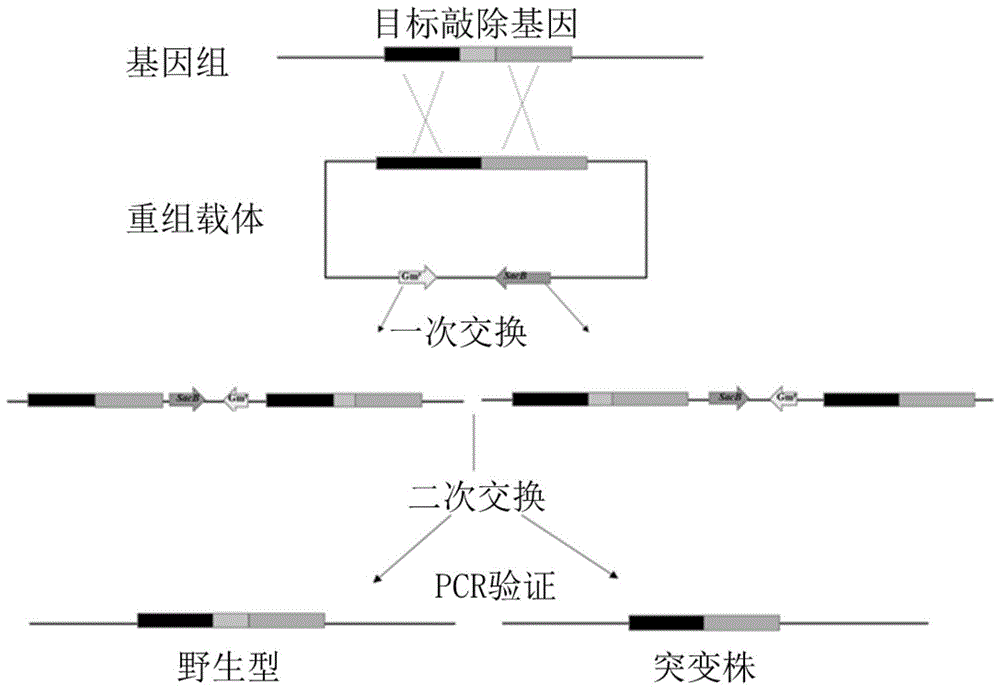
一种基于同源重组的变棕溶杆菌oh23基因敲除系统的构建方法,利用含有庆大霉素抗性基因的自杀质粒pjq200sk构建用于基因敲除的重组载体,将重组载体转入接合用s17-1λpir感受态细胞中,通过接合的方式将重组载体转入到具有利福平抗性的oh23中,并在含利福平和庆大霉素抗性培养基上进行一次交换子,进而通过pjq200sk载体所带的sacb蔗糖致死基因进行二次交换子的筛选,再利用pcr进行突变体筛选获得基因敲除突变体。
作为改进的是,所述的自杀质粒为质粒pjq200sk。
作为改进的是,上述构建方法包括以下步骤:
步骤1,构建重组载体
设计目的基因上游臂引物和下游臂引物,利用pcr扩增目的基因的上下游同源臂;将自杀质粒与上下游同源臂利用相应的内切酶进行酶切并连接得到用于基因敲除的重组载体;将重组载体以电转化方式转入s17-1λpir感受态细胞中,将细胞涂布于含庆大霉素抗性培养基上培养,至于37℃静置培养,进而获得含有重组载体的s17-1λpir大肠杆菌;
步骤2,重组载体转入变棕溶杆菌oh23
挑取含有重组载体的s17-1λpir大肠杆菌和变棕溶杆菌oh23单菌落分别在lb培养基和nbws培养基中活化,待生长至od600≈0.6-0.8,分别取2ml菌液离心后,用无菌水清洗三次后,在无菌滤膜上进行接合;接合条件:37℃1h后,正置放于28℃培养箱中培养12h;将滤膜上的细胞涂布于含利福平和庆大霉素抗性培养基上;挑取单菌落进行pcr筛选一次交换子;
步骤3,构建基因敲除突变体
挑取一次交换子单菌落于nbws培养基中进行二次交换培养;取上述二次交换培养液涂布于复配质量分数4%蔗糖的naws平板培养基,筛选二次交换子;挑取转化子进行pcr验证,获得基因敲除突变体。
作为改进的是,步骤1中所述含庆大霉素抗性培养基为含8μg/ml庆大霉素的lb平板。
作为改进的是,步骤2中所述含利福平和庆大霉素抗性培养基为含20μg/ml利福平和8μg/ml庆大霉素的naws平板。
作为改进的是,步骤3中所述二次交换培养的条件为:28°c,180rpm培养7h。
作为改进的是,所述nbws培养基为不含蔗糖的nb培养基;所述naws平板培养基为不含蔗糖的na固体培养基;所述na固体培养基为1lnb培养基,17g琼脂;所述lb培养基的配方为:10g蛋白胨,5g酵母粉,10g氯化钠,ph7.0-7.2,1l灭菌水;所述nb培养基的配方为:3g牛肉膏,1g酵母粉,5g蛋白胨,10g蔗糖,ph7.0-7.2,1l灭菌水。
上述方法获得的基因敲除突变体在小分子代谢产物生物合成、以及研究遗传调控好作用机制上的应用。
溶杆菌oh23(lysobacterbrunescensoh23)是一株从植物根际土壤中筛选到,对植物病原黄单胞菌具有特异性拮抗活性的溶杆菌,已于2017年02月12日保藏于中国微生物菌种保藏管理委员会普通微生物中心(简称cgmcc,地址:北京市朝阳区北辰西路1号院3号),保藏号为cgmccno.13677。
有益效果:
本发明针对变棕溶杆菌oh23基因敲除系统不能共用的问题,提出一种新的简单方便的方法,所得基因敲除突变体,适用于检测多种抗生素,具有通用性。因此,该基因敲除突变体特别是可为合成其小分子代谢产物生物、以及遗传调控和作用机制的研究奠定基础。
附图说明
图1为本发明构建方法的过程示意图;
图2为重组载体pcr验证及酶切验证,其中,m2000,markerdl2000;(a)1-7,基因luxi重组载体的pcr检测,-为阴性对照,即以pjq200sk为模板,引物luxi-f1/luxi-r2,重组载体pcr产物预期为1530bp;(b)1-8,基因luxr重组载体的pcr检测,-为阴性对照,以pjq200sk为模板,引物luxr-f1/luxr-r2,重组载体pcr产物预期为1486bp;(c)1-11,基因peg.2863重组载体的pcr检测,-为阴性对照,以pjq200sk为模板,引物peg.2863-f1/peg.2863-r2,重组载体pcr产物预期为935bp;
图3为基因luxi与luxr敲除突变体的构建,其中,(a)luxi敲除突变体利用引物luxi-deletion-f/luxi-deletion-r的pcr筛选。markerdl2000;+以oh23野生型dna模板,目的片段为2090bp;-,以重组载体pjq200sk-luxi为pcr模板,预期片段大小为1478bp;1、2、9、12、15、17、19、20和23为luxi基因的敲除突变体,预期片段大小1478bp;(b)luxr敲除突变体利用引物luxr-deletion-f/luxr-deletion-r的pcr筛选。m2000rkerdl2000;+以oh23野生型dna模板,目的片段为2239bp;-,以重组载体pjq200sk-luxr为pcr模板,预期片段大小为1486bp;7、11、12、13、14、15、16、19、20、22和23为luxr基因的敲除突变体,预期片段大小1486bp;(c)peg.2863敲除突变体利用引物peg.2863-deletion-f/peg.2863-deletion-r的pcr筛选。m2000rkerdl2000;+以oh23野生型dna模板,目的片段为2763bp;-,以重组载体pjq200sk-peg.2863为pcr模板,预期片段大小为1500bp;1、2、10、11、12、19和22为peg.2863基因的敲除突变体,预期片段大小1500bp;
图4为野生型菌株oh23与peg.2863缺失株拮抗病原黄单胞菌水稻细菌性条斑病菌(xanthomonasoryzaepv.oryzaers105)能力。
具体实施方式
下面结合具体实例对本发明的发明方法进行详细描述和说明。其内容是对本发明的解释而非限定本发明的保护范围。
需要的培养基为:oh23及其突变体培养于nb培养基(3g牛肉膏,1g酵母粉,5g蛋白胨,10g蔗糖,ph7.0-7.2,1l灭菌水);
nbws培养基(nb不含蔗糖);
固体培养基na(1lnb加17g琼脂);
naws培养基(na不含蔗糖);
含20μg/ml利福平和8μg/ml庆大霉素的naws平板;
naws+4%蔗糖培养基(1l培养基中含40g蔗糖)。
实施例1变棕溶杆菌oh23中luxi基因的的敲除
一种基于同源重组的变棕溶杆菌oh23基因敲除系统的构建方法,包括以下步骤:
步骤1,根据变棕溶杆菌oh23全基因组信息,设计目的基因luxi上游臂引物luxi-f1/luxi-r1和下游臂引物luxi-f2/luxi-r2及突变体验证引物luxi-deletion-f/luxi-deletion-r,序列如下表所示。
步骤2,pcr扩增上游臂774bp,下游臂774bp,利用overlappingpcr获得同源臂,片段长度为1530bp,酶切位点为bamhi/xbai,质粒pjq200sk利用bamhi/xbai酶切;
步骤3,酶切后的同源臂与相应酶切位点的质粒pjq200sk连接,转入大肠杆菌扩增后,获得重组载体pjq200sk-luxr,利用引物luxr-f1/luxr-r2进行pcr验证,获得1530bp片段,如图2a所示,2、3、5、6、7正确;
步骤4,重组载体转入变棕溶杆菌oh23:分别挑取含有重组载体的s17-1λpir大肠杆菌和变棕溶杆菌oh23单菌落在lb培养基和nbws培养基中活化,待生长至od600≈0.6-0.8左右,分别取2ml菌液离心后,用无菌水清洗三次后,在无菌滤膜上进行接合;接合条件:37℃1h后,正置放于28℃培养箱中培养12h;将滤膜上的细胞涂布于含利福平和庆大霉素抗性培养基上;挑取单菌落进行pcr筛选一次交换子;
步骤5,获得基因luxr一次交换子后,挑取一次交换子单克隆于装有25mlnbws的250ml三角瓶中,28℃,180rpm培养7h,取100µl培养液涂布于naws+4%蔗糖培养基,培养5天;
步骤6,pcr筛选luxi基因敲除突变体,利用引物luxi-deletion-f/luxi-deletion-r,预期片段大小为1478bp,获得1、2、9、12、15、17、19、20和23,共9个luxi基因敲除突变体(图3a)。
实施例2变棕溶杆菌oh23中luxr基因的敲除
一种基于同源重组的变棕溶杆菌oh23基因敲除系统的构建方法,包括以下步骤:
步骤1,根据变棕溶杆菌oh23全基因组信息,设计目的基因luxr上游臂引物luxr-f1/luxr-r1和下游臂引物luxr-f2/luxr-r2及突变体验证引物luxr-deletion-f/luxr-deletion-r,序列如下表所示。
步骤2,pcr扩增上游臂778bp,下游臂777bp,利用overlappingpcr获得同源臂,片段长度为1486bp,酶切位点为bamhi/xbai,质粒pjq200sk利用bamhi/xbai酶切;
步骤3,酶切后的同源臂与相应酶切位点的质粒pjq200sk连接,转入大肠杆菌扩增后,获得重组载体pjq200sk-luxr,利用引物luxr-deletion-f/luxr-deletion-r进行pcr验证,获得1486bp片段,3、6、9正确(图2b);
步骤4,重组载体转入变棕溶杆菌oh23:分别挑取含有重组载体的s17-1λpir大肠杆菌和变棕溶杆菌oh23单菌落在lb培养基和nbws培养基中活化,待生长至od600≈0.6-0.8左右,分别取2ml菌液离心后,用无菌水清洗三次后,在无菌滤膜上进行接合;接合条件:37℃1h后,正置放于28℃培养箱中培养12h;将滤膜上的细胞涂布于含利福平和庆大霉素抗性培养基上;挑取单菌落进行pcr筛选一次交换子;
步骤5,获得基因luxr一次交换子后,挑取一次交换子单克隆于装有25mlnbws的250ml三角瓶中,28℃,180rpm培养7h,取100µl培养液涂布于naws+4%蔗糖培养基,培养5天;
步骤6,pcr筛选luxr基因敲除突变体。利用引物luxr-deletion-f/luxr-deletion-r,预期片段大小为1486bp,获得7、11、12、13、14、15、16、19、20、22和23,共11个luxr基因敲除突变体(图3b)。
实施例3变棕溶杆菌oh23中次级代谢产物合成基因peg.2863基因的敲除
一种基于同源重组的变棕溶杆菌oh23基因敲除系统的构建方法,包括以下步骤:
步骤1,根据变棕溶杆菌oh23全基因组信息,设计目的基因peg.2863上游臂引物peg.2863-f1/peg.2863-r1和下游臂引物peg.2863-f2/peg.2863-r2及突变体验证引物peg.2863-deletion-f/peg.2863-deletion-r,序列如下表所示。
步骤2,pcr扩增上游臂473bp,下游臂477bp,利用overlappingpcr获得同源臂,片段长度为935bp,酶切位点为bamhi/xbai,质粒pjq200sk利用bamhi/xbai酶切;
步骤3,酶切后的同源臂与相应酶切位点的质粒pjq200sk连接,转入大肠杆菌扩增后,获得重组载体pjq200sk-peg.2863,利用引物peg.2863-deletion-f/peg.2863-deletion-r进行pcr验证,获得1500bp片段,1、2、3、4、5、6、7、10正确(图2b);
步骤4,重组载体转入变棕溶杆菌oh23:分别挑取含有重组载体的s17-1λpir大肠杆菌和变棕溶杆菌oh23单菌落在lb培养基和nbws培养基中活化,待生长至od600≈0.6-0.8左右,分别取2ml菌液离心后,用无菌水清洗三次后,在无菌滤膜上进行接合;接合条件:37℃1h后,正置放于28℃培养箱中培养12h;将滤膜上的细胞涂布于含利福平和庆大霉素抗性培养基上;挑取单菌落进行pcr筛选一次交换子;
步骤5,获得基因peg.2863一次交换子后,挑取一次交换子单克隆于装有25mlnbws的250ml三角瓶中,28℃,180rpm培养7h,取100µl培养液涂布于naws+4%蔗糖培养基,培养5天;
步骤6,pcr筛选peg.2863基因敲除突变体。利用引物peg.2863-deletion-f/peg.2863-deletion-r,预期片段大小为1486bp,获得1、2、10、11、12、19和22,共7个peg.2863基因敲除突变体(图3b)。
步骤7,检测野生型菌株oh23与peg.2863缺失株拮抗病原黄单胞菌水稻细菌性条斑病菌(xanthomonasoryzaepv.oryzaers105)能力。将108的xanthomonasoryzaepv.oryzaers105平铺在na平板上,将3×106的野生型菌株或peg.2863缺失株菌液点在na平板中央,置于28℃培养箱中培养两天,观察拮抗圈;野生型oh23处理后,具有较强的拮抗病原黄单胞菌水稻细菌性条斑病菌的能力,平板出现明显的拮抗圈;而δpeg.2863-1、δpeg.2863-2和δpeg.2863-9由于缺失了合成拮抗此生代谢产物合成途径关键基因peg.2863,都丧失了拮抗病原黄单胞菌水稻细菌性条斑病菌的能力,未能形成明显的拮抗圈(图4)。
以上所述的具体实施方式,对本发明的目的、技术方案和有益效果进行了进一步详细说明,所应理解的是,以上所述仅为本发明的具体实施方式而已,并不用于限制本发明,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
序列表
<110>江苏省农业科学院
<120>一种基于同源重组的变棕溶杆菌oh23基因敲除系统的构建方法与应用
<160>18
<170>siposequencelisting1.0
<210>1
<211>37
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>1
cagcccgggggatccatgtcgatgcggacgggcgtcc37
<210>2
<211>29
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>2
gacaccatgtgatcgaagcgcggcgacgc29
<210>3
<211>46
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>3
cttcgatcacatggtgtctccttggtgtggtgaggagacgccagtg46
<210>4
<211>35
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>4
agcggccgctctagactgggacgatgcgcaacagc35
<210>5
<211>21
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>5
gtcgatgcggacgggcgtccg21
<210>6
<211>20
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>6
tcgatcgccgcgtcgtcgat20
<210>7
<211>40
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>7
tcctgcagcccgggggatccccacgtgcaggccgaggtgg40
<210>8
<211>35
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>8
gggtcatgtgagcgcctgagcccgcgggcgcgatc35
<210>9
<211>28
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>9
gcgctcacatgacccctgttcccgattc28
<210>10
<211>40
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>10
gcggcagcggccgctctagacacggcgacgaaatcgacgc40
<210>11
<211>20
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>11
caggccgaggtggcgcagca20
<210>12
<211>31
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>12
acgaaatcgacgccgtcgcggtgcgcgcctt31
<210>13
<211>33
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>13
cagcccgggggatcccggagatggcagcgatgc33
<210>14
<211>27
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>14
ccgcgatgtgacccccgccggcaacag27
<210>15
<211>27
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>15
ggggtcacatcgcggggaactccaggt27
<210>16
<211>41
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>16
gcggcagcggccgctctagatcgccatcgtggtatccatcg41
<210>17
<211>22
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>17
gggagcgccacaggaggatcgc22
<210>18
<211>23
<212>dna
<213>artificialsequence(2ambystomalateralexambystomajeffersonianum)
<400>18
acaagctcaggcgtggcatgcag23
- 还没有人留言评论。精彩留言会获得点赞!