一种催化合成2,5-二甲基吡嗪的重组菌的制作方法
本发明涉及一种催化合成2,5-二甲基吡嗪的重组菌,属于生物合成技术领域。
背景技术:
烷基吡嗪是一类支链带有烷基基团的含氮杂环化合物,作为重要的风味物质,主要贡献食品中的坚果味,烤肉味和烤面包味。烷基吡嗪由于阈值低,可以显示出强烈的气味特性,是我国gb2760-86规定允许使用的香精物质,在食品工业中主要用作调味的食品添加剂和一些香料中间体。
烷基吡嗪除了具有独特的风味价值之外,在医药方面也具有重要价值,可用作药物或医药中间体。ttmp被发现可以作为药物治疗一些疾病,例如中风、心肌细胞损伤、膝骨关节炎等。另外,ttmp还可以作为其他生物药物的前体来源,例如,4-(2,3,5,6-ttmp-1)-4'-去甲表鬼臼是一种具有较强抗肿瘤活性的新型化合物,ttmp-2'o-阿魏酸钠可以提供神经保护,防止神经炎症和脑损伤。2,5-dmp可以作为抗菌类药物5-甲基吡嗪-2-羧酸的重要合成原料。
在烷基吡嗪的生产方面,其主要通过化学方法合成,以2,5-dmp的化学合成为例,目前2,5-dmp的化学合成方法主要包括液相法和气相法。由于化学合成普遍存在严峻的环保问题,并且可能存在不希望的副产物,使得分离提纯相对困难,另外,反应条件一般较剧烈,设备要求较高,且产品不是天然的,这些因素都促使风味化合物生产公司将注意力集中在生物来源的风味化合物上。
在风味化合物微生物合成研究领域遇到的第一个绊脚石便是普遍缺乏生化知识,虽然可能存在合理的假设,但通常缺少使用标记前体的证据以及所涉及的酶和基因的鉴定。烷基吡嗪作为一种重要的风味化合物,尽管对于烷基吡嗪微生物来源的研究已经探索了很长时间,但是对于其合成机制的认知还是非常有限的。目前,只有ttmp的微生物合成机制被阐明:首先,b.subtilis以d-葡萄糖为底物,经糖酵解途径生成丙酮酸,后者在α-乙酰乳酸合酶及α-乙酰乳酸脱羧酶催化作用下生成乙偶姻;同时,原材料蛋白在蛋白质水解酶作用下生成氨基酸,后者继而生成氨,或者在铵盐存在的情况下,乙偶姻与氨/铵可以通过非酶促反应生成2-氨基-3-丁酮,继而2-氨基-3-丁酮经过脱水缩合与氧化反应生成ttmp。
然而,ttmp和2,5-dmp的微生物合成途径完全不同,无法按照已有的ttmp合成途径解析确定2,5-dmp合成过程。
综上,目前市场上2,5-dmp的生产方法主要通过化学方法合成,但化学法合成存在缺陷。虽然也有微生物菌株能够合成潜在有价值的风味化合物,但是其产量往往很低;而且由于缺乏关于生化途径,酶和代谢调节的知识,使得利用生物技术生产风味化合物的发展受到阻碍。
技术实现要素:
为了解决上述至少一个问题,本发明提供了一种催化合成2,5-二甲基吡嗪的重组菌。该菌可以利用l-苏氨酸高产2,5-dmp的菌株,以5.83g/l的l-苏氨酸为底物发酵24h的产量可达616.04mg/l,生产强度可达25.67mg/(l·h),转化率可达10.6%;与野生型菌株相比,该菌株的产量提高了22.5倍,实现了2,5-dmp高效的生物转化。
本发明的第一个目的是提供一种催化合成2,5-二甲基吡嗪的重组菌,所述重组菌重组表达了苏氨酸脱氢酶(tdh)。
在一种实施方式中,所述重组菌是以枯草芽孢杆菌为宿主构建得到的。
在一种实施方式中,所述重组菌是以b.subtilis168为宿主构建得到的
在一种实施方式中,所述苏氨酸脱氢酶为b.subtilis、b.licheniformis、b.amyloliquefaciens、p.putida、e.coli、a.candidus或者a.uvarum来源的苏氨酸脱氢酶。
在一种实施方式中,所述苏氨酸脱氢酶tdh的序列与genebankid分别为np_389581(b.subtilis168)、wp_085959523(b.licheniformisatcc14580)、wp_014470388(b.amyloliquefaciensdsm7)、wp_064301272(p.putida)、np_418073(e.colik-12)、xp_024673913(a.candidus)、xp_025487049(a.uvarum)的序列相同。
在一种实施方式中,所述重组菌是利用pma0911载体表达苏氨酸脱氢酶。
在一种实施方式中,所述表达,是将苏氨酸脱氢酶基因tdh连接到表达载体pma0911上得到重组表达质粒pma0911-tdh,然后将表达质粒pma0911-tdh转入b.subtilis168中进行表达。
在一种实施方式中,所述表达还包括:将苏氨酸脱氢酶与nox进行共表达。可选地,所述共表达,是将nox编码基因nox与tdh编码基因tdh在同一质粒转录表达。
在一种实施方式中,所述共表达,包括:获取nox基因,然后将nox基因连接到酶切后的pma0911-tdh中,连接,得到共表达的重组质粒pma0911-tdh-nox;重组质粒转化宿主细胞,即得到苏氨酸脱氢酶与nox进行共表达的重组菌。
本发明的第二个目的是提供一种生物合成2,5-dmp的方法,所述方法是利用本发明的重组菌作为生产菌株。
在一种实施方式中,所述发酵是利用含有l-苏氨酸的培养基进行发酵生产。
在一种实施方式中,所述发酵是以l-苏氨酸为唯一底物进行发酵生产。
在一种实施方式中,所述发酵使用的培养基为含有l-苏氨酸的lbt液体培养基。可选地,所述lbt液体培养基中,含有蛋白胨10.0g/l、酵母粉5.0g/l、氯化钠10.0g/l、一定量l-苏氨酸。
本发明的优点和效果:
本发明构建了可以利用l-苏氨酸高产2,5-dmp的菌株,该菌株重组表达了苏氨酸脱氢酶(tdh)。优选地,共表达nox和e.coli来源的苏氨酸脱氢酶的重组枯草芽孢杆菌,在以5.83g/l的l-苏氨酸为底物发酵24h时,产量可达616.04mg/l,生产强度可达25.67mg/(l·h),转化率可达10.6%;该菌株与野生型菌株相比,产量提高了22.5倍,生产强度和转化率以大幅提升,实现了2,5-dmp高效的生物转化。
附图说明
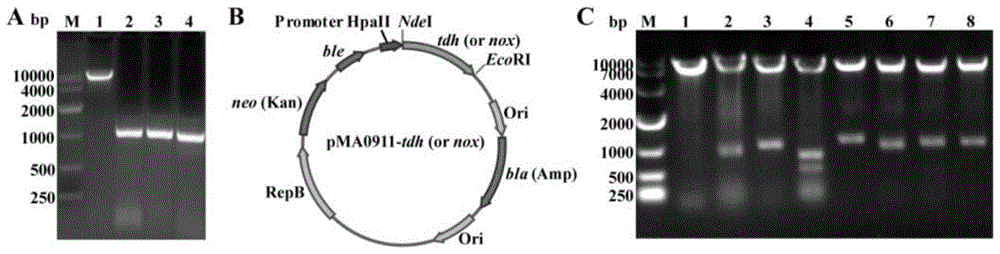
图1tdh表达载体pma0911-tdh的构建与酶切验证;其中,a:质粒pma0911的酶切与不同菌株的tdh编码基因tdh的pcr扩增,m:marker,1:pma0911,2:tdh(b.subtilis168),3:tdh(b.licheniformisatcc14580),4:tdh(b.amyloliquefaciensdsm7);b:重组质粒pma0911-tdh或pma0911-nox示意图;c:重组质粒pma0911-tdh的酶切验证,m:marker,1:pma0911,2:pma0911-tdh(b.s),3:pma0911-tdh(b.l),4:pma0911-tdh(b.a),5:pma0911-tdh(p.p),6:pma0911-tdh(e.c),7:pma0911-tdh(a.c),8:pma0911-tdh(a.u)。
图2基因工程菌株b.subtilis168/pma0911-tdh菌液pcr验证;其中,m:marker,1:b.subtilis168/pma0911,2:b.subtilis168/pma0911-tdh(b.s),3:b.subtilis168/pma0911-tdh(b.l),4:b.subtilis168/pma0911-tdh(b.a),5:b.subtilis168/pma0911-tdh(p.p),6:b.subtilis168/pma0911-tdh(e.c),7:b.subtilis168/pma0911-tdh(a.c),8:b.subtilis168/pma0911-tdh(a.u)。
图3基因工程菌株b.subtilis168/pma0911-tdh利用l-苏氨酸生物合成2,5-dmp;其中,control:b.subtilis168/pma0911,tdh(b.s):b.subtilis168/pma0911-tdh(b.s),tdh(b.l):b.subtilis168/pma0911-tdh(b.l),tdh(b.a):b.subtilis168/pma0911-tdh(b.a),tdh(p.p):b.subtilis168/pma0911-tdh(p.p),tdh(e.c):b.subtilis168/pma0911-tdh(e.c),tdh(a.c):b.subtilis168/pma0911-tdh(a.c),tdh(a.u):b.subtilis168/pma0911-tdh(a.u),***:差异显著(p≤0.001),**:差异显著(p≤0.01),*:差异显著(p≤0.05)。
图4基因工程菌株中tdh的sds-page分析图谱;其中,m:marker,1:b.subtilis168/pma0911,2:b.subtilis168/pma0911-tdh(b.s),3:b.subtilis168/pma0911-tdh(b.l),4:b.subtilis168/pma0911-tdh(b.a),5:b.subtilis168/pma0911-tdh(p.p),6:b.subtilis168/pma0911-tdh(e.c),7:b.subtilis168/pma0911-tdh(a.c),8:b.subtilis168/pma0911-tdh(a.u)。
图5tdh和nox共表达质粒pma0911-tdh(e.c)-nox的构建与酶切验证;其中:a:质粒pma0911-tdh(e.c)的酶切与目的基因nox的pcr扩增,m:marker,1:pma0911-tdh(e.c),2:nox;b:重组质粒pma0911-tdh(e.c)-nox示意图;c:重组质粒pma0911-nox和pma0911-tdh(e.c)-nox的酶切验证,m:marker,1:pma0911-nox,2:pma0911-tdh(e.c)-nox。
图6基因工程菌株b.subtilis168/pma0911-nox和b.subtilis168/pma0911-tdh(e.c)-nox菌液pcr验证。
图7基因工程菌株b.subtilis168/pma0911-nox和b.subtilis168/pma0911-tdh(e.c)-nox利用l-苏氨酸生物合成2,5-dmp;其中,tdh(e.c):b.subtilis168/pma0911-tdh(e.c),nox:b.subtilis168/pma0911-nox,tdh(e.c)-nox:b.subtilis168/pma0911-tdh(e.c)-nox,**:差异显著(p≤0.01),*:差异显著(p≤0.05)。
图8表达sds-page分析图谱;其中,m:marker,1:b.subtilis168/pma0911,2:b.subtilis168/pma0911-nox,3:b.subtilis168/pma0911-tdh(e.c)-nox。
图9根据2,5-dmp溶液浓度与峰面积的变化拟合的2,5-dmp标准曲线。
图10利用uplc定量检测发酵液中2,5-dmp的色谱图。
具体实施方案
1、2,5-dmp的检测:采用超高效液相色谱(uplc)对2,5-dmp进行定量检测,具体条件如下:
色谱柱watersbehc18(100mm×2.1mm,1.7μmparticle)对样品中的2,5-dmp进行液相分离。流动相a为0.1%甲酸溶液(如无特殊说明,涉及到的百分数为体积分数),流动相b为色谱级甲醇。流动相洗脱梯度:初始,31%b;0-3min,31%-69%b;3-10min,31%b;运行时间为10min。流速为0.20ml·min-1,紫外检测波长为275nm。上样量1μl。
2、l-苏氨酸的定量检测:
采用opa柱前在线衍生-agilent1200高效液相色谱(highperformanceliquidchromatography,hplc)对l-苏氨酸进行定量检测。流动相a为乙酸钠缓冲液(55mmol·l-1,ph7.2),流动相b为乙酸钠缓冲液(275mmol·l-1,ph7.2)/色谱级甲醇/色谱级乙腈=1/2/2(v/v/v)。流动相梯度变化为:初始,8%b;0-23min,8%-52.3%b;23-23.5min,52.3%-100%b;23.5-26.5min,100%b;26.5-28min,100%-8%b;运行时间为30min。流速为1ml·min-1,色谱柱为agilentsb-c18(250mm×4.6mm,5μmparticle),采用紫外检测器,检测波长为338nm。进样器程序(opa自动衍生):从样品瓶1吸取5μl硼酸,从样品瓶2吸取0μl水(洗针),从设定位置吸取1μl样品,从样品瓶2吸取0μl水(洗针),混合3次,从样品瓶3吸取1μlopa,从样品瓶2吸取0μl水(洗针),混合15次,从样品瓶4吸取33μl水,混合5次,进样10μl。
3、lb基本培养基(g·l-1):蛋白胨10.0,酵母粉5.0,氯化钠10.0,固体培养基添加2%的琼脂粉,121℃灭菌20min。根据需要添加抗性,氨苄青霉素100μg·ml-1、硫酸卡那霉素50μg·ml-1、壮观霉素100μg·ml-1。
4、lbt培养基:lb培养基中添加5g·l-1的l-苏氨酸。配制浓度浓缩1.11倍的lb基本培养基,按所需体积的9/10进行分装,121℃灭菌20min。配制50g·l-1的l-苏氨酸水溶液,采用灭菌的0.22μm水系针头式滤器过滤除菌,按所需体积的1/10添加至灭菌的浓缩1.11倍的lb培养基中。
5、基因或引物说明
表1本发明涉及的基因
表2相关引物
注:下划线标注部分为限制性内切酶识别序列;tdh,l-苏氨酸脱氢酶编码基因;nox,nadh氧化酶编码基因。
下面是对本发明进行具体描述。
实施例1:以l-苏氨酸为发酵底物的产2,5-dmp菌株的构建及发酵
1、不同来源的tdh表达载体的构建
本实施例以b.subtilis168为出发菌种,首先选取不同种属来源的tdh,并分别外源表达,通过检测2,5-dmp产量,来筛选具有催化优势的tdh,并获得2,5-dmp增产菌种。
通过在ncbi查找,确定了7种不同种属来源的tdh编码基因tdh(表1):b.subtilis168(np_389581)、b.licheniformisatcc14580(wp_085959523)、b.amyloliquefaciensdsm7(wp_014470388)、p.putida(wp_064301272)、e.colik-12(np_418073)、a.candidus(xp_024673913)、a.uvarums(xp_025487049)。
分别提取b.subtilis168、b.licheniformisatcc14580、b.amyloliquefaciensdsm7全基因组,并以此为模板分别进行tdh编码基因tdh的克隆。选用b.subtilis外源表达质粒pma0911为载体,双酶切(ndei/ecori)后的质粒与pcr扩增的tdh编码基因tdh(图1a)经胶回收纯化后进行连接,构建tdh表达重组质粒(图1b)。另外,因p.putida、e.colik-12、a.candidus、a.uvarums四株菌株的基因组难以获得,因此对上述4种菌株的tdh编码基因tdh密码子优化后进行全基因合成,并构建到表达载体pma0911上。
对构建的重组质粒(图1b)进行双酶切(ndei/ecori)验证,酶切结果如图1c所示,各酶切片段长度与表3中一致,表明tdh编码基因tdh成功与表达载体连接。为了进一步确定基因序列的正确性,将酶切结果正确的重组质粒进行测序,测序结果与原有序列进行比对,确定序列一致,表明各个来源的tdh编码基因tdh未发生突变,不同来源的tdh表达载体pma0911-tdh构建成功。
表3重组质粒的酶切片段长度
2、tdh表达菌株的构建
将验证正确的7种不同种属来源的tdh表达载体pma0911-tdh分别转入b.subtilis168感受态中,对阳性转化子进行菌液pcr验证,结果如图2所示,与理论上的pcr产物长度一致(表4),表明以b.subtilis168为宿主,7种不同种属来源的tdh外源表达菌株构建成功。将含有来源于b.subtilis168、b.licheniformisatcc14580、b.amyloliquefaciensdsm7、p.putida、e.colik-12、a.candidus、a.uvarums来源的tdh的重组菌,分别命名为,b.subtilis168/pma0911-tdh(b.s)、b.subtilis168/pma0911-tdh(b.l)、b.subtilis168/pma0911-tdh(b.a)、b.subtilis168/pma0911-tdh(p.p)、b.subtilis168/pma0911-tdh(e.c)、b.subtilis168/pma0911-tdh(a.c)、b.subtilis168/pma0911-tdh(a.u)。
表4基因工程菌株菌液pcr产物长度
3、重组菌的发酵验证
对构建成功的工程菌株进行发酵验证,在含有5.83g/l的l-苏氨酸的lbt液体培养基中发酵24h,测定细胞生长量(od600)、生成的2,5-dmp和消耗的l-苏氨酸。
将菌株在lb平板上进行活化,37℃过夜培养,挑取单菌落至lb试管中,37℃、200rpm摇床培养12h获取种子液。抗性为硫酸卡那霉素(50μg·ml-1),按1%接种量转接至50mllbt培养基(250ml摇瓶,50μg·ml-1硫酸卡那霉素)进行培养,24h后取样。样品处理:取50μl样品进行4倍稀释,测定od600nm,剩余样品进行离心,12000rpm,5min,取上清液,-20℃保存等待检测。
结果如图3所示,各基因工程菌的od600无明显差别(图3b),不同种属来源的tdh外源表达菌株积累2,5-dmp能力具有显著性差异,来源于e.colik-12的tdh外源表达菌株b.subtilis168/pma0911-tdh(e.c)利用l-苏氨酸消耗量最高,积累2,5-dmp浓度最高,发酵24h后,2,5-dmp的积累量高达527.43mg/l,21.98mg/(l·h),转化率可达9%。此外,来源于b.subtilis168、b.licheniformisatcc14580、b.amyloliquefaciensdsm7的tdh外源表达菌株发酵24h后,2,5-dmp的积累量分别约300mg/l、350mg/l、360mg/l。而来源于p.putida、a.candidus与a.uvarums的tdh外源表达菌株2,5-dmp积累量并无明显提高。
4、tdh的表达情况的sds-page分析
对各基因工程菌株中tdh的表达情况进行了sds-page分析。如图4所示,来源于b.subtilis168、b.licheniformisatcc14580、b.amyloliquefaciensdsm7和e.colik-12的tdh成功表达,该结果与基因工程菌株b.subtilis168/pma0911-tdh(b.s)、b.subtilis168/pma0911-tdh(b.l)、b.subtilis168/pma0911-tdh(b.a)和b.subtilis168/pma0911-tdh(e.c)利用l-苏氨酸产2,5-dmp的能力与对照菌株(b.subtilis168/pma0911)相比显著提高的实验结果相一致(图3)。然而,如图4所示,基因工程菌株b.subtilis168/pma0911-tdh(p.p)、b.subtilis168/pma0911-tdh(a.c)和b.subtilis168/pma0911-tdh(a.u)的细胞破碎液并未显示明显的tdh目的蛋白条带,与上述菌种利用l-苏氨酸产2,5-dmp的产量与对照菌株相比变化不大的实验结果相一致(图3)。虽进行密码子优化,但菌种p.putida、a.candidus及a.uvarums与宿主b.subtilis亲缘关系相距较远,可能导致目的蛋白tdh表达量较低甚至没有表达。
实施例2:tdh与nox共表达的重组菌的构建
在实施例2中,获得了一株可以利用l-苏氨酸高产2,5-dmp的基因工程菌株b.subtilis168/pma0911-tdh(e.c)。本实施例,在b.subtilis168/pma0911-tdh(e.c)的基础上进一步改进。本实施例实现了tdh与nox的共表达,通过外源表达nadh氧化酶nox,实现nad+的快速再生。
1、tdh与nox共表达载体的构建
本实施例将nox编码基因nox(表1)与tdh编码基因tdh在同一质粒转录表达。首先通过全基因合成获取携带nox转录基因nox的质粒pma0911-nox(图1b),并以此为模板,扩增出携带质粒pma0911同源臂的目的基因nox(图5a),将基因nox构建到酶切质粒pma0911-tdh(e.c)(图5a),重组质粒pma0911-tdh(e.c)-nox示意图如图5b所示。
具体如下:
通过基因合成获取重组质粒pma0911-nox,以此质粒为模板,设计引物nox-f/r,通过pcr扩增获得相应基因nox。pcr反应体系:2×primerstarmaxdnapolymerase25μl,引物f和r(20μm)各1μl,基因组dna模板2μl,无菌水21μl。pcr反应条件同98℃,3min;[98℃,30s;55℃,15s;72℃,(片段总长/1000)min],30个循环;72℃,10min。pcr扩增产物和酶切质粒pma0911-tdh(e.c)(ecori/bamhi,37℃,30min)经胶回收纯化后使用in-fusionhdcloningkit进行连接(50℃,15min)。将连接产物转入e.colidh5α感受态细胞。通过氨苄青霉素抗性(100μg·ml-1)筛选阳性克隆子,提取质粒,通过酶切(ndei/ecori/bamhi)和测序验证其正确性。
将构建好的重组质粒转入e.colidh5α感受态中进行克隆,对重组质粒进行酶切验证,结果如图5c所示,各酶切片段长度与表3中一致。为了进一步确定基因序列的正确性,将酶切结果正确的重组质粒进行测序,经过比对进一步确定了序列一致且读码框正确。以上结果说明双基因共转录质粒pma0911-tdh(e.c)-nox构建成功。
2、tdh与nox共表达菌株的构建
将重组质粒pma0911-nox和pma0911-tdh(e.c)-nox分别转入b.subtilis168感受态中,对阳性转化子进行菌液pcr验证,所用验证引物为pma0911-f/r,结果如图6所示,各菌株的菌液pcr产物长度如表4所示,经比对可以确定nox表达菌株和tdh与nox共表达菌株构建成功。得到b.subtilis168/pma0911-tdh(e.c)-nox和b.subtilis168/pma0911-tdh(e.c)。
3、tdh与nox共表达菌株的发酵验证
对构建成功的基因工程菌株进行发酵验证,在含有苏氨酸终浓度为5.83g/l的l-苏氨酸的lbt液体培养基(蛋白胨10.0g/l,酵母粉5.0g/l,氯化钠10.0g/l,l-苏氨酸5.83g/l)中,37℃,200rpm下发酵24h,对细胞生长量(od600)、生成的2,5-dmp以及消耗的l-苏氨酸进行测定,结果如图7所示。
从图7可以看出,虽然菌株b.subtilis168/pma0911-tdh(e.c)-nox的细胞生长量略低于菌株b.subtilis168/pma0911-tdh(e.c)(图7b),但对于2,5-dmp的积累量却高于菌株b.subtilis168/pma0911-tdh(e.c),且具有显著性差异(图7a),而两菌株对于l-苏氨酸的消耗量并不具有显著性差异(图7b)。因此,nox的存在对于过表达的tdh催化l-苏氨酸最终生成2,5-dmp有促进作用。
菌株b.subtilis168/pma0911-tdh(e.c)-nox利用5.83g/l的l-苏氨酸发酵24h,最终2,5-dmp的产量可达616.04mg/l,与对照菌株b.subtilis168/pma0911相比(图3a),产量提高了22.5倍,与基因工程菌株b.subtilis168/pma0911-tdh(e.c)相比,产量增加了88.61mg/l。
4、tdh和nox共表达情况的sds-page分析
对基因工程菌株b.subtilis168/pma0911-nox中nox的表达情况和b.subtilis168/pma0911-tdh(e.c)-nox中tdh和nox共表达情况进行sds-page分析。如图8所示,基因工程菌株b.subtilis168/pma0911-nox的破碎液中显示出nox外源表达目的条带。基因工程菌株b.subtilis168/pma0911-tdh(e.c)-nox的细胞破碎液中,均显示出tdh及nox外源表达目的条带,表明该菌株中tdh及nox均表达,与基因工程菌株b.subtilis168/pma0911-tdh(e.c)-nox具有更高的2,5-dmp产量结果相一致(7)。
基因工程菌株b.subtilis168/pma0911-nox利用l-苏氨酸产2,5-dmp的能力远低于b.subtilis168/pma0911-tdh(e.c)-nox(图7a),分析其原因,在tdh表达水平不高的情况下,细胞自身的还原力水平足以维持菌株自身的tdh催化所需要的还原力,也就是说,即使还原力水平提高,但是在催化过程中真正需要的还原力水平并没有提高,因此,该菌株b.subtilis168/pma0911-nox对于2,5-dmp的积累量接近于对照菌株b.subtilis168/pma0911(图3a)。
实施例3:利用uplc定量检测发酵液中2,5-dmp
2,5-dmp的定量分析:采用超高效液相色谱(uplc)对2,5-dmp进行定量检测,具体条件如下:色谱柱watersbehc18(100mm×2.1mm,1.7μmparticle)对样品中的2,5-dmp进行液相分离。流动相a为0.1%甲酸溶液(如无特殊说明,涉及到的百分数为体积分数),流动相b为色谱级甲醇。流动相洗脱梯度:初始,31%b;0-3min,31%-69%b;3-10min,31%b;运行时间为10min。流速为0.20ml·min-1,紫外检测波长为275nm。上样量1μl。
配制不同浓度梯度的2,5-dmp标准品溶液,根据浓度与峰面积的变化,利用origin软件拟合出2,5-dmp标准曲线,进而根据待测样品中相应出峰时间的峰面积计算出2,5-dmp的含量。结果如图9所示,利用此方法所获得的标准曲线为y=0.00004312*x-1.19126(其中,y代表2,5-dmp浓度,单位是mg/l,x代表峰面积,单位是μv*s,r2为1),标准品检测的线性范围:27mg/l-1725mg/l。
以如下发酵液为例:在添加1g/l的l-苏氨酸的lb培养基(lb的配方:5g/l酵母粉,10g/l蛋白胨,10g/l氯化钠)中,37℃、200rpm培养b.subtilis168至2d,离心取发酵上清液。结果如图10所示,可以明显看出,在复杂的发酵液体系中,2,5-dmp可以实现有效分离。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
sequencelisting
<110>江南大学
<120>一种催化合成2,5-二甲基吡嗪的重组菌
<160>14
<170>patentinversion3.3
<210>1
<211>33
<212>dna
<213>人工序列
<400>1
cgccggatccatgcagagtggaaagatgaaagc33
<210>2
<211>36
<212>dna
<213>人工序列
<400>2
cgccaagcttttatggaattaaaattacttttccgc36
<210>3
<211>35
<212>dna
<213>人工序列
<400>3
ggagcgatttacatatgatgcagagtggaaagatg35
<210>4
<211>41
<212>dna
<213>人工序列
<400>4
cgactctagaggatccttatggaattaaaattacttttccg41
<210>5
<211>35
<212>dna
<213>人工序列
<400>5
ggagcgatttacatatgatgcagagtggaaagatg35
<210>6
<211>41
<212>dna
<213>人工序列
<400>6
gcttgtcgacgaattcttatggaattaaaattacttttccg41
<210>7
<211>40
<212>dna
<213>人工序列
<400>7
ggagcgatttacatatgatgttgggaggaaagatgaaagc40
<210>8
<211>41
<212>dna
<213>人工序列
<400>8
gcttgtcgacgaattcttatggaatcagaatgactttgccg41
<210>9
<211>42
<212>dna
<213>人工序列
<400>9
ggagcgatttacatatgatgttggacggaaatatgaaagcgc42
<210>10
<211>41
<212>dna
<213>人工序列
<400>10
gcttgtcgacgaattcctacggtatcagtacgactttgccg41
<210>11
<211>37
<212>dna
<213>人工序列
<400>11
ctgggattaagaattcatgaaagttacggtggtcggc37
<210>12
<211>37
<212>dna
<213>人工序列
<400>12
cgactctagaggatcctcacgcattaacgctttgtgc37
<210>13
<211>20
<212>dna
<213>人工序列
<400>13
tgctgaataaaagatacgag20
<210>14
<211>20
<212>dna
<213>人工序列
<400>14
ttcaccgtcatcaccgaaac20
- 还没有人留言评论。精彩留言会获得点赞!