多替拉韦钠的晶型及其制备方法与流程
技术领域:
:本发明涉及多替拉韦钠的新晶型以及制备方法。
背景技术:
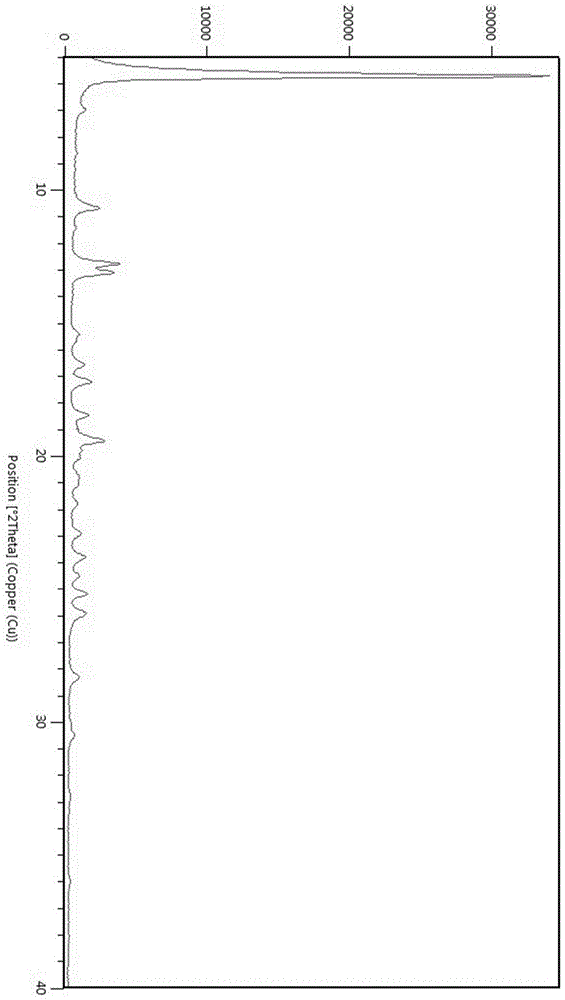
::hiv整合酶是一种在hiv复制中起重要作用的酶,是hiv感染的病原,如hiv逆转录酶和hiv蛋白酶。这种酶有助于将病毒dna整合到宿主细胞的dna中,抑制这一过程有助于抑制艾滋病毒感染。多替拉韦钠(式i)是由viivhlthcare研发的新一代hiv整合酶抑制剂,2013年8月12日获美国fda批准上市,用于hiv感染治疗,其单方片剂商品名“tivicay”。多替拉韦钠与其他抗逆转录病毒药物联合使用,可用于治疗感染hiv的体重至少40公斤的成人和12岁及12岁以上儿童,也可用于治疗未经治疗或治疗过的hiv感染患者,包括接受过其他整合酶链转移抑制剂治疗的成人。在两项大型研究中评估了治疗幼稚患者的多替拉韦,其治疗方案可有效降低病毒载量(即hiv-1rna<50copies/ml)。在其中一项研究中,患者接受了多替拉韦(50毫克一天一次)或多替拉韦(400毫克一天两次)联合双核苷逆转录酶抑制剂(nrti)治疗(阿巴卡韦+拉米夫定或恩曲他滨+替诺福韦),治疗48周后,采用多替拉韦和raltegravir方案的hivrna<50copies/ml患者的百分比分别为88%和86%;在这些研究中的第二个研究中,患者使用多替拉韦钠(50毫克一天一次)联合阿巴卡韦+拉米夫定或依法韦恩茨+恩曲他滨+替诺福韦治疗,48周后,两种方案治疗的hivrna<50copies/ml患者百分比分别为88%和81%。固体原料药在制备过程中,如果采用不同重结晶溶剂和精制工艺,可能会使同一药物的晶体具有两种或两种以上的空间结构和晶胞常数,即产生药物的多晶型现象。固体口服制剂原料药的多晶型问题可能会直接影响到药物稳定性、溶解性以及药物的吸收和疗效,进而导致药物临床疗效的差异。因此,研究固体口服制剂原料药的多晶型问题就显得非常重要。目前多篇专利文献报道了多替拉韦钠的水合物、无定型、溶剂化合物(n-甲基吡咯烷酮、正丁醇,1,2-丙二醇)、钙盐、钾盐等固体形式,但仍需要开发新的多替拉韦钠晶型来满足更高的制剂要求,因此,继续开发一种稳定性好,更适于制剂过程的多替拉韦钠晶型,是十分必要的。技术实现要素::本发明的目的在于提供稳定性好,纯度高,更适于制剂过程的多替拉韦钠新晶型,其命名为α晶型与β晶型以及两种新晶型的制备方法。本发明提供一种多替拉韦钠(式i)的α晶型,其特征在于,使用cu-kα辐射,该晶体的粉末x-射线衍射图在2θ为5.70±0.2°、6.98±0.2°、10.67±0.2°、12.77±0.2°、13.09±0.2°、19.41±0.2°、23.80±0.2°有特征吸收峰。所述的替拉韦钠α晶型,使用cu-kα辐射,该晶体的粉末x-射线衍射图在2θ为5.70±0.2°、6.98±0.2°、10.67±0.2°、12.77±0.2°、13.09±0.2°、15.40±0.2°、16.56±0.2°、17.20±0.2°、18.45±0.2°、19.41±0.2°、22.89±0.2°、23.80±0.2°、24.50±0.2°、25.17±0.2°、25.92±0.2°和28.32±0.2°有特征吸收峰。所述的替拉韦钠α晶型,其粉末x-射线衍射图中的峰位和峰强如表1所示:表1所述的替拉韦钠α晶型,其粉末x-射线衍射图基本如图1所示。所述的多替拉韦钠的晶型α,其热重分析(tga)谱图中,当加热至100~170℃时,产生失重,失重比例为13.6%,表明多替拉韦钠与正丁醇的摩尔比为1:1;当加热至350℃以上时,开始熔融分解。所述的多替拉韦钠的晶型α,为多替拉韦钠与一分子正丁醇的溶剂合物。差示扫描量热法(dsc)分析在加热至峰值温度为156℃时,出现第一个吸热峰;继续加热至273℃时,出现放热峰;当温度在350℃以上时,开始熔融分解,与热重分析谱图吻合。所述的多替拉韦钠的晶型α,其ir光谱在波数为3272、3070、2955、2930、2873、1650、1628、1529、1455和1318cm-1处有特征峰。本发明提供一种多替拉韦钠(式i)的β晶型,其特征在于,使用cu-kα辐射,该晶体的粉末x-射线衍射图在2θ为6.31±0.2°、7.87±0.2°、12.59±0.2°、14.29±0.2°、16.53±0.2°、18.80±0.2°、24.64±0.2°有特征吸收峰。所述的替拉韦钠β晶型,使用cu-kα辐射,粉末x-射线衍射图在2θ为6.31±0.2°、7.87±0.2°、9.25±0.2°、12.59±0.2°、14.29±0.2°、16.53±0.2°、18.12±0.2°、18.80±0.2°、22.39±0.2°、24.64±0.2°、27.01±0.2°和28.01±0.2有特征吸收峰。所述的替拉韦钠β晶型,其粉末x-射线衍射图中的峰位和峰强如表2所示:表2所述的替拉韦钠β晶型,其粉末x-射线衍射图基本如图5所示。所述的多替拉韦钠的晶型β,其热重分析(tga)谱图中,加热至200℃时,无失重现象当加热至340℃以上时,开始熔融分解。所述的多替拉韦钠的晶型β,其差示扫描量热法(dsc)分析在在加热至峰值温度为269℃时,出现放热峰;当温度在340℃以上时,开始熔融分解,与热重吻合。所述的多替拉韦钠的晶型β,为多替拉韦钠无水的固体形式。所述的多替拉韦钠的晶型β,其ir光谱在波数为3066、2973、2862、1640、1591、1533、1507、1428、1403和1319cm-1处有特征峰。本发明提供一种多替拉韦钠α晶型的制备方法,其特征在于,包括如下步骤:(1)将多替拉韦溶于有机混合溶剂,加热,所述有机混合溶剂为正丁醇和甲醇溶剂;(2)溶液热滤,加入甲醇洗涤,再次加热回流;(3)向多替拉韦溶液中缓慢加入naoh的甲醇溶液;(4)在20~40℃反应3~5h,抽滤,真空干燥步骤(1)中所述溶剂正丁醇和甲醇溶剂的体积比为0.5:1~5:1,更优选为2:1~3:1;步骤(1)中,所述溶解温度优选为30~80℃,更优选为60~70℃;步骤(2)中,所述加入甲醇溶液的用量为步骤(1)中加入量的一半;步骤(3)中,所述naoh的甲醇溶液优选浓度为10mg/ml;步骤(3)中,多替拉韦溶液温度优选为10~50℃加入naoh的甲醇溶液,更优选为30℃;步骤(4)中,所述反应温度优选为10~40℃,更优选为20~30℃;步骤(4)中,所述反应时间优选为3~5h,更优选为4h。步骤(4)中,所述干燥温度和干燥时间优选为20~40℃、10~30h,更优选为30℃、20h。本发明提供一种多替拉韦钠(式i)β晶型的制备方法,其特征在于,包括如下步骤:将多替拉韦钠晶型α于真空干燥或鼓风干燥,干燥条件为90~200℃、15min~60h,则转化为多替拉韦钠晶型β。优选地,在真空干燥箱中,真空干燥温度和时间为90~110℃、40~60h,更优选为100℃、50h。优选地,在鼓风干燥箱中,干燥温度和时间为190~210℃、15~25min,更优选为200℃、20min。附图说明图1为多替拉韦钠晶型α的粉末x-射线衍射图。图2为多替拉韦钠晶型α的dsc和tga图。图3为多替拉韦钠晶型α的扫描电镜图。图4为多替拉韦钠晶型α的ir光谱图。图5为多替拉韦钠晶型β的粉末x-射线衍射图。图6为多替拉韦钠晶型β的dsc和tga图。图7为多替拉韦钠晶型β的扫描电镜图。图8为多替拉韦钠晶型β的ir光谱图。图9为多替拉韦钠晶型α与m2晶型的的粉末x-射线衍射对比图。图10为多替拉韦钠晶型β与m4晶型的的粉末x-射线衍射对比图。具体实施方式下文结合具体实施例对本发明的多晶型及其制备方法做进一步的说明。下面实施例仅为示例性说明和解释本发明,凡是基于本发明上述内容实现的技术均为本发明保护范围内。除非另有说明,以下实施例中使用的原料和溶剂均为市售商品,或者通过已知方法制备。检测仪器:1.粉末x-射线衍射仪:型号:x’pertprompd型panalyticalx-射线粉末衍射仪。测试条件:铜靶,电压为40kv,电流40ma,扫描方式为连续,步长为发射狭缝1/8°,防散射狭缝1/4°,扫描范围3~40°,扫描速度8°/min。2.dsc/tga同步热分析仪型号:1staresystem型dsc/tga同步热分析仪。测试条件:氮气吹扫,升温速率为10℃/min。3.扫描电子显微镜:型号:zeisssigma300型扫描电子显微镜。4.傅里叶变换红外光谱仪(ir)型号:nicoletis50型傅里叶变换红外光谱仪测试条件:扫描范围:4000cm-1~400cm-1;分辨率:4cm-1;扫描次数:16。实施例1多替拉韦钠晶型α的制备称取0.5g多替拉韦于三口烧瓶中,并向其中加入15ml正丁醇和5ml甲醇,升温至80℃溶清,热滤溶液,并用2.5ml热甲醇洗涤,继续加热回流,向体系中半小时内缓慢加入浓度为10mg/ml的氢氧化钠的甲醇溶液5ml,并于20℃搅拌5h,抽滤,收集产物,其pxrd图谱基本为图1所示,收率97%。实施例2多替拉韦钠晶型α的制备称取1g多替拉韦于三口烧瓶中,并向其中加入30ml正丁醇和10ml甲醇,升温至70℃溶清,热滤溶液,并用5ml热甲醇洗涤,继续加热回流,向体系中半小时内缓慢加入浓度为10mg/ml的氢氧化钠的甲醇溶液10ml,并于20℃搅拌4h,抽滤,收集产物,其pxrd图谱基本为图1所示,收率96%。实施例3多替拉韦钠晶型α的制备称取5g多替拉韦于三口烧瓶中,并向其中加入150ml正丁醇和50ml甲醇,升温至75℃溶清,热滤溶液,并用25ml热甲醇洗涤,继续加热回流,向体系中半小时内缓慢加入浓度为10mg/ml的氢氧化钠的甲醇溶液50ml,并于25℃搅拌4h,抽滤,收集产物,其pxrd图谱基本为图1所示,收率96%。实施例4多替拉韦钠晶型β的制备将制备例3中多替拉韦钠晶型α在200℃条件下加热20min,得到晶型β,收集产物,其pxrd图谱基本为图5所示,收率99%。实施例5多替拉韦钠晶型β的制备将制备例3中多替拉韦钠晶型α在100℃条件下加热50h,得到晶型β,收集产物,收率99%,pxrd图谱基本为图5所示。实施例6多替拉韦钠晶型β的制备将制备例3中多替拉韦钠晶型α在90℃条件下加热60h,得到晶型β,收集产物,收率99%,其pxrd图谱基本为图5所示。对比例1参照wo2015118460专利,制备得到m2晶型:0.5g多替拉韦溶于15ml正丁醇和7.5ml甲醇,升温至80℃溶清,热滤溶液,在25~30℃加入少量正丁醇洗涤,随后并向其中加入0.25n的氢氧化钠的甲醇溶液5ml,并于30℃搅拌5h,抽滤,干燥,得到多替拉韦钠m2晶型。与本发明晶型α的2θ数据对比如表3所示,两者的粉末x-射线衍射对比图谱如图9所示。表3本申请晶型αwo2015118460中m2晶型位移变化5.70-6.986.23+0.75-7.87-9.8010.67-12.7712.46,12.88+0.31,-0.1113.09-18.4518.42,18.74+0.03,-0.2919.4119.06,19.54+0.35,-0.13-21.3423.8023.84-0.04对比例2参照wo2015118460专利,制备得到m4晶型:1g多替拉韦钠m2晶型,于90℃真空干燥中干燥30小时,得到多替拉韦钠m4晶型。与本发明晶型β的2θ数据对比如表4所示,两者的粉末x-射线衍射对比图谱如图10所示。表4当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1